")
Back to Journals » International Journal of Nanomedicine » Volume 12
The acute pulmonary and thrombotic effects of cerium oxide nanoparticles after intratracheal instillation in mice
Authors Nemmar A, Al-Salam S, Beegam S, Yuvaraju P, Ali BH
Received 9 November 2016
Accepted for publication 2 December 2016
Published 10 April 2017 Volume 2017:12 Pages 2913—2922
DOI https://doi.org/10.2147/IJN.S127180
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Abderrahim Nemmar,1 Suhail Al-Salam,2 Sumaya Beegam,1 Priya Yuvaraju,2 Badreldin H Ali3
1Department of Physiology, 2Department of Pathology, College of Medicine and Health Sciences, United Arab Emirates University, Al Ain, UAE; 3Department of Pharmacology and Clinical Pharmacy, College of Medicine & Health Sciences, Sultan Qaboos University, Muscat, Al-Khod, Sultanate of Oman
Abstract: Cerium oxide nanoparticles (CeO2 NPs), used as a diesel fuel catalyst, can be emitted into the ambient air, resulting in exposure to humans by inhalation. Recent studies have reported the development of lung toxicity after pulmonary exposure to CeO2 NPs. However, little is known about the possible thrombotic effects of these NPs. The present study investigated the acute (24 hours) effect of intratracheal (IT) instillation of either CeO2 NPs (0.1 or 0.5 mg/kg) or saline (control) on pulmonary and systemic inflammation and oxidative stress and thrombosis in mice. CeO2 NPs induced a significant increase of neutrophils into the bronchoalveolar lavage (BAL) fluid with an elevation of tumor necrosis factor α (TNFα) and a decrease in the activity of the antioxidant catalase. Lung sections of mice exposed to CeO2 NPs showed a dose-dependent infiltration of inflammatory cells consisting of macrophages and neutrophils. Similarly, the plasma levels of C-reactive protein and TNFα were significantly increased, whereas the activities of catalase and total antioxidant were significantly decreased. Interestingly, CeO2 NPs significantly and dose dependently induced a shortening of the thrombotic occlusion time in pial arterioles and venules. Moreover, the plasma concentrations of fibrinogen and plasminogen activator inhibitor-1 were significantly elevated by CeO2 NPs. The direct addition of CeO2 NPs (1, 5, or 25 µg/mL) to mouse whole blood, collected from the inferior vena cava, in vitro neither caused significant platelet aggregation nor affected prothrombin time or partial thromboplastin time, suggesting that the thrombotic events observed in vivo may have resulted from systemic inflammation and/or oxidative stress induced by CeO2 NPs. This study concludes that acute pulmonary exposure to CeO2 NPs induces pulmonary and systemic inflammation and oxidative stress and promotes thrombosis in vivo.
Keywords: cerium oxide nanoparticles, coagulation, lung inflammation, oxidative stress, thrombosis, platelet aggregation, bronchoalveolar lavage
Introduction
The use of nanotechnology has resulted in some novel and interesting medical and/or industrial uses, but also probable danger for human and environmental health.1–3 Accidental or involuntary contact during production or use of nanoparticles (NPs) may occur via various routes such as inhalation, ingestion, or skin penetration.3,4 Actually, the potential of NPs to pass across the alveolar–capillary barrier and to reach the circulating blood and thus have access to other organs is a reason for disquiet.3,5 It has been shown that NPs may penetrate the cells and be more biologically active than microsized particles because of their small size and large surface to volume ratio.3,5,6
Cerium oxide (CeO2) is an important nanomaterial with a vast array of applications, including use in solar and fuel cells, gas sensors, and oxygen pumps and use as a fuel additive.7,8 CeO2 NPs have been widely used as a diesel fuel additive since 1999, because of their enhanced fuel-burning efficiency and decreased levels of greenhouse gases and particle numbers in vehicle exhaust.7,9–11 However, it has been shown that, along with diesel exhaust particles (DEPs), CeO2 NPs are emitted in diesel exhaust, resulting in exposure to humans by inhalation.8
Experimental studies in rodents have shown that acute (1 day) and subacute (28 days) intratracheal (IT) instillation of CeO2 NPs in rats induced lung inflammation, alveolar macrophage toxicity, air–blood barrier damage, and phospholipidosis.12,13 It has also been demonstrated that either acute inhalation or IT instillation of CeO2 NPs in mice caused pulmonary inflammation and small granulomas.14,15 More recently, it has been shown that CeO2 NPs induce pulmonary fibrosis and that the amorphous silica coating of CeO2 NPs reduced the inflammation, phospholipidosis, and fibrosis.16,17
It is well-established that particulate air pollution and some engineered NPs affect the cardiovascular system either through inflammatory mediators produced in the lungs and released into the circulation or via their ability to cross the alveolar–capillary barrier and reach the cardiovascular system.5,18 Since CeO2 NPs have been shown to cross the alveolar–capillary barrier to reach extrapulmonary sites19,20 and cause lung inflammation,14,15,20 the present study hypothesized that these NPs can plausibly interact with the circulating platelets and vasculature and induce thrombotic events. This has not been investigated before, and, consequently, the aim of this study was to assess the acute (24 hours) effect of CeO2 NPs on pulmonary and systemic inflammation and oxidative stress in mice and to evaluate their effects on coagulation in vivo and in vitro.
Material and methods
Particles
CeO2 NPs, 10%, w/w, in water with an average diameter of ~20 nm, were obtained from Sigma-Aldrich (St Louis, MO, USA). The CeO2 NP samples were diluted in sterile saline (0.9% NaCl). To minimize aggregation, particle suspensions were sonicated for 5 minutes in a Clifton ultrasonic bath (Clifton, NJ, USA) before dilution and IT administration. Particle suspensions were prepared immediately before use and were vigorously vortexed to obtain a well-mixed suspension prior to each instillation. The same particles from the same source were characterized and used recently by Ma et al.12,21
Animals and IT instillation
This project was reviewed and approved by the Institutional Review Board of the United Arab Emirates University, College of Medicine and Health Sciences, and the experiments were performed in accordance with the protocols approved by the Institutional Animal Care and Research Advisory Committee.
Both male and female BALB/c mice (Taconic Farms Inc., Germantown, NY, USA) weighing 23±2 g were housed in light- (12-hour light:12-hour dark cycle) and temperature-controlled (22°C ±1°C) rooms. The animals had free access to tap water and commercial laboratory chow.
Mice were anesthetized with sodium pentobarbital (60 mg/kg, i.p.) and placed supine with extended neck on an angled board. A Becton Dickinson (Sparks, MD, USA) 24-gauge cannula was inserted via the mouth into the trachea. Either CeO2 NP suspensions (0.1 or 0.5 mg/kg) or saline only were instilled IT (0.1 mL) via a sterile syringe, followed by an air bolus of 0.1 mL.
