Back to Journals » Medical Devices: Evidence and Research » Volume 9
The activL® Artificial Disc: a next-generation motion-preserving implant for chronic lumbar discogenic pain
Authors Yue J, Garcia R, Miller LE ![]()
Received 22 December 2015
Accepted for publication 18 February 2016
Published 10 May 2016 Volume 2016:9 Pages 75—84
DOI https://doi.org/10.2147/MDER.S102949
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
James J Yue,1 Rolando Garcia Jr,2 Larry E Miller3
1Department of Orthopaedic Surgery, Yale School of Medicine, New Haven, CT, 2Orthopedic Care Center, Miami, FL, 3Miller Scientific Consulting, Inc., Asheville, NC, USA
Abstract: Degeneration of the lumbar intervertebral discs is a leading cause of chronic low back pain in adults. Treatment options for patients with chronic lumbar discogenic pain unresponsive to conservative management include total disc replacement (TDR) or lumbar fusion. Until recently, only two lumbar TDRs had been approved by the US Food and Drug Administration - the Charité Artificial Disc in 2004 and the ProDisc-L Total Disc Replacement in 2006. In June 2015, a next-generation lumbar TDR received Food and Drug Administration approval - the activL® Artificial Disc (Aesculap Implant Systems). Compared to previous-generation lumbar TDRs, the activL® Artificial Disc incorporates specific design enhancements that result in a more precise anatomical match and allow a range of motion that better mimics the healthy spine. The results of mechanical and clinical studies demonstrate that the activL® Artificial Disc results in improved mechanical and clinical outcomes versus earlier-generation artificial discs and compares favorably to lumbar fusion. The purpose of this report is to describe the activL® Artificial Disc including implant characteristics, intended use, surgical technique, postoperative care, mechanical testing, and clinical experience to date.
Keywords: activL® Artificial Disc, artificial disc, degenerative disc disease, discogenic, implant, lumbar, motion preservation, pain
Introduction
Degeneration of the lumbar intervertebral discs is a leading cause of chronic low back pain in adults1 and responsible for 62 million annual physician visits in the US.2 Internal disc disruption is identified in 42% of patients reporting persistent low back symptoms.3,4 Most patients with definitive imaging evidence of disc degeneration coupled with chronic low back pain have a poor prognosis for recovery with conservative management alone.5–8 When comprehensive multimodal conservative treatment options have been exhausted, lumbar fusion or total disc replacement (TDR) may be considered to alleviate chronic discogenic pain.
The goal of lumbar fusion is to eliminate motion and instability at the painful motion segment, which may relieve discogenic pain. However, fusion is associated with a 10%–15% risk of reoperation within 5 years9,10 and a 30%–80% risk of adjacent level disease due to increased stress at the adjacent segment.11,12 Additionally, since patients undergoing fusion for lumbar degenerative disc disease (DDD) are notably younger than the typical spine patient,1 the risk of future complications must be carefully considered since revision surgery is technically demanding, associated with greater surgical risk, and results in mixed clinical success.
TDR is an alternative to spinal fusion in well-selected patients with chronic symptomatic lumbar DDD. The basic premise of TDR is to eliminate pain and improve function by eliminating the painful disc and restoring disc height while preserving segmental range of motion which may lower risk of adjacent segment degeneration. Numerous meta-analyses of randomized controlled studies have shown that TDR yields comparable or superior outcomes versus lumbar fusion through 2 years.13–16 Additionally, the long-term risk of adjacent segment degeneration is lower with TDR17 since functional movement is preserved, not obliterated, as with spinal fusion.
Until recently, only two lumbar TDRs had been approved by the US Food and Drug Administration (FDA) − the Charité Artificial Disc (DePuy Spine, Raynham, MA, USA) in 200418 and the ProDisc-L Total Disc Replacement (Synthes Spine, West Chester, PA, USA) in 2006.19 The Charité disc was subsequently removed from the market in 2012. In June 2015, a next-generation lumbar TDR received FDA approval − the activL® Artificial Disc (Aesculap Implant Systems, Center Valley, PA, USA).20 Compared to previous-generation lumbar TDRs, the activL® Artificial Disc incorporates specific design enhancements that result in a more precise anatomical match and allow a range of motion that better mimics the healthy spine. The purpose of this report is to describe the activL® Artificial Disc including implant characteristics, intended use, surgical technique, postoperative care, mechanical testing, and clinical experience to date.
