Back to Journals » Neuropsychiatric Disease and Treatment » Volume 14
The abnormal activation of D1R/Shp-2 complex involved in levodopa-induced dyskinesia in 6-hydroxydopamine-lesioned Parkinson’s rats
Authors Wu N, Wan Y, Song L, Qi C, Liu Z, Gan J
Received 15 January 2018
Accepted for publication 2 May 2018
Published 5 July 2018 Volume 2018:14 Pages 1779—1786
DOI https://doi.org/10.2147/NDT.S162562
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Wai Kwong Tang
Na Wu, Ying Wan, Lu Song, Chen Qi, Zhenguo Liu, Jing Gan
Department of Neurology, Xinhua Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200092, China
Background: Levodopa-induced dyskinesia (LID) is a troublesome problem in the treatment of Parkinson’s disease (PD). The mechanisms of LID are still mysterious. Recently, the interaction between Shp-2 and D1 dopamine receptor (D1R) has been identified to be indispensable in the D1R-mediated extracellular signal-regulated kinases 1 and 2 (ERK1/2) activation and the occurrence of LID. However, the role of Shp-2 in the D1R-mediated signaling pathway of dyskinetic rat models is not fully clear. We designed this study with the purpose of exploring the role of D1R/Shp-2 complex in the D1R-mediated signaling pathway in the occurrence of LID.
Materials and methods: The 6-hydroxydopamine (6-OHDA) was injected unilaterally to produce the rat models of PD. Successful PD rat models were randomly divided into three groups to receive the treatment with L-3,4-dihydroxyphenylalanine (L-DOPA) + benserazide, L-DOPA + benserazide + D1R antagonist (SCH23390) or D1R agonist (SKF38393). Abnormal involuntary movements were assessed in different groups during the treatment. The interaction between D1R and Shp-2 was confirmed in the sham and LID rats through the methods of coimmunoprecipitation. In addition, the levels of p-Shp-2, p-ERK1/2 and p-mTOR were determined by Western blot in different groups.
Results: After the treatment with L-DOPA + benserazide for 22 days, PD rats presented with dyskinesia. D1R agonist, SKF38393, induced similar involuntary movements in PD rats. In contrast, the dyskinetic movements were not induced by coadministration of L-DOPA + D1R antagonist (SCH23390). The interaction between D1R and Shp-2 in the normal rats was kept stable after the long-term use of L-DOPA. Moreover, we found that the pulsatile levodopa administration induced hyperphosphorylation of Shp-2, ERK1/2 and mTOR, while the coadministration of L-DOPA and D1R antagonist, SCH23390, did not induce the hyperphosphorylation of these proteins.
Conclusion: These data verified the existence of D1R/Shp-2 complex and its crucial role in the D1R-mediated signaling pathway in dyskinetic rats. Focus on the D1R/Shp-2 complex might be a potential treatment of LID in the future.
Keywords: Parkinson’s disease, dyskinesia, levodopa, D1 receptors, Shp-2
Introduction
Parkinson’s disease (PD) is a debilitating neurodegenerative disorder characterized by the progressive loss of dopaminergic neurons in the substantia nigra.1 This disease is the second major neurodegenerative disorder after Alzheimer’s disease.2 The clinical traits of PD are bradykinesia, resting tremor, muscle rigidity and postural instability. To the best of our knowledge, L-3,4-dihydroxyphenylalanine (L-DOPA) continues to be the gold standard therapy for PD.3 However, the long-term use of L-DOPA leads to a severe motor side effect known as levodopa-induced dyskinesia (LID), which depletes its efficacy gradually.4 LID interferes with PD patients’ daily activities and social interactions, resulting in a significant decrease in patients’ quality of life.5 The risk factors for LID include the age of PD onset, duration of L-DOPA, dosage of L-DOPA, etc.6 Despite great progress in basic research on LID in recent years, the mechanisms of LID are still not fully understood.
