Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 12
Th17 profile in COPD exacerbations
Authors Ponce-Gallegos MA ![]() , Ramírez-Venegas A
, Ramírez-Venegas A ![]() , Falfán-Valencia R
, Falfán-Valencia R ![]()
Received 9 March 2017
Accepted for publication 19 April 2017
Published 22 June 2017 Volume 2017:12 Pages 1857—1865
DOI https://doi.org/10.2147/COPD.S136592
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Marco Antonio Ponce-Gallegos,1–3 Alejandra Ramírez-Venegas,4 Ramcés Falfán-Valencia1
1HLA Laboratory, Instituto Nacional de Enfermedades Respiratorias Ismael Cosío Villegas, Mexico City, Mexico; 2Medicine Academic Unit, Universidad Autónoma de Nayarit. Tepic, Nayarit, Mexico; 3Interinstitutional Program for Strengthening Research and the Postgraduate in the Pacific (Dolphin), Tepic, Nayarit, México; 4Tobacco Smoking and COPD Research Department, Instituto Nacional de Enfermedades Respiratorias Ismael Cosío Villegas, Mexico City, Mexico
Abstract: COPD is characterized by an ongoing inflammatory process of the airways that leads to obstruction or limitation of airflow. It is mainly associated with exposure to cigarette smoke. In addition, it is considered, at present, a serious public health problem, ranking fourth in mortality worldwide. Many cells participate in the pathophysiology of COPD, the most important are neutrophils, macrophages and CD4+ and CD8+ T cells. Neutrophil migration to the inflammation area could be mediated largely by cytokines related to CD4+ Th17 lymphocytes, because it has been shown that IL-17A, IL-17F and IL-22 act as inducers for CXCL8, CXCL1, CXCL5, G-CSF, and GM-CSF secretion by epithelial cells of the airways. The aims of these molecules are differentiation, proliferation and recruitment of neutrophils. Furthermore, it is believed that CD4+ lymphocytes Th17 may be involved in protection against pathogens for which Th1 and Th2 are not prepared to fight. In COPD exacerbations, there is an increased cellularity in the lung region and respiratory tract. Therefore, the increase in the number of neutrophils and macrophages in the airways and the increase in proinflammatory cytokines are directly related to the severity of exacerbations and that is the importance of the functions of Th17 profile in this entity.
Keywords: IL-17A, bacteria, virus, IL-17F, IL-22, tobacco smoking
Introduction
COPD is characterized by a continuous inflammatory process of the airways, leading to obstruction or limitation of airflow, which is mainly associated with exposure to cigarette smoke.1,2 However, only 10%–20% of smokers will develop the disease at some day in their life. These data lead to the belief that this disease is due to multiple factors that interact with each other. Among the most prominent are genetic factors that can condition patients to have a certain susceptibility to COPD. For example, deficiency of alpha1-antitrypsin (AAT) (responsible for inhibiting proteases, whose function is to deplete the extracellular matrix) is present in at least 1% of the carriers of the disease in Europe and the United States and other regions around the world such as Africa and Australia.3
Many cells participate in the pathophysiology of COPD, the most important are neutrophils, macrophages, CD4+ and CD8+ T lymphocytes and the chemical mediators they produce (cytokines, chemokines, enzymes, growth factors, etc.). For example, neutrophil elastase (NE) is an enzyme that degrades the extracellular matrix to facilitate the migration of the neutrophil through the lung parenchyma to the inflammation area. People harboring AAT deficiency alleles have a greater susceptibility to presenting the disease or presenting it at an earlier age.1,4 On the other hand, cytokines such as TNF-α, IL-1β, IL-6, CXCL8 (also called IL-8), IL-22, IL-23, IL-17A, IL-17F and TGF-β among others are increased in samples from patients with COPD, suggesting an important role in its pathophysiology and in the exacerbations that may occur due to infections by pathogenic microorganisms, with bacteria and viruses being the most prevalent.4,5