Blood collection and bronchoalveolar lavage (BAL) fluid analysis
Twenty-four hours after the IT administration of either CeO2 NPs or saline, the mice were anesthetized as described earlier, and blood was drawn from the inferior vena cava in ethylenediaminetetraacetic acid (4%). The collected blood was centrifuged at 4°C for 15 minutes at 900× g, and the plasma samples were stored at −80°C pending analysis.
Mice were then sacrificed with an overdose of sodium pentobarbital. The trachea was cannulated and the left bronchus was clamped. The right bronchi and right lungs were lavaged three times with 0.7 mL of sterile saline (NaCl 0.9%). The recovered fluid aliquots were pooled. No difference in the volume of collected fluid was observed between the different groups. BAL fluid was centrifuged (1,000× g ×10 minutes, 4°C). Cells were counted in a Thoma hemocytometer after resuspension of the pellets and staining with 1% gentian violet. The cell differentials (n=6–8 per group) were microscopically determined on cytocentrifuge preparations fixed in methanol and stained with Diff Quick (Dade, Brussels, Belgium). The supernatant was stored at −80°C until further analysis.
Histopathology
The unlavaged left lungs obtained from the animals above (n=6–8 per group) were fixed with 10% buffered formalin, excised, washed with ice-cold saline, blotted with filter paper, and weighed. Each left lung was sectioned, put in a cassette, and dehydrated in increasing concentrations of ethanol, cleared with xylene, and embedded in paraffin. Sections of 3 μm were prepared from paraffin blocks and stained with hematoxylin and eosin.22,23 The stained sections were evaluated by using light microscopy by a histopathologist participating in this project.
Determination of levels of tumor necrosis factor-α (TNFα), C-reactive protein (CRP), fibrinogen, and plasminogen activator inhibitor-1 (PAI-1) and activities of catalase and total antioxidant
In BAL fluid supernatant (n=6–8 per group), TNFα level was measured with a commercially available ELISA kit (Duo Set; R & D Systems, Minneapolis, MN, USA), and catalase activity was quantified by using a colorimetric assay kit (Cayman Chemical Company, Ann Arbor, MI, USA).
In plasma (n=6–8 per group), besides measuring TNFα level and catalase activity as described earlier, the levels of CRP (GenWay Biotech, Inc., San Diego, CA, USA), fibrinogen (Molecular Innovation, Southfield, MI, USA), and PAI-1 (Molecular Innovation) were measured by using ELISA kits. The total antioxidant activity was quantified with a colorimetric assay kit (Cayman Chemicals, Ann Arbor, MI, USA).
Experimental pial cerebral arteriole thrombosis model
In a separate experiment (n=6–8 per group), in vivo pial arteriolar and venular thrombogenesis was assessed 24 hours after IT instillation of either CeO2 NPs or saline according to a previously described technique.24–27
In vitro platelet aggregation in mouse whole blood
The platelet aggregation assay in whole blood (n=4–6 per group) was performed as described in previous studies.28,29 After anesthesia, blood from untreated mice was withdrawn from the inferior vena cava and placed in citrate (3.2%), and 0.1 mL aliquots were added to the well of a Merlin coagulometer (MC 1 VET; Merlin, Lemgo, Germany). The blood samples were incubated at 37°C with either saline (control) or CeO2 NPs (1, 5, or 25 μg/mL) for 3 minutes and then stirred for another 3 minutes. At the end of this period, 0.025 mL samples were removed and fixed on ice in 225 mL cellFix (Becton Dickinson). After fixation, single platelets were counted in a VET ABX Micros with a mouse card (ABX, Montpellier, France). The degree of platelet aggregation following CeO2 NP exposure was calculated as a fall in the number of single platelets counted and expressed as a % of control (saline-treated blood).27
In vitro measurement of prothrombin time (PT) and activated partial thromboplastin time (aPTT)
After anesthesia, blood from untreated mice was withdrawn, as described earlier. The PT was measured on freshly collected, platelet-poor plasma with human, relipidated, recombinant thromboplastin (Recombiplastin; Instrumentation Laboratory, Orangeburg, NY, USA) in combination with an MC 1 VET (Merlin). Briefly, the plasma (n=4–6 per group) was incubated for 3 minutes at 37°C. After that, either CeO2 NPs (1, 5, or 25 μg/mL) or saline (control) was added for another 3 minutes, and then PT was measured as previously described.25,30,31 The aPTT was measured with automated aPTT reagent (BioMerieux, Durham, NC, USA) by using an MC 1 VET (Merlin). Briefly, the plasma (n=4–6 per group) was incubated for 3 minutes at 37°C. After that, either CeO2 NPs (1, 5, or 25 μg/mL) or saline (control) was added for another 3 minutes, and then aPTT was measured as previously described.25,30,31 Normal plasma was used as reference for both the PT and aPTT.
Statistics
All statistical analyses were performed with GraphPad Prism Software version 5 (San Diego, CA, USA). To determine whether parameters were normally distributed, the Kolmogorov–Smirnov statistic normality test was applied. Comparisons between groups were performed by one-way analysis of variance (ANOVA), followed by Newman–Keuls multiple range tests. Polymorphonuclear neutrophils (PMNs) in BAL fluid and TNFα in BAL fluid and plasma, which were not normally distributed, were analyzed with the Kruskal–Wallis test, followed by Dunn’s multiple comparison test. P-values <0.05 were considered as significant. All the data in the figures were expressed as mean ± standard error of the mean.
Results
Effect of CeO2 NPs on lung histology
Histological sections from lungs of control mice had normal appearance (Figure 1A–D). In mice exposed to 0.1 mg/kg CeO2 NPs, particles were found inside the alveolar macrophages (Figure 1E and G) as well as within alveolar air spaces (Figure 1F). Particles were also seen within the alveolar interstitial space (Figure 1F). There was focal damage of the alveolar wall and foci of expansion of the alveolar interstitial space due to neutrophil and macrophage infiltration of the interstitium (Figure 1G and H). In the group IT instilled with 0.5 mg/kg CeO2 NPs, particles were also found engulfed by alveolar macrophages (Figure 1L), and some particles were seen within macrophages within the interstitium (Figure 1K). There were foci of severe expansion of the alveolar interstitial space due to heavy neutrophil and macrophage infiltration of the interstitium (Figure 1I–K).