To the best of our knowledge, the substantia nigra pars compacta provides dopamine input to the striatum, which contains two subtypes of GABAergic efferent neurons that express different dopamine receptors.7 In other words, there are two pathways in those neurons: the direct (motor activation) and the indirect (motor depression) basal ganglia pathways of motor control. D1R is one of the G protein-coupled receptors with seven transmembrane domains.8 Accumulating evidence supported that LID was closely associated with the sensitivity of D1R located on the medium spiny neurons (MSNs).9 It is widely accepted that in rodent and nonhuman primate models, the hypersensitivity of D1 receptor and the abnormal D1R-mediated signaling pathways play an important role in the occurrence of LID.10 In particular, repeated L-DOPA administration led to persistent hyperactivation of cyclic AMP (cAMP) signaling cascade.11 The impulsive L-DOPA stimulation resulted in the overexpression of D1R and the enhanced combination between D1R and G protein, resulting in the increased level of adenylyl cyclase 5/6 and cAMP synthesis.12 An abnormally increased cAMP activated cAMP-dependent protein kinase (PKA) and the activated PKA led to an aberrant phosphorylation of downstream dopamine (DA)- and cAMP-regulated phosphorus–protein 32k DA (DARPP-32).13 Then, the activation of DARPP-32 further inhibited the dephosphorylation of extracellular signal-regulated kinases 1 and 2 (ERK1/2).14
However, studies suggested that in the ventral striatum and nucleus accumbens, the D1R-mediated ERK1/2 activation was dependent on the activation of phosphatase inhibitor, DARPP-32, and in the dorsal striatum, the activation of D1R-induced phosphorylation of ERK1/2 was not related to the activation of DARPP-32.15 It is indicated that other mechanisms might be involved in the D1R-mediated activation of ERK1/2. Recently, Fiorentini et al16 found a new mechanism of D1R-mediated ERK1/2 activation. It was reported that Shp-2, which is a tyrosine phosphatase, interacted with D1R and played a crucial role in the D1R/Shp-2/ERK1/2 signaling pathway. The results were confirmed in both transfected human embryonic kidney cells (HEK293) and dyskinetic rat models.17 There were few studies focused on the interaction of Shp-2 and D1R in the D1R-mediated ERK1/2 activation in LID rat models. Hence, we attempted to corroborate whether Shp-2 interacted with D1R as a complex in dyskinetic rats and whether the interaction of Shp-2 and D1R affected the activation of D1R/Shp-2/ERK1/2 signaling pathway, which was a key mechanism underlying LID.
Furthermore, mTOR, a downstream molecule of D1R-mediated ERK1/2 activation, modulated the mRNA translation and protein production in the occurrence of LID.18 Hence, we explored whether mTOR would be consequently activated as a downstream molecular event of ERK1/2 signaling pathway after the activation of D1R/Shp-2 complex.
Materials and methods
Animals
We used adult male rats (Sprague–Dawley, 180–220 g) in this study. Animals were housed under a 12/12-hour light/dark cycle in a controlled environment at a constant temperature of 23°C and humidity of 50% ± 10%. The rats had free access to food and water and were acclimated for at least 1 week before the L-DOPA/benserazide injections. Animal experiments were performed according to the guidelines of the National Institutes of Health (publication No 80-23). All protocols in the studies were approved by the institutional review board of Xinhua Hospital affiliated to the Shanghai Jiao Tong University Medical School.
6-Hydroxydopamine (6-OHDA) lesion
The 6-OHDA (8 μg dissolved in 4 μL of 0.9% physiological saline containing 0.2% ascorbic acid) was injected into the right medial forebrain bundle of the rats through a stereotaxic apparatus (Narishige, Tokyo, Japan). The coordinates were calculated according to the rat brain atlas as follows: anterior–posterior (AP), −4.4 mm; medial–lateral (ML), −1.2 mm and dorsal–ventral (DV), −7.8 mm; AP, −3.7 mm; ML −1.7 mm; DV, −7.8 mm. The tooth bar was set to −2.4 mm. Three weeks later, the 6-OHDA-lesioned rats received the injection of apomorphine (0.5 mg/kg, intraperitoneal [i.p.]). The rats that exhibited at least seven full turns per minute away from the lesioned side were considered as the successful PD rat models.