Prosthesis characteristics
The activL® Artificial Disc is a next-generation biomimetic implant that incorporates several innovative features not found in previous-generation lumbar TDRs that are intended to accommodate a wider range of anatomical variations and more accurately replicate the kinematic patterns of the human lumbar spine. The activL® Artificial Disc consists of two metal endplates and one polyethylene inlay (Figure 1). The superior and inferior endplates consist of a cobalt chromium alloy with a Plasmapore™ μ-CaP surface coating, composed of titanium and a microscopic dicalcium phosphate overcoating. Each endplate incorporates three anterior horizontal spikes that ensure secure initial fixation. For lumbosacral arthroplasty, an optional inferior endplate design is available for patients with an ovoid S1 footprint (Figure 2). The S1 endplate has rounded posterior edges and can be placed close to the posterior rim of S1 without the endplate edges protruding into the spinal canal. In some cases, this may allow the surgeon to use a larger size compared to the standard endplate shape, thereby reducing the risk of subsidence and nerve root irritation. For each disc design, endplates are available in four sizes (26×31, 28×34.5, 30×39, and 33×40 mm) (anteroposterior × lateral dimensions). Incremental sizing allows the surgeon to select the device that provides maximal endplate coverage. Superior endplates are available in 6° or 11° lordotic angle options and inferior endplates are provided in 0° or 5° lordotic angle options, allowing for constructing lordotic angle options of 6°, 11°, or 16°.
 | Figure 1 activL® Artificial Disc. |
 | Figure 2 Footprint of the activL® Artificial Disc standard (left) and S1 (right) inferior endplate. |
The activL® Artificial Disc inlay is manufactured from ultra-high molecular weight polyethylene and includes an integrated tantalum radiographic marker. The inlays are available in four heights, resulting in total device heights of 8.5, 10, 12, and 14 mm, and each inlay accommodates any of the available endplates. The pocketed design of the endplate prevents anterior expulsion of the inlay. The range of available device sizes and configurations is greater with activL® Artificial Disc versus ProDisc-L, the only other lumbar TDR currently available in the US market, which allows the surgeon to provide a customized anatomical fit (Table 1). Importantly, the activL® Artificial Disc is the only lumbar TDR that offers an 8.5 mm total device height (measured posteriorly), which was the ideal implant size for 87% of patients in a clinical trial.21 In contrast, the ProDisc-L disc is only available in 10, 12, and 14 mm heights.
 | Table 1 Available configurations of activL® Artificial Disc and ProDisc-L Total Disc Replacement |
Earlier-generation Charité artificial discs with no lateral translation restraint resulted in abnormal kinetics and early device failures. The ProDisc-L disc utilizes a fixed center of rotation that restrains a physiological range of motion. In contrast, the semiconstrained ultra high molecular weight polyethylene core of the activL® Artificial Disc supports translation only in the anteroposterior direction to more closely replicate natural physiological motion, potentially minimizing biomechanical stress at the facet joint and adjacent levels.
Intended use
The success of any lumbar TDR procedure is highly dependent on proper patient selection. The activL® Artificial Disc is intended for treatment of skeletally mature adults with single-level symptomatic DDD at L4–L5 or L5–S1 who have failed at least 6 months of nonoperative treatment. The main contraindications to use include active infection, osteoporosis or osteopenia, allergy or sensitivity to implant materials, isolated lumbar or chronic radiculopathy, disc extrusion with sequestration, myelopathy, spinal stenosis, spinal deformity, spondylolisthesis grade II to IV, vertebral body pathology at the affected level, facet ankyloses or facet joint degeneration, disc height <3 mm, symptoms attributable to more than one level, abdominal pathology that would preclude an anterior retroperitoneal approach, and involved vertebral endplate <31 mm medial–lateral and/or <26 mm anteroposterior. Clinical findings should closely correlate with radiologic imaging findings from magnetic resonance imaging, standing plain X-ray studies, discography, or computed tomography in order to correctly identify the lumbar disc as the primary pain generator.
Surgical technique
The patient is placed on a fluoroscopic imaging table in a supine position. Fluoroscopic views are obtained so that with the C-arm in zero degree rotation (anteroposterior view) of the spine, the spinous process is equidistant from the medial pedicle edges. On the lateral view, the anterior and posterior vertebral body cortices should be easily identifiable. A standard anterior retroperitoneal approach to the lower lumbar spine is utilized. Once the anterior disc has been exposed and the appropriate level of dissection verified, the midpoint of the disc space is marked under fluoroscopic imaging. A complete discectomy and mobilization are then performed. Special care should be taken to preserve the subchondral bone. The entire posterior longitudinal ligament does not need to be removed unless removal of extruded disc material or greater intervertebral disc mobilization is required.