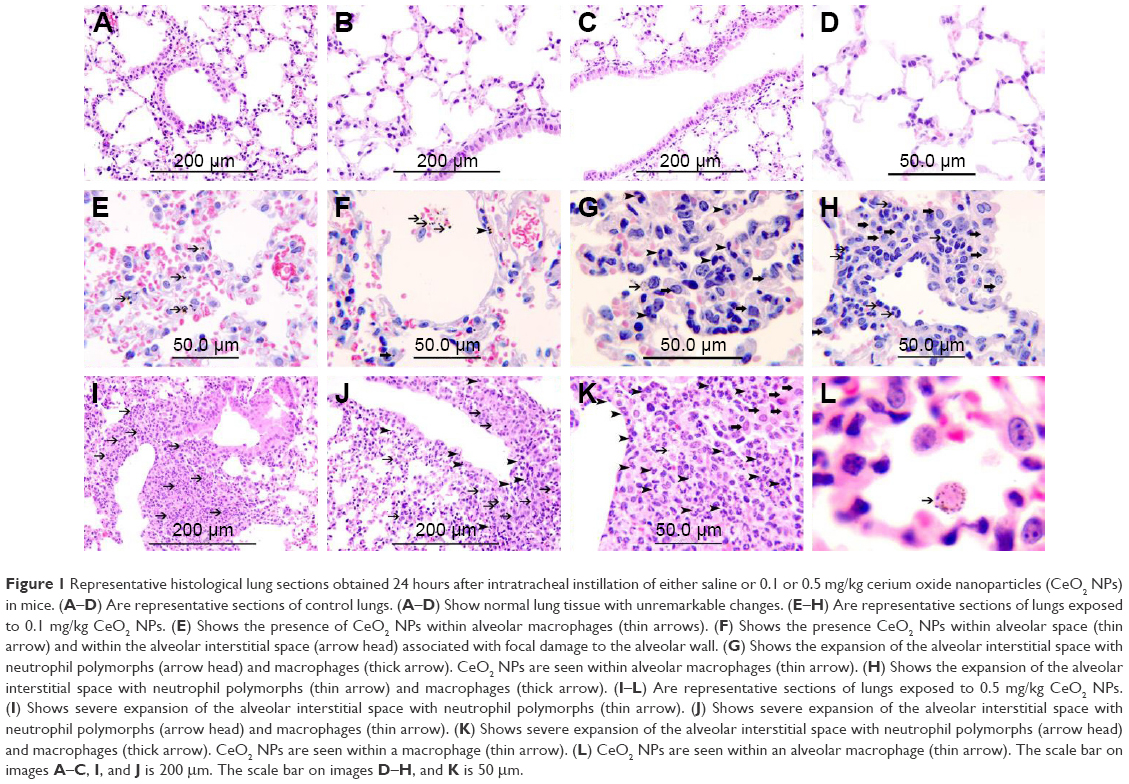 | Figure 1 Representative histological lung sections obtained 24 hours after intratracheal instillation of either saline or 0.1 or 0.5 mg/kg cerium oxide nanoparticles (CeO2 NPs) in mice. (A–D) Are representative sections of control lungs. (A–D) Show normal lung tissue with unremarkable changes. (E–H) Are representative sections of lungs exposed to 0.1 mg/kg CeO2 NPs. (E) Shows the presence of CeO2 NPs within alveolar macrophages (thin arrows). (F) Shows the presence CeO2 NPs within alveolar space (thin arrow) and within the alveolar interstitial space (arrow head) associated with focal damage to the alveolar wall. (G) Shows the expansion of the alveolar interstitial space with neutrophil polymorphs (arrow head) and macrophages (thick arrow). CeO2 NPs are seen within alveolar macrophages (thin arrow). (H) Shows the expansion of the alveolar interstitial space with neutrophil polymorphs (thin arrow) and macrophages (thick arrow). (I–L) Are representative sections of lungs exposed to 0.5 mg/kg CeO2 NPs. (I) Shows severe expansion of the alveolar interstitial space with neutrophil polymorphs (thin arrow). (J) Shows severe expansion of the alveolar interstitial space with neutrophil polymorphs (arrow head) and macrophages (thin arrow). (K) Shows severe expansion of the alveolar interstitial space with neutrophil polymorphs (arrow head) and macrophages (thick arrow). CeO2 NPs are seen within a macrophage (thin arrow). (L) CeO2 NPs are seen within an alveolar macrophage (thin arrow). The scale bar on images A–C, I, and J is 200 μm. The scale bar on images D–H, and K is 50 μm. |
Effect of CeO2 NPs on cell composition and number in BAL fluid
Figure 2A represents the total cell numbers in the different groups, and Figure 2B shows the effects of CeO2 NPs on the influx of PMNs in BAL 24 hours after IT instillation. Compared with the saline-treated group (0.1±0.03×106 cells/mL), the total number of cells in BAL fluid was significantly increased following the IT administration of 0.1 mg/kg (0.5±0.09×106 cells/mL, P<0.01) and 0.5 mg/kg (0.6±0.09×106 cells/mL, P<0.01) CeO2 NPs. In controls, PMNs made up 0.6%±0.4% of the total cells, the remaining cells being macrophages. The IT instillation of CeO2 NPs led to a PMN influx at 0.1 mg/kg (61%±10%, P<0.01) and at 0.5 mg/kg (57%±9%, P<0.01).
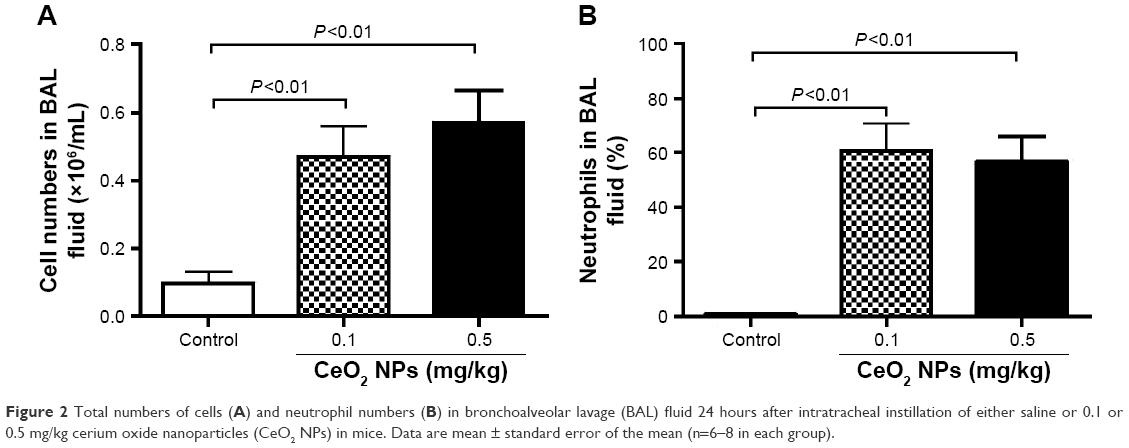 | Figure 2 Total numbers of cells (A) and neutrophil numbers (B) in bronchoalveolar lavage (BAL) fluid 24 hours after intratracheal instillation of either saline or 0.1 or 0.5 mg/kg cerium oxide nanoparticles (CeO2 NPs) in mice. Data are mean ± standard error of the mean (n=6–8 in each group). |
Effect of CeO2 NPs on TNFα concentration and catalase activity in BAL fluid
As shown in Figure 3A, compared with the control group, the IT instillation of CeO2 NPs induced significant increases in TNFα concentrations in BAL fluid at 0.1 and 0.5 mg/kg (P<0.05). On the contrary, IT instillation of CeO2 NPs caused a significant decrease of catalase activity in BAL fluid at 0.1 mg/kg (P<0.05) and 0.5 mg/kg (P<0.05) compared with the saline-treated group (Figure 3B).