Drug treatments
Two sets of experiments were conducted in our study. In the first set of experiments, six PD rats were treated with levodopa (25 mg/kg, i.p.) plus benserazide (6.25 mg/kg, i.p.) for 22 days to induce LID. Meanwhile, we chose six normal rats as control (n = 6). The two groups were compared by coimmunoprecipitation to confirm the existence of D1R/Shp-2 complex. In the second set of experiments, the successful PD rats were randomly divided into three groups: 1) L-DOPA group: PD rats were treated with levodopa (25 mg/kg, i.p.) plus benserazide (6.25 mg/kg, i.p.) twice daily (9:00 and 17:00) for 22 days (n = 6), 2) SCH23390 + L-DOPA group: rats were administered with levodopa/benserazide and D1R antagonist, SCH23390 (0.25 mg/kg, i.p.), for 22 days (n = 6), 3) SKF38393 group: the remaining PD rats were treated with D1R agonist, SKF38393 (1.5 mg/kg, i.p.), for 22 days (n = 6). Furthermore, there was a sham group including six rats who underwent sham surgery (injected with a vehicle solution) and were then treated with intraperitoneal saline twice daily for 22 days (n = 6).
Abnormal involuntary movement (AIM) rating
Rats were observed for L-DOPA-induced AIMs as reported previously, which were performed by an investigator who was unaware of the pharmacological intervention. The assessment of AIMs was immediately conducted after the intervention on day 2, day 7, day 12, day 17 and day 22. Briefly, on test days, rats were individually placed in plastic trays 5 min before the drug treatment. After injections, each rat was assessed for exhibition of orolingual, limb, axial and locomotor movements. The assessment of AIMs was observed for 60 seconds and conducted every 20 minutes during the 120 minutes of the drug treatment. Each of the subitem of AIMs was rated from 0 to 4 according to the degree of rats’ AIMs. Subitem score 0 was assigned for the absence of AIMs; 1 for occasional AIMs (<50% of observation time); 2 for frequent AIMs (>50% of observation time); 3 for continuous AIMs interrupted by strong sensory stimuli, and 4 for continuous uninterrupted AIMs.
Coimmunoprecipitation and immunoblot
We chose six normal rats and six LID rats for coimmunoprecipitation. Rat striatal tissue was homogenized by sonication in an immunoprecipitation lysis buffer containing 50 mM Tris (pH 7.4), 150 mM NaCl, 1% Nonidet P-40 (Thermo Fisher Scientific, Waltham, MA, USA) plus a protease inhibitor cocktail and a phosphatase inhibitor cocktail (Hoffman-La Roche Ltd., Basel, Switzerland) and then centrifuged at 800× g for 10 minutes at 4°C. The supernatant was collected and centrifuged at 11,000× g for 30 minutes at 4°C. Then, the pellet we collected was used for coimmunoprecipitation. Samples were incubated with a mouse antibody against D1R (EMD Millipore, Billerica, MA, USA) or a rabbit antibody against Shp-2 (Proteintech, Rosemont, IL, USA) overnight at 4°C. The complex was precipitated with protein G agarose beads or protein A agarose beads by gentle rocking for 3 hours at 4°C; then, the beads were washed five times. Elution buffer was added into the pellet; each pellet was added with 60 μL of elution buffer and incubated for 5 minutes at room temperature. After the pellets were centrifuged, we collected the supernatant. Samples were suspended in a buffer containing 0.5% sodium dodecyl sulfate (SDS) and boiled for 5 minutes. Proteins were resolved by SDS-polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride membrane (EMD Millipore). Membranes were blocked in 5% nonfat milk for 1 hour at room temperature and incubated with a mouse primary antibody against D1R (EMD Millipore) or a rabbit primary antibody against Shp-2 (Proteintech) overnight at 4°C. Membranes were then washed and incubated with horseradish peroxidase conjugated secondary antibodies (1:1,000) for 1 hour at room temperature. Immunoblots were developed with the enhanced electrochemiluminescence reagent (GE Healthcare Bio-Sciences Corp., Piscataway, NJ, USA) and captured by a Bio-Rad molecular imager (Bio Rad, Hercules, CA, USA).