After endplate preparation and disc space mobilization are complete, implant trialing is performed. Disc and plate size height and lordotic angles should be reconfirmed. Under anteroposterior and lateral X-ray views, trial plates are inserted to confirm correct device sizing. The largest possible endplate coverage should be chosen in all cases. Care must be exercised to avoid placing the plates too deeply. The disc space is distracted to a point where the implant will be held firmly in place – the height measurement is then observed on the distractor. Next, the implant is assembled and attached to the inserter. During implantation, it is imperative that the artificial disc does not deviate from a central position. Unlike other lumbar TDRs that require the endplates to be inserted first followed by disc space distraction to seat the core, the activL® Artificial Disc is inserted en bloc which lowers the risk of overdistraction and reduces operative time. Finally, the position of the implant is confirmed under fluoroscopy and the inserter is disconnected from the implant. The set contains an impactor which can be utilized to manipulate the individual endplates posteriorly following insertion.
Postoperative care
Patients are generally permitted to ambulate on the day of surgery, as tolerated, with an elastic bandage or lumbosacral orthosis to support the abdominal musculature. Patients are advised to avoid hyperextension for 3 weeks. Lumbar stabilization therapy can typically be initiated 2 to 4 weeks following surgery. Aerobic walking is emphasized for the first six postoperative weeks after which progressive resistance exercise may be undertaken. Median time to return to work following implant with the activL® Artificial Disc is 68 days, which compares favorably to ProDisc-L (median 97 days).21 Patients are typically allowed to return to nonstrenuous activities by 6 weeks and normal activities by 3 months.
Mechanical testing
The activL® Artificial Disc has been extensively tested in nonclinical studies. Where applicable, the International Organization for Standardization and American Society for Testing and Materials standards for testing artificial discs were followed using finished discs. Device testing included worst-case modes and loading conditions that were anticipated in an in vivo environment. A summary of these tests is provided below.
Static endplate expulsion
Five activL® Artificial Disc endplates were inserted onto custom grade 15 polycarbonate urethane foam blocks with a 1 mm thickness of grade 80 foam on the surface to simulate the denser bone of the endplate. Under a 450 N axial load, shear loading was applied to the endplate at 5 mm/minute and the force necessary to dislodge the endplates was measured. The acceptance criteria for this test was at least 400 N, which is the maximum shear force encountered in the lumbar spine.22 The maximum shear force measured was 1,259±60 N. For comparison, the maximum sheer force required to dislodge ProDisc-L endplates was 933 N under identical testing conditions.
Subsidence
Five activL® Artificial Disc endplates were compressed into custom grade 15 polycarbonate urethane foam blocks with a 1 mm thickness of grade 80 foam on the surface to simulate the denser bone of the endplate. The load was applied at 0.1 mm/minute and the maximum subsidence load was measured. The acceptance criteria for this test was a subsidence load ≥3,400 N, the maximum in vivo axial force.23 The maximum observed subsidence load was 5,761±391 N.
Static compression shear
Five activL® Artificial Disc endplates were tested under static compression-shear (10° angle) in saline at 37°C at a rate of 50 N/s until failure. The acceptance criteria for this test was a value ≥5,500 N, which is equivalent to the fracture load of the L5 vertebral body.24 The mean yield load of the specimens was 6,626±272 N.
Dynamic compression shear
Five activL® Artificial Disc specimens were tested under compression shear loads (10° angle) in saline at 37°C using a sinusoidal wave form with R=10 at 5 Hz until 10 million cycles or failure. The acceptance criterion for this test was a value ≥3,400 N, the maximum in vivo axial force.23 Four activL® Artificial Disc specimens completed 10 million cycles at 4,000 N with no failure. In contrast, when subjected to the same test conditions, the ProDisc-L specimen suffered a fractured inferior endplate at 4,000 N.
Creep characterization
Six activL® Artificial Disc specimens with the tallest (14 mm) and six with the shortest (8.5 mm) ultra-high-molecular-weight polyethylene inlays were loaded in compression shear (10° angle) in saline at 37°C using the following sequential test protocol: 1) static: 300 N for 4 hours; 2) dynamic: 300–1,000 N (1 Hz) for 16 hours; 3) static: 300 N for 8 hours; 4) dynamic: 300–2,000 N (1 Hz) for 16 hours; 5) static: 300 N for 8 hours; 6) dynamic: 300–3,000 N (1 Hz) for 16 hours; and 7) static: 300 N for 8 hours. The acceptance criterion for this test was observed plastic deformations less than 1.5 mm, which represents the diurnal change of the intervertebral disc.25 Under worst-case conditions, the maximum observed displacements were 0.5 mm after the 3,000 N cyclic loading and maximum observed plastic deformations were 0.16 mm for the 14 mm inlay.