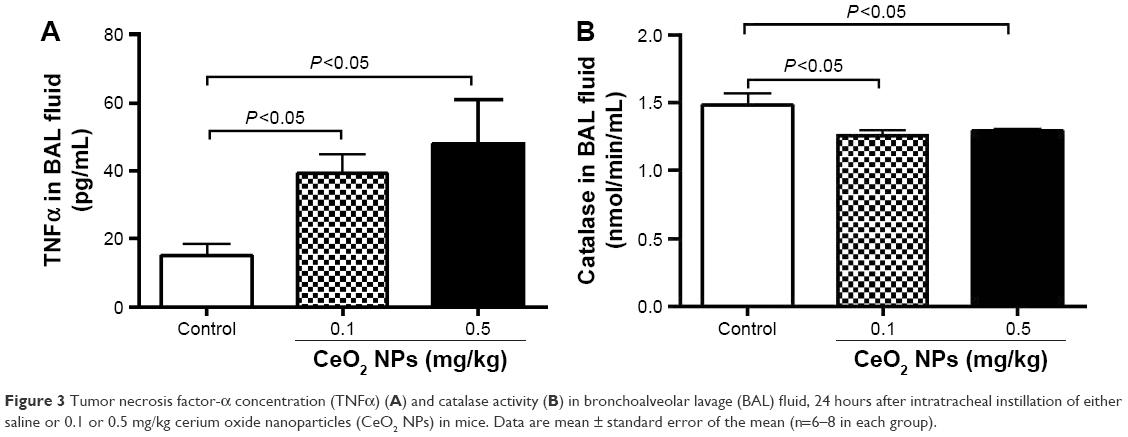 | Figure 3 Tumor necrosis factor-α concentration (TNFα) (A) and catalase activity (B) in bronchoalveolar lavage (BAL) fluid, 24 hours after intratracheal instillation of either saline or 0.1 or 0.5 mg/kg cerium oxide nanoparticles (CeO2 NPs) in mice. Data are mean ± standard error of the mean (n=6–8 in each group). |
Effect in plasma of CeO2 NPs on concentrations of CRP and TNFα, catalase activity, and antioxidant capacity
Pulmonary exposure to CeO2 NPs induced a slight but a statistically significant increase in CRP concentration at 0.1 mg/kg (P<0.001) and 0.5 mg/kg (P<0.001) compared with the control group (Figure 4A). TNFα was dose dependently increased by CeO2 NPs, but the level of significance was only achieved at 0.5 mg/kg (P<0.05) (Figure 4B). On the other hand, compared with the control group, catalase activity was significantly decreased by the 0.5 mg/kg dose (P<0.01) (Figure 4C), whereas both doses of CeO2 NPs induced a significant decrease (P<0.05) of total antioxidant capacity (Figure 4D).
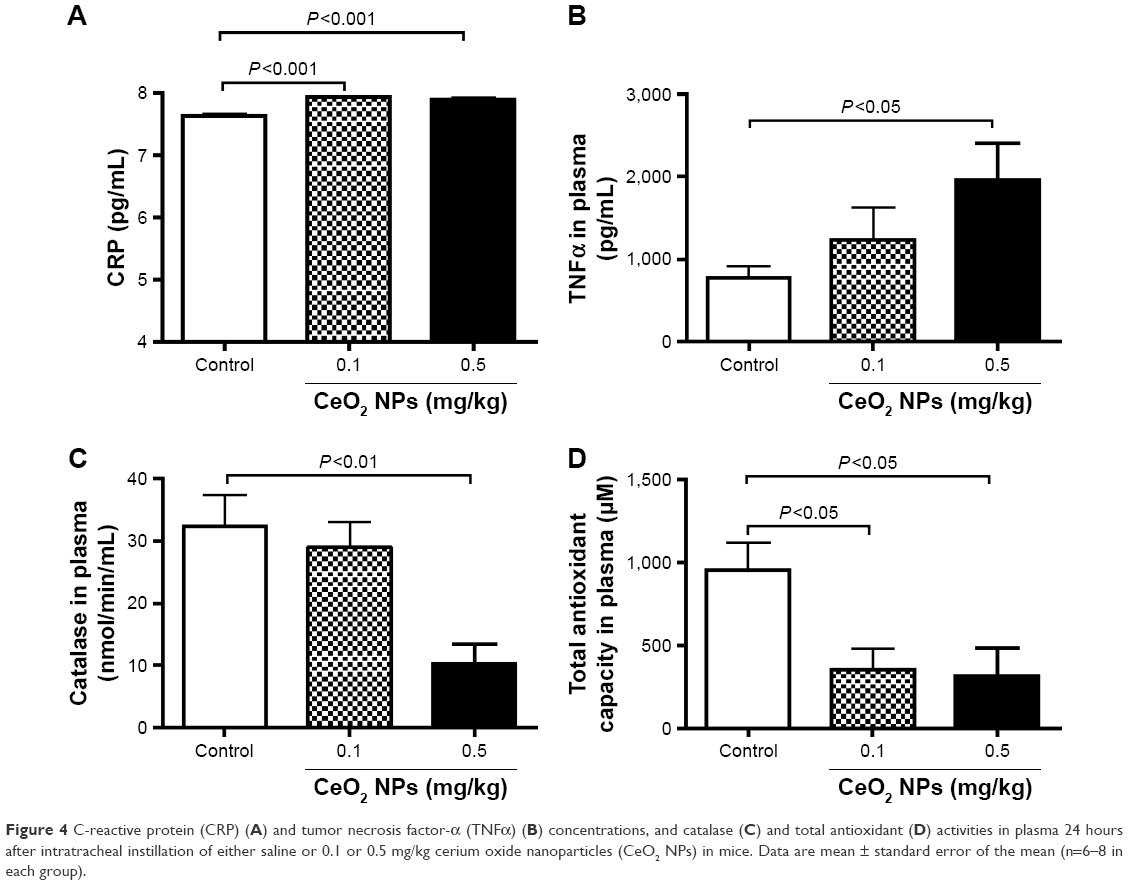 | Figure 4 C-reactive protein (CRP) (A) and tumor necrosis factor-α (TNFα) (B) concentrations, and catalase (C) and total antioxidant (D) activities in plasma 24 hours after intratracheal instillation of either saline or 0.1 or 0.5 mg/kg cerium oxide nanoparticles (CeO2 NPs) in mice. Data are mean ± standard error of the mean (n=6–8 in each group). |
Effect of CeO2 NPs on pial arteriole and venule thrombosis
Figure 5A illustrates that the pulmonary exposure to CeO2 NPs induced a significant (P<0.001) and dose-dependent shortening of the thrombotic occlusion time in pial arterioles in photochemically injured vessels. Similarly, in pial venules, CeO2 NP administration caused a dose-dependent and significant (P<0.001) shortening of the thrombotic occlusion time compared with the saline-treated group.