Western blots
The rats were sacrificed immediately by decapitation. Corpus striatum of the lesioned side from each rat was obtained. We also removed the midbrain to detect the expression of tyrosine hydroxylase (TH) at substantia nigra. Tissues were homogenized in lysis buffer at 4°C containing Tris at pH 7.4 (50 mM) and NaCl (50 mM), 1 mM each of ethylenediaminetetraacetic acid, ethylene glycol bis(2-aminoethyl ether)tetraacetic acid, phenylmethanesulfonyl fluoride, sodium orthovanadate, sodium fluoride, 1% SDS and a protease inhibitor cocktail (Hoffman-La Roche Ltd.). The supernatant was collected after centrifugation at 12,000× g for 10 minutes at 4°C. After protein concentration measurement with a bicinchoninic acid (BCA) assay kit (Pierce, Appleton, WI, USA), samples containing 40 μg proteins were resolved by SDS-polyacrylamide gel (10%), transferred onto polyvinylidene difluoride membrane (Bio-Rad Laboratories Inc., Hercules, CA, USA) and then blotted for 2 hours at room temperature in Tris-buffered saline containing 0.1% Tween 20 and 5% nonfat powdered milk. The membrane was incubated overnight at 4°C with polyclonal rabbit antibodies (dilutions: TH 1:5,000; D1R 1:1,000; Shp-2 1:1,000; phospho-Shp-2 1:1,000; ERK 1:1,000; phospho-ERK 1:1,000; m-TOR 1:1,000; phospho-mTOR 1:1,000; Scr 1:1,000; phospho-Scr 1:1,000; GAPDH 1:1,000; EMD Millipore). The membrane was then incubated with anti-rabbit horseradish peroxidase-conjugated IgG (dilutions: 1:1,000; Cell Signaling, Danvers, MA, USA) for 2 hours at room temperature. Immunoreactive bands were scanned with Image-Pro plus 6.0 analyses software; the optical density (OD) values were calculated with a computerized image analysis system (Image Lab; Bio Rad).
Statistics analyses
Data were expressed as mean ± SD. An analysis of variance (ANOVA) followed by least significant difference post hoc comparison tests were used to compare the experimental data between groups. Statistical significance was set at p < 0.05. Statistical tests were performed with SPSS 17.0 software (SPSS Inc., Chicago, IL, USA).
Results
Effects of 6-OHDA on the expression of TH
PD is characterized by severe loss of dopaminergic neurons and depletion of dopamine in substantia nigra. TH is one of the key markers in the metabolism of dopaminergic production. The expression of TH in substantia nigra in PD rats could validate the lesion of dopamine neurons with 6-OHDA. We found that the injection of 6-OHDA significantly reduced the expression of TH in PD rats and LID rats. Therefore, in our study, the injection of 6-OHDA into medial forebrain bundle resulted in diminished dopamine neurons; thus, it showed that the models of PD rats were successful (Figure 1).
 | Figure 1 Diminished expression of TH in substantia nigra in PD and LID rats. |
PD rats show different AIM scores after administration of L-DOPA, D1R antagonist or agonist
We divided PD rats into three groups: 1) L-DOPA group, 2) SCH23390 + L-DOPA group, 3) SKF38393 group. Then AIMs were assessed including axial, limb, orolingual and locomotor movements.