Wear testing
Six activL® Artificial Disc specimens were wear tested to 10 million cycles. A complex loading profile combining flexion/extension, lateral bending, axial rotation, and axial load was applied at a frequency of 1 Hz. Specimens were tested in calf serum and deionized water solution with ethylenediaminetetraacetic acid. Specimens were weighed prior to testing and at each 500,000 cycle increment. The acceptance criterion for this test was wear debris consistent with that reported for other lumbar devices. Average cumulative wear was 2.7 mg per 1 million cycles, with no observable wear of the polyethylene inlay.26 Compared to the activL® Artificial Disc, average cumulative wear was six-fold greater (16.6 mg per 1 million cycles) with ProDisc-L and seven-fold greater (19.3 mg per 1 million cycles) with the Charité artificial discs under an identical testing protocol.27 These marked improvements in wear rate with the activL® Artificial Disc are likely attributable to the beta sterilization process, which results in less oxidative damage and reduces delamination compared to the gamma sterilization process used with other lumbar TDRs.28
Wear debris animal study
An animal study was conducted to characterize the local and systemic reactions that may be caused by ultra-high-molecular-weight polyethylene wear debris implanted into the epidural space of New Zealand white rabbits. Animals were injected with solution containing no particles (n=12), 10 million particles (n=12), or 25 million particles (n=12) and sacrificed at 3 or 6 months. Assessments included clinical and neurological observations, and hematological, histological, and gross pathologic methods. The study showed no evidence of neurotoxicity, systemic toxicity, or local effects associated with wear debris.
Clinical experience
The activL® Artificial Disc has been in commercial use in Europe since 2005, with nearly 8,000 discs implanted to date. The commercial experience has been favorable with only four device explants, one device migration, and no device expulsions reported during this time.
Early clinical experience with the activL® Artificial Disc included an unpublished multicenter prospective study conducted in Germany in 2005. At 6 months postsurgery, back pain decreased 51%, Oswestry Disability Index (ODI) scores decreased 39%, and employment increased from 61% to 87%. Another unpublished European multicenter study treated 50 patients with the activL® Artificial Disc and reported an 87% reduction in pain severity and a 92% reduction in ODI at 1 year. A subsequent study reported that segmental motion preservation was superior with activL® Artificial Disc versus fusion in patients with single-level lumbar DDD.29 Based on these favorable initial results, an FDA-investigational device exemption (IDE) trial of the activL® Artificial Disc was initiated in 2007.
The FDA-IDE study of the activL® Artificial Disc was a prospective, multicenter, randomized, single-blind, controlled study (ClinicalTrials.gov NCT00589797).21 Eligible patients reported lumbar pain due to a radiographically confirmed diagnosis of DDD at either L4–L5 or L5–S1 despite at least 6 months of nonsurgical management. Patients were randomly allocated (2:1) to activL® Artificial Disc (n=218) or Control (n=106), consisting of ProDisc-L or Charité based on surgeon preference. The overall treatment success rate at 2 years with activL® Artificial Disc was superior to the Control group (P=0.02). Patients treated with the activL® Artificial Disc also had higher rates of radiographic success (59% vs 43%, P<0.01) and ODI success (75% vs 66%, P=0.08) compared to Controls (Figure 3). The percentage of patients reporting improvement in back pain severity ≥20 mm (90% vs 83%), ODI ≥15 points (88% vs 81%), and Medical Outcomes Study Questionnaire Short Form 36 Physical Component Summary score ≥15% (88% vs 81%) all favored the activL® Artificial Disc group. Patient satisfaction with treatment was over 90% in each of the TDR groups at 2 years. Return to work was 1 month sooner with activL® Artificial Disc (68 days) compared to Controls (97 days) (P=0.08) (Figure 4). Change in range of motion in lateral flexion–extension radiographs was statistically greater with activL® Artificial Disc compared to Controls in segmental rotation (+0.9° vs −1.4°, P<0.01) and translation (+0.6 vs +0.2 mm, P<0.001), while no differences were noted in lateral rotation on side bending radiographs (+0.6° vs +0.8°, P=0.52). Serious adverse events, regardless of cause, were less common in patients treated with activL® Artificial Disc versus Controls through 2 years (30% vs 43%, P=0.02). Serious adverse events related to the TDR were also less common with activL® Artificial Disc (12% vs 19%, P=0.13) (Figure 5). Heterotopic ossification interfering with range of motion was rare (activL® Artificial Disc 1.6%, Controls 1.1%). No evidence of osteolysis or aseptic loosening was identified in postoperative imaging or explant analysis in either group. The percentage of patients undergoing surgical reintervention at the index level was comparable between groups through 2 years (activL® Artificial Disc 2.3%, Control 1.9%).