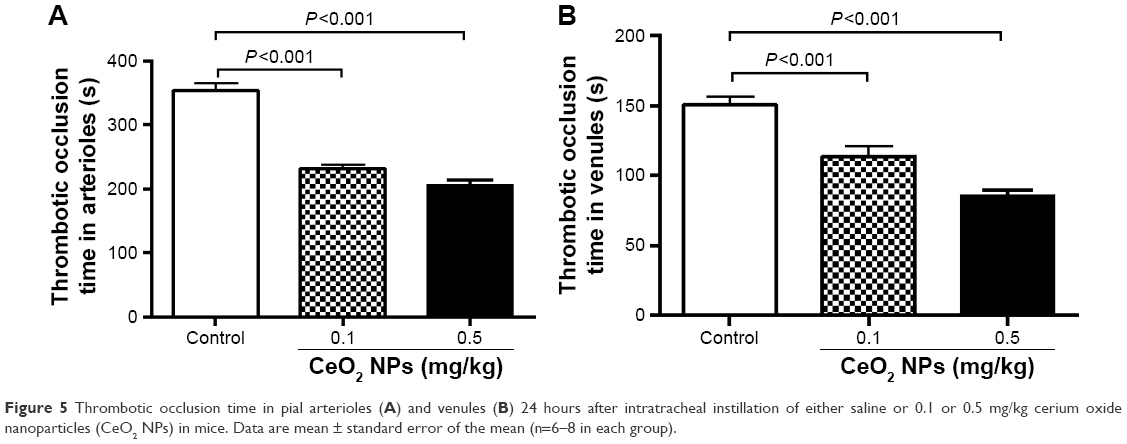 | Figure 5 Thrombotic occlusion time in pial arterioles (A) and venules (B) 24 hours after intratracheal instillation of either saline or 0.1 or 0.5 mg/kg cerium oxide nanoparticles (CeO2 NPs) in mice. Data are mean ± standard error of the mean (n=6–8 in each group). |
Effect of CeO2 NPs on fibrinogen and PAI-1 concentrations in plasma
Figure 6A shows that the IT instillation of CeO2 NPs induced a dose-dependent and significant increase of fibrinogen at doses of 0.1 mg/kg (P<0.001) and 0.5 mg/kg (P<0.001) compared with the control group. The concentration of PAI-1 was, in the same way, increased by the IT instillation of CeO2 NPs at 0.1 mg/kg (P<0.01) and 0.5 mg/kg (P<0.001) compared with the control group.
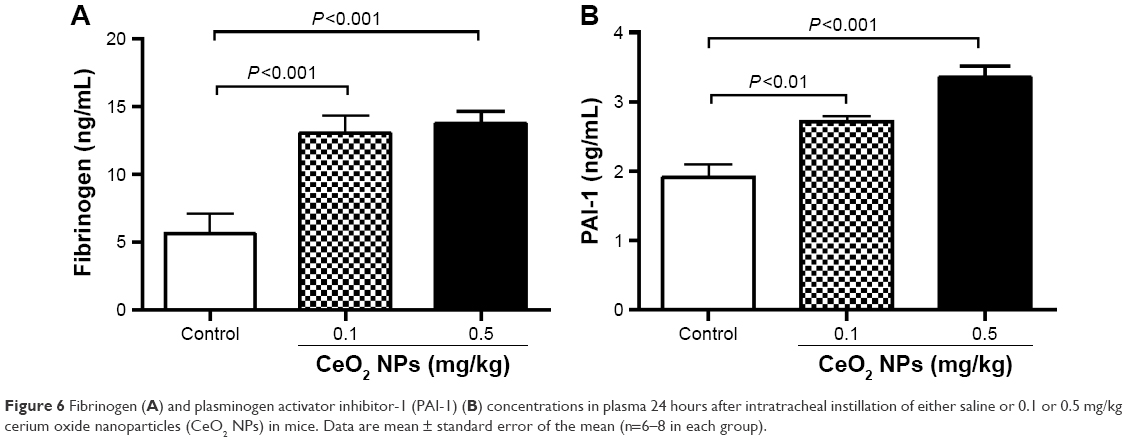 | Figure 6 Fibrinogen (A) and plasminogen activator inhibitor-1 (PAI-1) (B) concentrations in plasma 24 hours after intratracheal instillation of either saline or 0.1 or 0.5 mg/kg cerium oxide nanoparticles (CeO2 NPs) in mice. Data are mean ± standard error of the mean (n=6–8 in each group). |
Effect of CeO2 NPs on platelet aggregation in vitro in whole blood, PT, and aPTT
Figure 7 shows that the direct addition of various concentrations of CeO2 NPs (1–25 μg/mL) to untreated whole blood in vitro did not induce any platelet aggregation. Also, compared with the control group, the incubation of untreated plasma with CeO2 NPs (1–25 μg/mL) neither altered the PT (Figure 7B) nor the aPTT (Figure 7C).
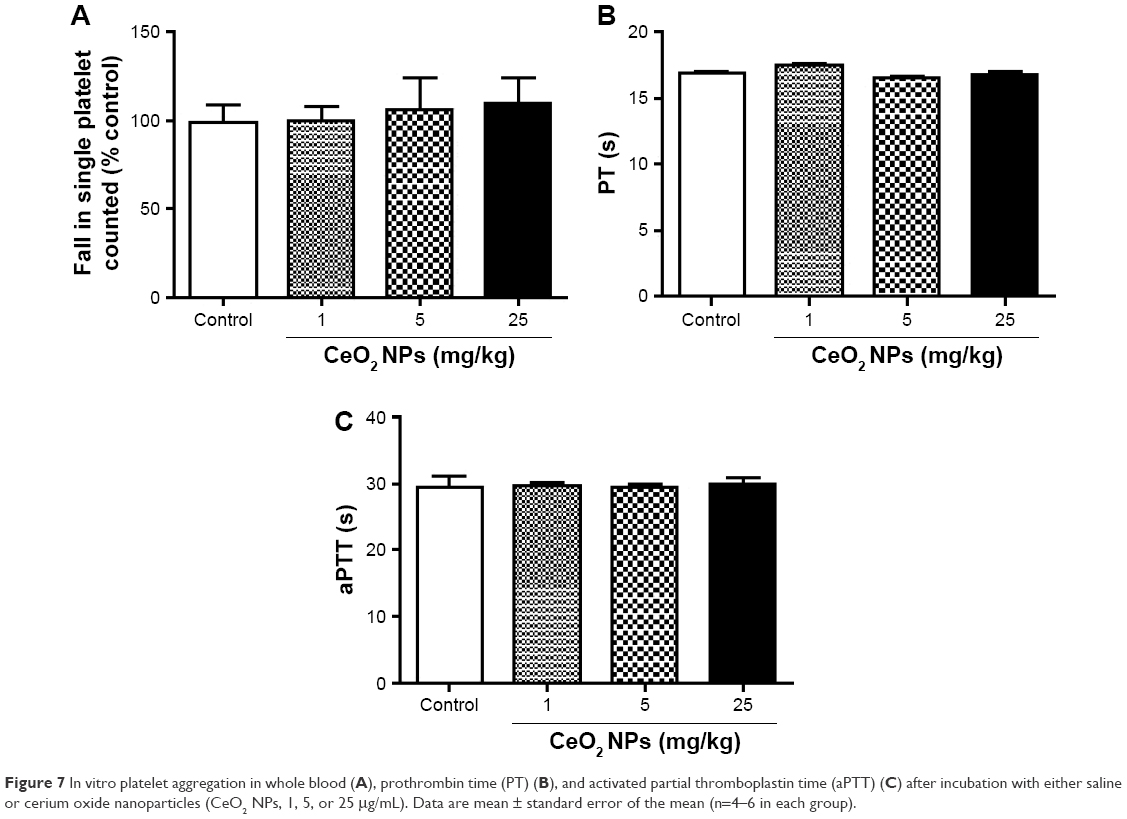 | Figure 7 In vitro platelet aggregation in whole blood (A), prothrombin time (PT) (B), and activated partial thromboplastin time (aPTT) (C) after incubation with either saline or cerium oxide nanoparticles (CeO2 NPs, 1, 5, or 25 μg/mL). Data are mean ± standard error of the mean (n=4–6 in each group). |
Discussion
In this work, a histological analysis of the lung 24 hours after the IT instillation of CeO2 NPs revealed an expansion of the alveolar interstitial space due to infiltration of neutrophils and macrophages into the interstitium; particles were also seen within macrophages, and others were found within the alveolar interstitial space. BAL fluid analysis confirmed the influx of neutrophils and an increase of TNFα concentration and decrease in catalase activity. Likewise, in the plasma, the concentrations of CRP and TNFα were increased, whereas the activities of catalase and total antioxidant capacity were decreased. CeO2 NPs induced prothrombotic events in the pial arterioles and venules in vivo and increased fibrinogen and PAI-1 in plasma. However, the direct addition of CeO2 NPs to whole blood failed to induce platelet aggregation in vitro. Likewise, neither PT nor aPTT was affected by the direct addition of CeO2 NPs in vitro.