Rats in the L-DOPA group were treated with levodopa plus benserazide twice daily for 22 days. As shown in Figure 2, a continuous increase in AIM scores in PD rats was observed during the period of L-DOPA treatment. The axial AIMs increased from 2.53 ± 0.22 (day 2) to 3.36 ± 0.18 (day 22; p < 0.05; Figure 2A), and the limb AIMs increased from 1.73 ± 0.30 to 2.67 ± 0.26 (p < 0.05; Figure 2B). The orolingual AIMs increased from 1.77 ± 0.25 (day 2) to 2.63 ± 0.30 (day 22; p < 0.05; Figure 2C), and the locomotor AIMs increased from 2.73 ± 0.35 to 3.60 ± 0.15 (p < 0.05; Figure 2D). A significant rise was also observed in all the subitems of AIMs in the rats of the SKF38393 group. The axial AIMs showed a significant increment from 2.45 ± 0.18 (day 2) to 2.83 ± 0.20 (day 22; p < 0.01; Figure 2A), and the limb AIMs showed a significant increment from 2.03 ± 0.32 (day 2) to 2.20 ± 0.32 (day 22; p < 0.05; Figure 2B). The orolingual AIMs showed a significant increment from 1.70 ± 0.14 (day 2) to 2.20 ± 0.14 (day 22; p < 0.05; Figure 2C), and the locomotor AIMs showed a significant increment from 2.57 ± 0.15 (day 2) to 3.47 ± 0.18 (day 22; p < 0.05; Figure 2D). Moreover, axial AIMs, limb AIMs, orolingual AIMs and locomotor AIMs of the SCH23390 + L-DOPA group were lower than AIMs of the L-DOPA group, especially on day 12 and day 17 (p < 0.05). In contrast, such an increase in AIMs scores was not observed in the rats of the SCH23390 + L-DOPA group. The axial AIMs score on day 2 (2.13 ± 0.18) was not statistically different from the score on day 22 (2.07 ± 0.09; p > 0.05; Figure 2A). No difference was detected in the score of the limb AIMs, orolingual AIMs and locomotor AIMs between day 2 and day 22 (p > 0.05; Figure 2B–D).
 | Figure 2 PD rats show different AIMs scores after the administration of L-DOPA, D1R antagonist or agonist. |
D1R interacts with Shp-2 in the striatal neurons
Then, we wanted to confirm the interaction between D1R and Shp-2 in striatal neurons of the normal and LID groups through the method of coimmunoprecipitation. By using anti-D1R antibodies, we detected the band of Shp-2 in the protein complex extracted from the striatal neurons of the sham and LID rats (Figure 3A). Meanwhile, we also found the D1R band in the protein complex extracted from the striatal neurons of the normal and LID rats with the anti-Shp-2 antibodies (Figure 3B). Our results indicated that D1R was constitutively associated with Shp-2 in the striatal neurons of normal rats and the interaction between D1R and Shp-2 would not be affected by the long-term pulsatile L-DOPA stimulation in the occurrence of LID.
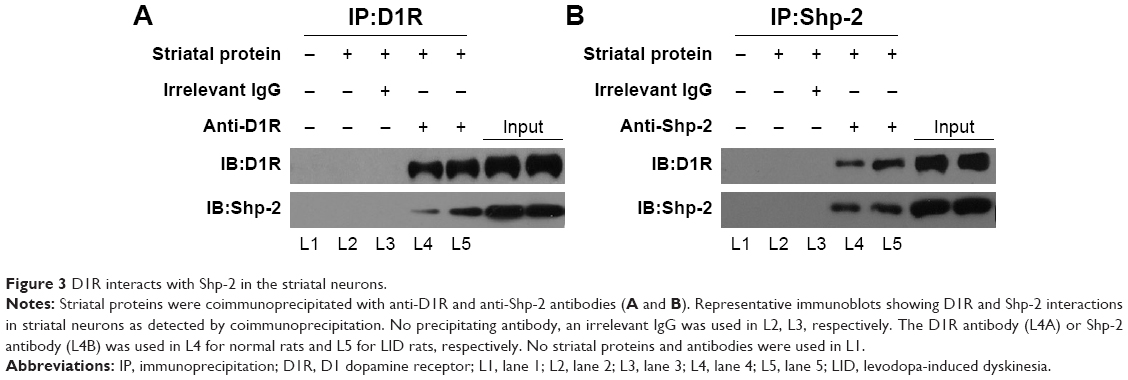 | Figure 3 D1R interacts with Shp-2 in the striatal neurons. |
Molecular events underlying LID involving D1R/Shp-2 complex and its downstream signaling factors, such as ERK1/2 and mTOR
Western blot was used to detect the key elements of D1R/ERK1/2 pathway, including Shp-2, ERK1/2 and mTOR. First, we detected p-Shp-2 expression levels. As shown in Figure 4A, L-DOPA + benserazide injected twice daily for 22 days (group 2) induced an elevated expression compared to the sham group (group 1; p < 0.05). However, a lower expression of p-Shp-2 was detected after L-DOPA plus SCH23390 injection for 22 days (group 3) than in group 2. We also found that D1R agonist, SKF38393 (group 4), increased phosphorylation of Shp-2 levels significantly. There were no differences of total mTOR level among these groups (Figure 4A). This implied that the aberrant phosphorylation of Shp-2 could be activated by D1R.