 | Figure 3 Clinical and radiographic success rates in a randomized controlled trial comparing activL® Artificial Disc to control total disc replacements (ProDisc-L or Charité). |
 | Figure 4 Time to return to work in a randomized controlled trial comparing activL® Artificial Disc to control total disc replacements (ProDisc-L or Charité). |
 | Figure 5 Incidence of complications in a randomized controlled trial comparing activL® Artificial Disc to control total disc replacements (ProDisc-L or Charité). |
Longer term data with activL® Artificial Disc have recently become available. A single-site study reported 6-year outcomes of 32 patients treated with activL® Artificial Disc or ProDisc-L.30 Back pain decreased 89% from baseline (from 87±12 to 9±20) with activL® Artificial Disc and 71% (from 84±8 to 24±29) with ProDisc-L, with superior improvement observed with activL® Artificial Disc (P<0.05) (Figure 6). ODI scores decreased 76% from baseline (from 71±14 to 17±17) with activL® Artificial Disc and 57% (from 64±14 to 27±19) with ProDisc-L, with superior improvement observed with activL® Artificial Disc (P=0.04) (Figure 7). Serious device-related complication rates were 10% for activL® Artificial Disc and 45% for ProDisc-L (P=0.03), with lumbar/leg pain (6.9% vs 15.1%) and implant subsidence (1.4% vs 1.9%) most commonly reported. Reoperation rates at the index level were 5% for activL® Artificial Disc and 27% for ProDisc-L (P=0.11).
 | Figure 6 Changes in back pain severity over 6-year mean follow-up with activL® Artificial Disc versus ProDisc-L. |
 | Figure 7 Changes in Oswestry Disability Index over 6-year mean follow-up with activL® Artificial Disc versus ProDisc-L. |
Additional long-term studies with the activL® Artificial Disc are underway to further characterize the durability of treatment effect. A 7-year postapproval study with 376 patients is ongoing with the objective to evaluate the long-term safety and effectiveness of the activL® Artificial Disc compared to ProDisc-L or Charité. Representative 7-year follow-up imaging from this series is presented in Figure 8. A 10-year global safety study is also planned that will characterize adverse events and complaints in patients where the activL® Artificial Disc is used as intended. The main outcomes of this study will include reoperations, heterotopic ossification, device malfunction, and device removals as well as analysis of all device explants.
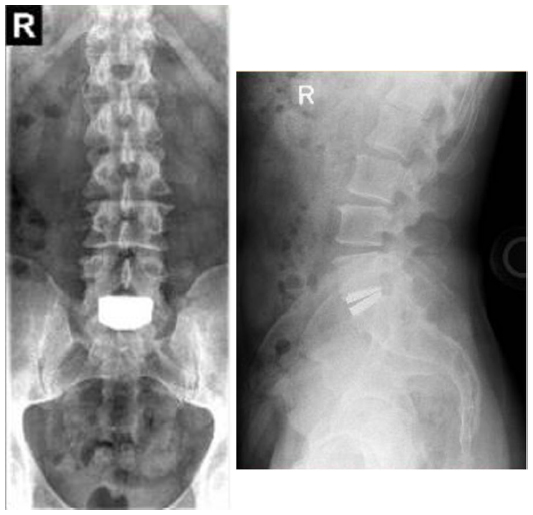 | Figure 8 Anterioposterior (left) and lateral (right) radiographs 7 years following implant with the activL® Artificial Disc. |
Overall, mid- to long-term data with the activL® Artificial Disc demonstrate outcomes that are at least comparable and, in some cases, superior versus previous-generation artificial lumbar discs. Additionally, patient outcomes with activL® Artificial Disc compare favorably to lumbar fusion. An analysis of the 2-year activL® Artificial Disc IDE data compared to 2-year outcomes from randomized controlled trials of TDR versus lumbar fusion31–36 shows lower back pain severity, lower ODI scores, and lower reoperation rates compared to other TDRs and fusion (Figures 9–11).