The present study used an IT instillation method as a mode of exposure to CeO2 NPs. The IT instillation is considered a valid, yet admittedly not perfect, mode of exposure to foreign compounds.32,33 The exact dose delivered to the lungs of each mouse can be established precisely, and this technique is simpler than inhalation, thus allowing the introduction of a range of doses to the lung in a short time.32,33 Moreover, IT instillation provides more accurate dosing, given that mice are nose breathers that filter most inhaled particles.32,33 The doses of particles used in the present study are comparable with those employed in previous animal models of IT administration of CeO2 NPs.12,34
In a previous study, it has been demonstrated that acute (24 hours) exposure of mice to DEPs causes impairment of cardiovascular homeostasis.5,29 This is in agreement with clinical and epidemiological studies that reported the development of myocardial complications within 24 hours of exposure to elevated levels of particulate air pollution,35 and hence, the effect of CeO2 NPs 24-hour postexposure was assessed. Moreover, this time point is identical to the one used recently to assess the role of mast cells in vascular reactivity and ischemia reperfusion injury following IT administration of CeO2 NPs.34
These data show that acute administration of CeO2 NPs caused a dose-dependent infiltration of neutrophils and macrophages, which caused an expansion of the alveolar interstitial space. Some CeO2 NPs were engulfed by macrophages, and others gained access to the interstitium. BAL fluid analysis confirmed the latter finding, showing a significant increase in neutrophils after the IT instillation of both doses of CeO2 NPs. Moreover, a significant and dose-dependent increase in the concentration of TNFα and a decrease in the activity of catalase were found, suggestive of a depletion of this antioxidant. These findings indicate the development of pulmonary inflammation and oxidative stress following acute exposure to CeO2 NPs. These results corroborate earlier findings which showed an induction of inflammation, cytotoxicity, and air–blood barrier damage, 24-hour post-IT instillation of CeO2 NPs in rats.12 The findings of this study are also in agreement with previous studies that demonstrated an increase in neutrophil numbers in BAL fluid and the concentration of cytokine-induced neutrophil chemoattractant (CINC)-1, CINC-2, chemokine for neutrophil, and heme oxygenase-1, 3 days after the IT instillation of either 0.2 mg or 1 mg CeO2 NPs in rats, which persisted for up to 3 months postexposure.36 Comparable findings were obtained, in the same study, following inhalation of CeO2 NPs (2 or 10 mg/m3) for 4 weeks (6 hours per day, 5 days per week).36
Previous reports showed that pulmonary exposure to pollutant particles and some engineered NPs induce systemic and cardiovascular events.22,24,37,38 The latter effects were explained by the capacity of NPs to induce lung inflammation, which causes systemic inflammation and oxidative stress, and/or the aptitude of NPs to traverse the air–blood barrier to enter the blood and affect cardiovascular endpoints.5,18 It has been reported that IV administration of CeO2 NPs induce liver injury and oxidative stress.39,40 More recently, it has been shown that pulmonary, IV, and gastric exposures to CeO2 NPs cause endothelium-dependent and -independent arteriolar dysfunction.20,41 Nevertheless, the systemic effects of CeO2 NPs following pulmonary exposure are not fully understood, particularly their possible impact on thrombosis. The data of the present study show a significant increase in CRP caused by both doses of NPs. Moreover, a dose-dependent increase in TNFα concentrations and a decrease in catalase and total antioxidant activities following the acute exposure to CeO2 NPs were observed. It has been shown that exposure to particulate air pollution increases pulmonary inflammatory mediators that translocate to the circulation, contributing to systemic inflammation, with downstream effects such as cardiovascular dysfunction.42 It has been shown that IT instillation of DEPs induces lung inflammation and oxidative stress responsible for systemic inflammation and oxidative stress at the 18–24-hour time point.24,37 It has been recently demonstrated that aortic RNA expression of endothelial nitric oxide synthase, tissue factor, and TNFα were significantly increased by 4-week inhalation exposure to whole CeO2 NPs added to diesel fuel (DECe) or gas-phase components of DECe.19 However, the latter results are in conflict with the data from another study which implied that the addition of a Ce-based fuel-borne catalyst may decrease the atherosclerotic burden induced by exposure to diesel fuel in atherosclerosis-prone mice.43 Also, it has been shown that CeO2 NPs improve microvascular reactivity in hypertensive rats.44
This study evaluated the impact of CeO2 NPs on coagulation by measuring a set of relevant endpoints, ie, thrombosis in pial arterioles and venules in vivo and measurement of plasma concentrations of fibrinogen and PAI-1 and in vitro platelet aggregation, PT, and aPTT. As far as we are aware, the effect of CeO2 NPs on an animal model of thrombosis in vivo has not been reported so far. The present study used a well-established model of photochemical-induced thrombosis in pial arterioles and venules.27,37,45 In this model, the damage to endothelial cells causes the platelets to adhere at the site of endothelial damage and then aggregate.27,37,45 The data of the present study show that acute IT administration of CeO2 NPs induced a significant and dose-dependent shortening of the thrombotic occlusion time in pial arterioles and venules, indicating that CeO2 NPs possess prothrombotic effects. Along with thrombosis in vivo, a dose-dependent and significant increase in the concentrations of the coagulation factor, fibrinogen, and PAI-1 in the plasma following acute exposure to CeO2 NPs was found. PAI-1 is a potent endogenous inhibitor of fibrinolysis and has been reported to increase following exposure to DEPs, silica NPs, and carbon nanotubes.46–48 It has been shown that a 4-week inhalation exposure to DECe in rats induced an increase of tissue factor, but not in PAI-1 concentration.19 The discrepancy between the latter study showing the absence of an increase of PAI-1 and this study could be related to the duration of exposure (24 hours versus 4 weeks), mode of exposure (IT instillation versus inhalation), or experimental animals used (mice versus rats). Additional studies are needed to clarify this point.