 | Figure 4 Molecular events underlying LID involving D1R/Shp-2 complex and its downstream signaling factors, such as ERK1/2 and mTOR. |
Next, we detected the levels of ERK, which is a biochemical marker of LID. The results showed that the long-term use of L-DOPA increased the levels of p-ERK in group 2 (p < 0.05). In contrast, we found reduced levels of p-ERK in group 3 compared to group 2. Furthermore, SKF38393, as an agonist of D1R, showed an enhanced expression of p-ERK in group 4 similar to group 2. Then, the levels of m-TOR and p-mTOR in the striatum were analyzed by Western blot. As shown in Figure 4C, after repeated administration of L-DOPA, p-mTOR levels of group 2 were increased in the striatum compared to vehicle-treated rats in group 1. Similarly, rats in group 4 showed an increased level of p-mTOR administered with SKF38393. In contrast, the administration of SCH23390 and L-DOPA prevented the increase of p-mTOR in group 3. Moreover, as shown in Figure 4B and C, no significant difference in ERK and mTOR levels was found in the four groups.
Discussion
Although L-DOPA has been the most effective medical treatment for PD, it could trigger troublesome AIMs known as LID in PD patients after years of medical treatment.19 Since LID significantly impairs PD patients’ motor ability and their quality of life,20 effective strategies are urgently required to solve the problems. The clarification of mechanisms underlying the occurrence of LID would help to provide more information for effective clinical intervention.
We compared the severity of involuntary movements among the PD rats treated with different interventions. We found that chronic L-DOPA administration or D1R agonist, SKF38393, readily induced dyskinesia in the 6-OHDA-lesioned rats. In contrast, the coadministration of D1R antagonist, SCH23390, and L-DOPA significantly reduced the subitems of AIM scores.
It is universally known that the occurrence of LID has been considered to be mainly attributed to the overresponse of D1R induced by the pulsatile dopamine stimulation.21 It was reported that there were two pathways in striatum convey the D1R signaling to ERK1/2. One was that D1R potentiated the cAMP/PKA/DARPP-32 signaling pathway and then decreased ERK1/2 dephosphorylation and signal amplification; the other was that D1R interacted with the tyrosine phosphatase Shp-2 as a complex which initiated ERK1/2 activation and then amplified the ERK1/2 signaling.15 The D1R/cAMP/DARPP-32/ERK1/2 pathway was widely studied in the LID rat model, revealing that D1R-mediated Shp-2-ERK1/2 activation underlying the LID pathway was a novel molecular mechanism. Fiorentini et al16 proved that the activation of ERK signaling pathway was dependent on the enhanced phosphorylation of Shp-2 in HEK293 cells. The overexpression of inactive Shp-2 (C/S) in cultured striatal neurons, or silencing Shp-2 expression by small interfering RNA, abolished the aberrant phosphorylation of ERK1/2. In vivo, it was found that the association of D1R with Shp-2 was disturbed by knocking down Shp-2. Then, Shp-2 knockdown obviously decreased ERK1/2 phosphorylation and attenuated the severity of LID.22 All the results mentioned earlier indicated that the activation of Shp-2 might be necessary for the D1R-mediated ERK1/2 activation and the induction of LID. Currently, we have confirmed the interaction between D1R and Shp-2 by coimmunoprecipitation. Moreover, we found an increased phosphorylation of Shp-2 induced by pulsatile stimulation of L-DOPA or D1R agonist. The aberrant phosphorylation of Shp-2 could be significantly inhibited by D1R antagonist. This suggested that, in LID rats, the hyperphosphorylation of Shp-2 required D1R activation. Accordingly, we proposed that agonism or antagonism toward D1R might affect the D1R/Shp-2 complex.