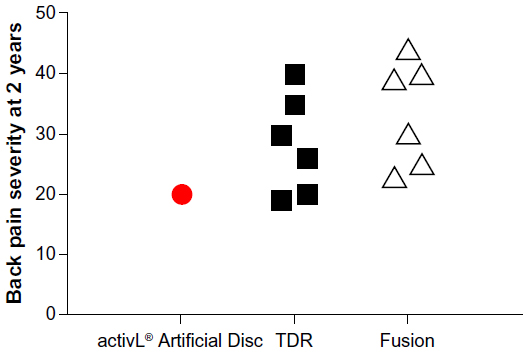 | Figure 9 Comparison of 2-year back pain severity with activL® Artificial Disc versus randomized controlled trial outcomes of lumbar total disc replacement (TDR) or fusion. |
 | Figure 10 Comparison of 2-year Oswestry Disability Index (ODI) with activL® Artificial Disc versus randomized controlled trial outcomes of lumbar total disc replacement (TDR) or fusion. |
 | Figure 11 Comparison of 2-year reoperation rates with activL® Artificial Disc versus randomized controlled trial outcomes of lumbar total disc replacement (TDR) or fusion. |
Discussion
Since the last lumbar TDR approval in the US, nearly a decade passed until the activL® Artificial Disc received FDA approval in 2015. Despite early enthusiasm following approval of the first lumbar TDRs, utilization rates have remained low over the last 10 years,37 which may be attributable to lack of TDR selection, strict indications for use, challenging instrumentation, mixed clinical outcomes, and reimbursement challenges. With recent advances in TDR technology and further refinements to patient eligibility criteria, TDR utilization may increase, particularly given that clinical outcomes with the activL® Artificial Disc demonstrate that next-generation lumbar TDRs perform better than their previous-generation counterparts. As more lumbar TDRs become available and favorable long-term (≥10 years) data continue to accumulate,38–40 this technology may become a mainstay in the physician’s armamentarium of therapeutic options of lumbar DDD.
Although the specific contributors to long-term treatment success with the activL® Artificial Disc have yet to be identified, the introduction of incremental device sizing and replication of physiological range of motion allow the artificial disc to closely mimic the anatomy and function of a healthy intervertebral disc. The range of available device configurations and ability for translational movement are not available in other TDRs. The influence of specific device-related characteristics on patient outcomes deserves further study.
Aside from implant characteristics, appropriate patient selection is arguably the most important factor in determining TDR treatment success.41 At one point in life, almost every individual will suffer an episode of low back pain of sufficient severity to disrupt normal daily activities, including work and recreation.42 Careful differential diagnosis to identify the lumbar disc as the primary pain generator is mandatory. Once a diagnosis of single-level symptomatic lumbar DDD is made, careful attention must be given to potential contraindications to TDR. Patients over 55 years have a higher risk of TDR contraindications, most commonly due to concomitant spinal stenosis, facet arthrosis, high-grade spondylolisthesis, and osteopenia.43 Therefore, although the benefits of TDR are generally comparable between older and younger patients,44 TDR eligibility is higher in the younger population. Lumbar TDR may also be more attractive from the patient’s perspective compared to fusion since recovery from surgery is quicker. Additionally, implant with the activL® Artificial Disc does not interfere with future surgical options if necessary. Specific repositioning and revisions instruments are available that allow for anterior implant translation, removal and replacement of the polyethylene inlay, or complete removal of the implant. Given this, an appropriate continuum of care for the patient with single-level lumbar DDD would begin with nonsurgical treatment and, in patients who fail conservative measures, TDR represents the next therapeutic step in eligible patients, with lumbar fusion as the last resort. Given the superiority of control TDRs versus fusion in previous reports14–17,45 and the favorable outcomes of activL® Artificial Disc versus control TDRs in FDA-IDE study, the benefits of the activL® Artificial Disc over fusion surgery can be inferred, thereby providing additional support for this proposed treatment pathway in patients with chronic lumbar discogenic pain.
In conclusion, the next-generation activL® Artificial Disc is a promising new therapeutic option for patients with single-level lumbar DDD unresponsive to nonsurgical management. Randomized controlled trials confirm improved mid-term effectiveness of this artificial disc compared to earlier-generation TDRs as well as favorable outcomes versus lumbar fusion. Additional safety and effectiveness data are anxiously awaited to confirm long-term treatment durability of the activL® Artificial Disc.
Acknowledgment
This research was funded by Aesculap Implant Systems, LLC.
Disclosure
The authors report no conflicts of interest in this work.