Recent studies have reported that pulmonary exposure to CeO2 NPs either by IT instillation or by inhalation resulted in systemic translocation and accumulation in various major organs such as the liver.19,49 This study wanted to verify whether the CeO2 NPs can directly induce platelet aggregation and affect PT and aPTT in vitro. The concentration of 1 μg/mL CeO2 NPs used here in vitro is similar to that employed recently to induce prothrombotic effects of DEPs in vitro.29,50 In addition to the latter concentration, two higher concentrations, that is, 5 and 25 μg/mL CeO2 NPs were used. These data show that CeO2 NPs (1–25 μg/mL) neither caused platelet aggregation nor affected either PT or aPTT. The latter findings suggest that the prothrombotic effects of CeO2 NPs observed in vivo may have resulted from systemic inflammation and oxidative stress rather than the direct contact of CeO2 NPs with platelets. These results also indicate that the in vitro effects of CeO2 NPs are different from those of DEPs reported previously, which showed that DEPs induced platelet aggregation in vitro and shortened PT and aPTT.29,51 These study data highlight the importance of conducting in vivo toxicity studies with NPs and suggest that the absence of toxicity of NPs in vitro does not necessarily mean that they are devoid of in vivo adverse effects.
This study has limitations. It did not thoroughly study the mechanisms underlying the systemic effects of CeO2 NPs and has investigated only the acute effects of these NPs. Further studies are warranted to assess the time effects (subacute and chronic) and the possible mechanism underlying the pulmonary and systemic effects of CeO2 NPs.
In conclusion, the data of this study provide novel evidence that pulmonary exposure to CeO2 NPs induces pulmonary and systemic inflammation and oxidative stress and promotes thrombosis in vivo.
Acknowledgments
This work was supported by funds of the College of Medicine and Health Sciences grant and United Arab Emirates University UPAR (31M189) and center-based interdisciplinary (31R052) grants. The authors wish to thank Professor Gerald Blunden (University of Portsmouth, Portsmouth, UK) for critically reading the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Radomska A, Leszczyszyn J, Radomski MW. The nanopharmacology and nanotoxicology of nanomaterials: new opportunities and challenges. Adv Clin Exp Med. 2016;25:151–162. | ||
Thorley AJ, Tetley TD. New perspectives in nanomedicine. Pharmacol Ther. 2013;140:176–185. | ||
Oberdorster G. Safety assessment for nanotechnology and nanomedicine: concepts of nanotoxicology. J Intern Med. 2010;267:89–105. | ||
Hoet PH, Bruske-Hohlfeld I, Salata OV. Nanoparticles – known and unknown health risks. J Nanobiotechnol. 2004;2:12. | ||
Nemmar A, Holme JA, Rosas I, Schwarze PE, Alfaro-Moreno E. Recent advances in particulate matter and nanoparticle toxicology: a review of the in vivo and in vitro studies. Biomed Res Int. 2013;2013:279371. | ||
Sotiriou GA, Pratsinis SE. Antibacterial activity of nanosilver ions and particles. Environ Sci Technol. 2010;44:5649–5654. | ||
HEI. Evaluation of human health risk from cerium added to diesel fuel. In: Hibbs JB Jr, editor. HEI Communication 9. Boston, MA, USA: Health Effects Institute; 2001. | ||
Cassee FR, van Balen EC, Singh C, et al. Exposure, health and ecological effects review of engineered nanoscale cerium and cerium oxide associated with its use as a fuel additive. Crit Rev Toxicol. 2011;41:213–229. | ||
Park B, Donaldson K, Duffin R, et al. Hazard and risk assessment of a nanoparticulate cerium oxide-based diesel fuel additive – a case study. Inhal Toxicol. 2008;20:547–566. | ||
Wakefield G, Wu X, Gardener M, Park B, Anderson S. Envirox™ fuel-borne catalyst: developing and launching a nano-fuel additive. Tech Anal Strat Manag. 2008;127–136. | ||
Selvan AM, Anand V, Udayakumar MRB. Effects of cerium oxide nanoparticle addition in diesel and diesel–biodiesel–ethanol blends on the performance and emission characteristics of a CI engine. J Eng Appl Sci. 2009;4(7):1–6. | ||
Ma JY, Zhao H, Mercer RR, et al. Cerium oxide nanoparticle-induced pulmonary inflammation and alveolar macrophage functional change in rats. Nanotoxicology. 2011;5:312–325. | ||
Ma J, Mercer RR, Barger M, et al. Effects of amorphous silica coating on cerium oxide nanoparticles induced pulmonary responses. Toxicol Appl Pharmacol. 2015;288:63–73. | ||
Park EJ, Cho WS, Jeong J, Yi J, Choi K, Kim Y. Induction of inflammatory responses in mice treated with cerium oxide nanoparticles by intratracheal instillation. J Health Sci. 2010;56:387–396. | ||
Srinivas A, Rao PJ, Selvam G, Murthy PB, Reddy PN. Acute inhalation toxicity of cerium oxide nanoparticles in rats. Toxicol Lett. 2011;205:105–115. | ||
Ma JY, Mercer RR, Barger M, et al. Induction of pulmonary fibrosis by cerium oxide nanoparticles. Toxicol Appl Pharmacol. 2012;262:255–264. | ||
Ma J, Mercer RR, Barger M, et al. Effects of amorphous silica coating on cerium oxide nanoparticles induced pulmonary responses. Toxicol Appl Pharmacol. 2015;288:63–73. | ||
Mills NL, Donaldson K, Hadoke PW, et al. Adverse cardiovascular effects of air pollution. Nat Clin Pract Cardiovasc Med. 2009;6:36–44. | ||
Snow SJ, Mcgee J, Miller DB, et al. Inhaled diesel emissions generated with cerium oxide nanoparticle fuel additive induce adverse pulmonary and systemic effects. Toxicol Sci. 2014;142:403–417. | ||
Minarchick VC, Stapleton PA, Porter DW, et al. Pulmonary cerium dioxide nanoparticle exposure differentially impairs coronary and mesenteric arteriolar reactivity. Cardiovasc Toxicol. 2013;13: 323–337. | ||
Ma JY, Young SH, Mercer RR, et al. Interactive effects of cerium oxide and diesel exhaust nanoparticles on inducing pulmonary fibrosis. Toxicol Appl Pharmacol. 2014;278:135–147. | ||