Then, we detected two important factors in the D1R/ERK1/2 signaling pathway, ERK1/2 and mTOR. Many studies evidenced that the activation of ERK1/2 was required in the gene transcription and the activation of the mTORC1-dependent signaling pathway,23,24 thus speeding up the translation process of mRNA led to an abnormal protein synthesis and the occurrence of LID.25 Our results showed that D1R antagonist, SCH23390, significantly decreased phosphorylation of ERK1/2 in 6-OHDA-lesioned PD rats, induced by L-DOPA. However, L-DOPA and D1R agonist, SKF38393, had the opposite effects. Then, we found that after the long-term use of L-DOPA, p-mTOR levels were increased in the striatum. Similarly, rats showed an increase in p-mTOR after treatment with SKF38393. Therefore, the coadministration of SCH23390 and L-DOPA suppressed the increase in p-mTOR expression. The results further proved that the aberrant phosphorylated expression of Shp-2 by D1R activation could lead to hyperphosphorylation of ERK1/2 and mTOR, both of which played crucial roles in the D1R-mediated signaling pathway.
Conclusion
Overall, our results verified the existence of D1R/Shp-2 complex and its crucial role in the D1R-mediated signaling pathway in dyskinetic rats. Disturbing the association of D1R with Shp-2 may be a valuable therapeutic strategy for LID. Moreover, the results reported in this study are the basis for future study.
Acknowledgment
The study was supported by the National Key R&D Program of China (2017YFC1310300, 2016YFC1306600), Projects of National Science Foundation of China (81400925, 81471148, 81671273, 81771211, 81703852), Projects of the Shanghai Committee of Science and Technology (17401901000) and SHSMU-ION Research Center for Brain Disorders (2015NKX007).
Disclosure
The authors report no conflicts of interest in this work.
References
Mosharov EV, Borgkvist A, Sulzer D. Presynaptic effects of levodopa and their possible role in dyskinesia. Mov Disord. 2015;30(1):45–53. | ||
Manoharan S, Guillemin GJ, Abiramasundari RS, Essa MM, Akbar M, Akbar MD. The role of reactive oxygen species in the pathogenesis of Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease: a mini review. Oxid Med Cell Longev. 2016;2016:8590578. | ||
Rizek P, Kumar N, Jog MS. An update on the diagnosis and treatment of Parkinson disease. CMAJ. 2016;188(16):1157–1165. | ||
Voon V, Napier TC, Frank MJ, et al. Impulse control disorders and levodopa-induced dyskinesias in Parkinson’s disease: an update. Lancet Neurol. 2017;16(3):238–250. | ||
Ferrazzoli D, Carter A, Ustun FS, et al. Dopamine replacement therapy, learning and reward prediction in Parkinson’s disease: implications for rehabilitation. Front Behav Neurosci. 2016;10:121. | ||
Oertel W, Schulz JB. Current and experimental treatments of Parkinson disease: a guide for neuroscientists. J Neurochem. 2016;139(Suppl 1):325–337. | ||
Feyder M, Bonito-Oliva A, Fisone G. L-DOPA-induced dyskinesia and abnormal signaling in striatal medium spiny neurons: focus on dopamine D1 receptor-mediated transmission. Front Behav Neurosci. 2011;5:71. | ||
Sharma S, Singh S, Sharma V, Singh VP, Deshmukh R. Neurobiology of l-DOPA induced dyskinesia and the novel therapeutic strategies. Biomed Pharmacother. 2015;70:283–293. | ||
Cenci MA. Presynaptic mechanisms of l-DOPA-induced dyskinesia: the findings, the debate, and the therapeutic implications. Front Neurol. 2014;5:242. | ||