References
DePalma MJ, Ketchum JM, Saullo T. What is the source of chronic low back pain and does age play a role? Pain Med. 2011;12(2):224–233. | |
Licciardone JC. The epidemiology and medical management of low back pain during ambulatory medical care visits in the United States. Osteopath Med Prim Care. 2008;2:11. | |
Schwarzer AC, Aprill CN, Derby R, Fortin J, Kine G, Bogduk N. The prevalence and clinical features of internal disc disruption in patients with chronic low back pain. Spine. 1995;20(17):1878–1883. | |
Manchikanti L, Singh V, Pampati V, et al. Evaluation of the relative contributions of various structures in chronic low back pain. Pain Physician. 2001;4(4):308–316. | |
Carey TS, Garrett JM, Jackman AM. Beyond the good prognosis. Examination of an inception cohort of patients with chronic low back pain. Spine. 2000;25(1):115–120. | |
Rhyne AL, Smith SE, Wood KE, Darden BV. Outcome of unoperated discogram-positive low back pain. Spine. 1995;20(18):1997–2000. | |
Von Korff M. Studying the natural history of back pain. Spine. 1994; 19(18 Suppl):2041S–2046S. | |
Peng B, Fu X, Pang X, et al. Prospective clinical study on natural history of discogenic low back pain at 4 years of follow-up. Pain Physician. 2012;15(6):525–532. | |
Martin BI, Mirza SK, Comstock BA, Gray DT, Kreuter W, Deyo RA. Are lumbar spine reoperation rates falling with greater use of fusion surgery and new surgical technology? Spine. 2007;32(19):2119–2126. | |
Greiner-Perth R, Boehm H, Allam Y, Elsaghir H, Franke J. Reoperation rate after instrumented posterior lumbar interbody fusion: a report on 1680 cases. Spine. 2004;29(22):2516–2520. | |
St-Pierre GH, Jack A, Siddiqui MM, Henderson RL, Nataraj A. Non-fusion does not prevent adjacent segment disease: Dynesys long term outcomes with minimum five years follow-up. Spine. 2016;41(3):265–273. | |
Litrico S, Lonjon N, Riouallon G, et al. Adjacent segment disease after anterior cervical interbody fusion: a multicenter retrospective study of 288 patients with long-term follow-up. Orthop Traumatol Surg Res. 2014;100(6 Suppl):S305–S309. | |
Jacobs WC, van der Gaag NA, Kruyt MC, et al. Total disc replacement for chronic discogenic low back pain: a Cochrane review. Spine. 2013;38(1):24–36. | |
Rao MJ, Cao SS. Artificial total disc replacement versus fusion for lumbar degenerative disc disease: a meta-analysis of randomized controlled trials. Arch Orthop Trauma Surg. 2014;134(2):149–158. | |
Nie H, Chen G, Wang X, Zeng J. Comparison of total disc replacement with lumbar fusion: a meta-analysis of randomized controlled trials. J Coll Physicians Surg Pak. 2015;25(1):60–67. | |
Wei J, Song Y, Sun L, Lv C. Comparison of artificial total disc replacement versus fusion for lumbar degenerative disc disease: a meta-analysis of randomized controlled trials. Int Orthop. 2013;37(7):1315–1325. | |
Ren C, Song Y, Liu L, Xue Y. Adjacent segment degeneration and disease after lumbar fusion compared with motion-preserving procedures: a meta-analysis. Eur J Orthop Surg Traumatol. 2014; 24 (Suppl 1):S245–S253. | |
Food and Drug Administration. CHARITé™ Artificial Disc – P040006. 2004. Available from: http://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/DeviceApprovalsandClearances/Recently-ApprovedDevices/ucm080693.htm. Accessed September 26, 2015. | |
Food and Drug Administration. PRODISC®-L Total Disc Replacement – P050010. 2006. Available from: http://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/DeviceApprovalsandClearances/Recently-ApprovedDevices/ucm077620.htm. Accessed September 26, 2015. | |
Food and Drug Administration. activL® Artificial Disc – P120024. 2015. Available from: http://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/DeviceApprovalsandClearances/Recently-ApprovedDevices/ucm455656.htm. Accessed September 26, 2015. | |
Garcia R, Yue JJ, Blumenthal S, et al. Lumbar total disc replacement for discogenic low back pain: two-year outcomes of the activL multicenter randomized controlled IDE clinical trial. Spine. 2015;40(24):1873–1881. | |
Han JS, Goel VK, Ahn JY, et al. Loads in the spinal structures during lifting: development of a three-dimensional comprehensive biomechanical model. Eur Spine J. 1995;4(3):153–168. | |
Nachemson A. The load on lumbar disks in different positions of the body. Clin Orthop Relat Res. 1966;45:107–122. | |
Panjabi MM, White AA, 3rd. Basic biomechanics of the spine. Neurosurgery. 1980;7(1):76–93. | |
Adams MA, Dolan P, Hutton WC, Porter RW. Diurnal changes in spinal mechanics and their clinical significance. J Bone Joint Surg Br. 1990;72(2):266–270. | |