Nemmar A, Melghit K, Al-Salam S, et al. Acute respiratory and systemic toxicity of pulmonary exposure to rutile Fe-doped TiO(2) nanorods. Toxicology. 2011;279:167–175. | ||
Nemmar A, Al-Salam S, Yuvaraju P, Beegam S, Ali BH. Emodin mitigates diesel exhaust particles-induced increase in airway resistance, inflammation and oxidative stress in mice. Respir Physiol Neurobiol. 2015;215:51–57. | ||
Nemmar A, Al Salam S, Dhanasekaran S, Sudhadevi M, Ali BH. Pulmonary exposure to diesel exhaust particles promotes cerebral microvessel thrombosis: protective effect of a cysteine prodrug l-2-oxothiazolidine-4-carboxylic acid. Toxicology. 2009;263:84–92. | ||
Nemmar A, Zia S, Subramaniyan D, Fahim MA, Ali BH. Exacerbation of thrombotic events by diesel exhaust particle in mouse model of hypertension. Toxicology. 2011;285:39–45. | ||
Nemmar A, Raza H, Subramaniyan D, et al. Short-term systemic effects of nose-only cigarette smoke exposure in mice: role of oxidative stress. Cell Physiol Biochem. 2013;31:15–24. | ||
Nemmar A, Beegam S, Yuvaraju P, et al. Ultrasmall superparamagnetic iron oxide nanoparticles acutely promote thrombosis and cardiac oxidative stress and DNA damage in mice. Part Fibre Toxicol. 2016;13:22. | ||
Nemmar A, Melghit K, Ali BH. The acute proinflammatory and prothrombotic effects of pulmonary exposure to rutile TiO2 nanorods in rats. Exp Biol Med (Maywood). 2008;233:610–619. | ||
Nemmar A, Al DR, Alamiri J, et al. Diesel exhaust particles induce impairment of vascular and cardiac homeostasis in mice: ameliorative effect of emodin. Cell Physiol Biochem. 2015;36:1517–1526. | ||
Nemmar A, Al Salam S, Zia S, Dhanasekaran S, Shudadevi M, Ali BH. Time-course effects of systemically administered diesel exhaust particles in rats. Toxicol Lett. 2010;194:58–65. | ||
Nemmar A, Yuvaraju P, Beegam S, John A, Raza H, Ali BH. Cardiovascular effects of nose-only water-pipe smoking exposure in mice. Am J Physiol Heart Circ Physiol. 2013;305:H740–H746. | ||
Driscoll KE, Costa DL, Hatch G, et al. Intratracheal instillation as an exposure technique for the evaluation of respiratory tract toxicity: uses and limitations. Toxicol Sci. 2000;55:24–35. | ||
Morimoto Y, Izumi H, Yoshiura Y, et al. Comparison of pulmonary inflammatory responses following intratracheal instillation and inhalation of nanoparticles. Nanotoxicology. 2016;10:607–618. | ||
Wingard CJ, Walters DM, Cathey BL, et al. Mast cells contribute to altered vascular reactivity and ischemia-reperfusion injury following cerium oxide nanoparticle instillation. Nanotoxicology. 2011;5:531–545. | ||
Brook RD, Rajagopalan S, Pope CA, et al. Particulate matter air pollution and cardiovascular disease. An update to the scientific statement from the American Heart Association. Circulation. 2010;121:2331–2378. | ||
Morimoto Y, Izumi H, Yoshiura Y, et al. Pulmonary toxicity of well-dispersed cerium oxide nanoparticles following intratracheal instillation and inhalation. J Nanopart Res. 2015;17:442. | ||
Nemmar A, Al-Salam S, Zia S, et al. Contrasting actions of diesel exhaust particles on the pulmonary and cardiovascular systems and the effects of thymoquinone. Br J Pharmacol. 2011;164:1871–1882. | ||
Budinger GRS, Mckell JL, Urich D, et al. Particulate matter-induced lung inflammation increases systemic levels of PAI-1 and activates coagulation through distinct mechanisms. Plos One. 2011;6(4):e18525. | ||
Tseng MT, Lu X, Duan X, et al. Alteration of hepatic structure and oxidative stress induced by intravenous nanoceria. Toxicol Appl Pharmacol. 2012;260:173–182. | ||
Yokel RA, Au TC, MacPhail R, et al. Distribution, elimination, and biopersistence to 90 days of a systemically introduced 30 nm ceria-engineered nanomaterial in rats. Toxicol Sci. 2012;127:256–268. | ||
Minarchick VC, Stapleton PA, Fix NR, Leonard SS, Sabolsky EM, Nurkiewicz TR. Intravenous and gastric cerium dioxide nanoparticle exposure disrupts microvascular smooth muscle signaling. Toxicol Sci. 2015;144:77–89. | ||
Kido T, Tamagawa E, Bai N, et al. Particulate matter induces IL-6 translocation from the lung to the systemic circulation. Am J Respir Cell Mol Biol. 2011;44(2):197–204. | ||
Cassee FR, Campbell A, Boere AJ, et al. The biological effects of subacute inhalation of diesel exhaust following addition of cerium oxide nanoparticles in atherosclerosis-prone mice. Environ Res. 2012;115:1–10. | ||
Minarchick VC, Stapleton PA, Sabolsky EM, Nurkiewicz TR. Cerium dioxide nanoparticle exposure improves microvascular dysfunction and reduces oxidative stress in spontaneously hypertensive rats. Front Physiol. 2015;6:339. | ||
Nemmar A, Yuvaraju P, Beegam S, Ali BH. Short-term nose-only water-pipe (shisha) smoking exposure accelerates coagulation and causes cardiac inflammation and oxidative stress in mice. Cell Physiol Biochem. 2015;35:829–840. | ||
Nemmar A, Subramaniyan D, Ali BH. Protective effect of curcumin on pulmonary and cardiovascular effects induced by repeated exposure to diesel exhaust particles in mice. PLoS One. 2012;7:e39554. | ||
Erdely A, Hulderman T, Salmen R, et al. Cross-talk between lung and systemic circulation during carbon nanotube respiratory exposure. Potential biomarkers. Nano Lett. 2009;9:36–43. | ||
Nemmar A, Albarwani S, Beegam S, et al. Amorphous silica nanoparticles impair vascular homeostasis and induce systemic inflammation. Int J Nanomedicine. 2014;9:2779–2789. | ||
He X, Zhang H, Ma Y, et al. Lung deposition and extrapulmonary translocation of nano-ceria after intratracheal instillation. Nanotechnology. 2010;21:285103. | ||
Tabor CM, Shaw CA, Robertson S, et al. Platelet activation independent of pulmonary inflammation contributes to diesel exhaust particulate-induced promotion of arterial thrombosis. Part Fibre Toxicol. 2016;13:6. | ||
Radomski A, Jurasz P, Alonso-Escolano D, et al. Nanoparticle-induced platelet aggregation and vascular thrombosis. Br J Pharmacol. 2005;146:882–893. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.