Francardo V, Cenci MA. Investigating the molecular mechanisms of L-DOPA-induced dyskinesia in the mouse. Parkinsonism Relat Disord. 2014;20(Suppl 1):S20–S22. | ||
Schaeffer E, Pilotto A, Berg D. Pharmacological strategies for the management of levodopa-induced dyskinesia in patients with Parkinson’s disease. CNS Drugs. 2014;28(12):1155–1184. | ||
Santini E, Sgambato-Faure V, Li Q, et al. Distinct changes in cAMP and extracellular signal-regulated protein kinase signalling in L-DOPA-induced dyskinesia. PLoS One. 2010;5(8):e12322. | ||
Svenningsson P, Nishi A, Fisone G, Girault JA, Nairn AC, Greengard P. DARPP-32: an integrator of neurotransmission. Annu Rev Pharmacol Toxicol. 2004;44:269–296. | ||
Valjent E, Pascoli V, Svenningsson P, et al. Regulation of a protein phosphatase cascade allows convergent dopamine and glutamate signals to activate ERK in the striatum. Proc Natl Acad Sci U S A. 2005;102(2):491–496. | ||
Gerfen CR, Paletzki R, Worley P. Differences between dorsal and ventral striatum in Drd1a dopamine receptor coupling of dopamine- and cAMP-regulated phosphoprotein-32 to activation of extracellular signal-regulated kinase. J Neurosci. 2008;28(28):7113–7120. | ||
Fiorentini C, Savoia P, Savoldi D, Barbon A, Missale C. Persistent activation of the D1R/Shp-2/Erk1/2 pathway in l-DOPA-induced dyskinesia in the 6-hydroxy-dopamine rat model of Parkinson’s disease. Neurobiol Dis. 2013;54:339–348. | ||
Fiorentini C, Mattanza C, Collo G, Savoia P, Spano P, Missale C. The tyrosine phosphatase Shp-2 interacts with the dopamine D (1) receptor and triggers D(1) -mediated Erk signaling in striatal neurons. J Neurochem. 2011;117(2):253–263. | ||
Santini E, Heiman M, Greengard P, Valjent E, Fisone G. Inhibition of mTOR signaling in Parkinson’s disease prevents L-DOPA-induced dyskinesia. Sci Signal. 2009;2(80):ra36. | ||
Calabresi P, Di Filippo M, Ghiglieri V, Tambasco N, Picconi B. Levodopa-induced dyskinesias in patients with Parkinson’s disease: filling the bench-to-bedside gap. Lancet Neurol. 2010;9(11):1106–1117. | ||
Müller-Rebstein S, Trenkwalder C, Oertel WH, Culmsee C, Eckermann G, Höglinger GU. Pharmacotherapy of Parkinson’s disease: aspects of drug safety. Nervenarzt. 2017;88(8):888–894. | ||
Song L, Zhang Z, Hu R, et al. Targeting the D1-N-methyl-D-aspartate receptor complex reduces L-dopa-induced dyskinesia in 6-hydroxydopamine-lesioned Parkinson’s rats. Drug Des Devel Ther. 2016;10:547–555. | ||
Fiorentini C, Savoia P, Savoldi D, et al. Shp-2 knockdown prevents l-dopa-induced dyskinesia in a rat model of Parkinson’s disease. Mov Disord. 2016;31(4):512–520. | ||
Santini E, Feyder M, Gangarossa G, Bateup HS, Greengard P, Fisone G. Dopamine- and cAMP-regulated phosphoprotein of 32-kDa (DARPP-32)-dependent activation of extracellular signal-regulated kinase (ERK) and mammalian target of rapamycin complex 1 (mTORC1) signaling in experimental parkinsonism. J Biol Chem. 2012;287(33):27806–27812. | ||
Santini E, Valjent E, Fisone G. mTORC1 signaling in Parkinson’s disease and L-DOPA-induced dyskinesia: a sensitized matter. Cell Cycle. 2010;9(14):2713–2718. | ||
Brugnoli A, Napolitano F, Usiello A, Morari M. Genetic deletion of Rhes or pharmacological blockade of mTORC1 prevent striato-nigral neurons activation in levodopa-induced dyskinesia. Neurobiol Dis. 2016;85:155–163. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.