Grupp TM, Yue JJ, Garcia R, Jr, et al. Biotribological evaluation of artificial disc arthroplasty devices: influence of loading and kinematic patterns during in vitro wear simulation. Eur Spine J. 2009;18(1):98–108. | |
Nechtow W, Hintner M, Bushelow M, Kaddick C. IVD replacement mechanical performance depends strongly on input parameters. 52nd Annual Meeting of the Orthopaedic Research Society; 2006; Chicago, IL. | |
American Academy of Orthopedic Surgeons. What modifications can be made to materials to improve wear behavior? In: Wright TM, Goodman SB, editors. Implant Wear in Total Joint Replacement: Clinical and Biologic Issues, Material and Design Considerations: American Academy of Orthopedic Surgeons; 2001. | |
Nabhan A, Al-Yhary A, Ishak B, Steudel WI, Kollmar O, Steimer O. Analysis of spinal kinematics following implantation of lumbar spine disc prostheses versus fusion: radiological study. J Long Term Eff Med Implants. 2007;17(3):207–216. | |
Yue JJ. Long-term prospective randomized outcomes following lumbar total disc replacement: a single site experience with 6 years follow-up. Paper presented at: ISASS16; April 6, 2016, 2016; Las Vegas, NV. | |
Blumenthal S, McAfee PC, Guyer RD, et al. A prospective, randomized, multicenter Food and Drug Administration investigational device exemptions study of lumbar total disc replacement with the CHARITE artificial disc versus lumbar fusion: part I: evaluation of clinical outcomes. Spine. 2005;30(14):1565–1575; discussion E387-391. | |
Berg S, Tullberg T, Branth B, Olerud C, Tropp H. Total disc replacement compared to lumbar fusion: a randomised controlled trial with 2-year follow-up. Eur Spine J. 2009;18(10):1512–1519. | |
Delamarter R, Zigler JE, Balderston RA, Cammisa FP, Goldstein JA, Spivak JM. Prospective, randomized, multicenter Food and Drug Administration investigational device exemption study of the ProDisc-L total disc replacement compared with circumferential arthrodesis for the treatment of two-level lumbar degenerative disc disease: results at twenty-four months. J Bone Joint Surg Am. 2011;93(8):705–715. | |
Delamarter RB, Bae HW, Pradhan BB. Clinical results of ProDisc-II lumbar total disc replacement: report from the United States clinical trial. Orthop Clin North Am. 2005;36(3):301–313. | |
Gornet MF, Burkus JK, Dryer RF, Peloza JH. Lumbar disc arthroplasty with Maverick disc versus stand-alone interbody fusion: a prospective, randomized, controlled, multicenter investigational device exemption trial. Spine. 2011;36(25):E1600–E1611. | |
Zigler J, Delamarter R, Spivak JM, et al. Results of the prospective, randomized, multicenter Food and Drug Administration investigational device exemption study of the ProDisc-L total disc replacement versus circumferential fusion for the treatment of 1-level degenerative disc disease. Spine. 2007;32(11):1155–1162; discussion 1163. | |
Yoshihara H, Yoneoka D. National trends in the surgical treatment for lumbar degenerative disc disease: United States, 2000 to 2009. Spine J. 2015;15(2):265–271. | |
Siepe CJ, Heider F, Wiechert K, Hitzl W, Ishak B, Mayer MH. Mid- to long-term results of total lumbar disc replacement: a prospective analysis with 5- to 10-year follow-up. Spine J. 2014;14(8):1417–1431. | |
Lu SB, Hai Y, Kong C, et al. An 11-year minimum follow-up of the Charite III lumbar disc replacement for the treatment of symptomatic degenerative disc disease. Eur Spine J. 2015;24(9):2056–2064. | |
David T. Long-term results of one-level lumbar arthroplasty: minimum 10-year follow-up of the CHARITE artificial disc in 106 patients. Spine. 2007;32(6):661–666. | |
Pettine K, Ryu R, Techy F. Why lumbar artificial disc replacements (LADRs) fail. J Spinal Disord Tech. Epub 2015 Jun 26. | |
Deyo RA, Cherkin D, Conrad D, Volinn E. Cost, controversy, crisis: low back pain and the health of the public. Annu Rev Public Health. 1991;12:141–156. | |
Chin KR. Epidemiology of indications and contraindications to total disc replacement in an academic practice. Spine J. 2007;7(4):392–398. | |
Guyer RD, Geisler FH, Blumenthal SL, McAfee PC, Mullin BB. Effect of age on clinical and radiographic outcomes and adverse events following 1-level lumbar arthroplasty after a minimum 2-year follow-up. J Neurosurg Spine. 2008;8(2):101–107. | |
Yajun W, Yue Z, Xiuxin H, Cui C. A meta-analysis of artificial total disc replacement versus fusion for lumbar degenerative disc disease. Eur Spine J. 2010;19(8):1250–1261. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.