Epidemiology
COPD is now considered a serious public health problem, reaching the fourth place in mortality worldwide and in Mexico as well.6,7 Globally, this disease is present in ~600 million of people, especially in low- and middle-income countries. It is estimated that in 2020, it will rank among the first three causes of death worldwide.8 COPD and asthma are the airway obstructive pathologies more prevalent.9
COPD
The Global Initiative for Chronic Obstructive Lung Disease (GOLD) defines COPD as a common and treatable disease characterized by the persistent limitation of airflow, which is usually progressive and associated with an increase in the inflammatory response in the airways and in the lungs by harmful particles and gases.10 Also, this guide defines exacerbations as an acute event characterized by worsening of patient’s respiratory symptoms that go beyond daily variations and leads to a change in medication.6,10,48
In addition, the guide also points out that a patient with frequent exacerbating COPD will be considered when he has two or more exacerbations per year, where the best predictor will be the history of previous treatments for exacerbations.10 Indeed, about half of COPD patients die after 4 years of their first episode of exacerbation.11 The symptomatology of COPD is diverse, depending on the phenotype (emphysema and chronic bronchitis, the most common). However, there is a range of signs and symptoms that occur more frequently, such as dyspnea of small efforts, chronic cough and increased sputum production. In addition, other clinical data that may appear are hypoxemia, hypercapnia, the so-called “chest in barrel” (which appears due to lack of elasticity in the lungs and does not retract properly), weight loss, etc.6,10,48
Molecular mechanisms in COPD
Innate immunity
COPD is characterized by a chronic inflammatory process caused by tobacco smoke, harmful particles or gases that are capable of activating the cascade of inflammatory reactions that cause tissue destruction in the airways.5 Pathological changes that are induced as a consequence of chronic exposure to cigarette smoke are observed more early in airway epithelium, which acts as a barrier that protects the lungs from environmental factors and also from >4,000 toxic compounds present in tobacco smoke.12
Toxic particles contained in the cigarette smoke generate tissue remodeling (Figure 1) as a consequence of the damage they cause to airway epithelium.13 The most pronounced changes are shortening of the cilia (resulting in less mobility of the mucus produced by goblet cells and less elimination of pathogens), metaplasia (change from one cell type to another, which in normal situations would not be present in a specific tissue) of squamous epithelial cells, hyperplasia of goblet-producing goblet cells and basal cells (a type of cells capable of differentiating into ciliated epithelial cells and goblet cells, caused by the action of growth factors such as the epidermal growth factor [EGF]), in addition to the loss of tight junctions that, under normal conditions, work as an impermeable barrier and protect the respiratory tract from pathogens, xenobiotics and other harmful particles.9,13
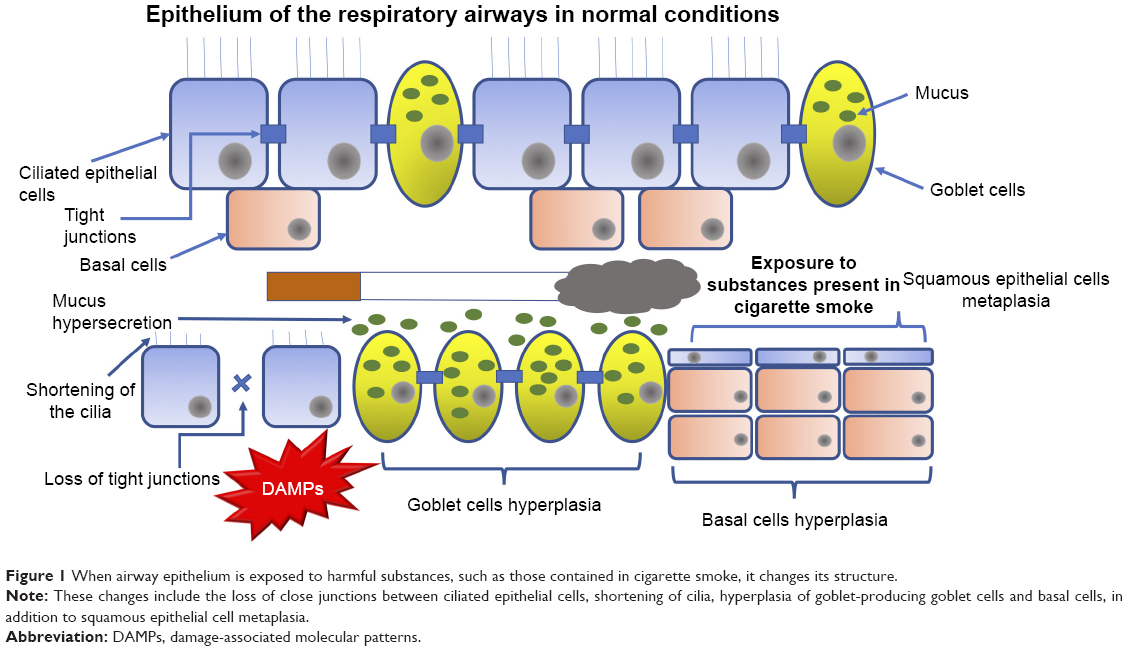 | Figure 1 When airway epithelium is exposed to harmful substances, such as those contained in cigarette smoke, it changes its structure. |
Once the damage is generated in airway epithelial cells, they secrete chemical mediators (chemokines, cytokines, etc.) in order to generate and maintain an inflammatory response against foreign agents.5 Cell death produced releases damage-associated molecular patterns (DAMPs) such as heat shock proteins, S100 protein and high mobility group box 1 (HMGB1),14 which are recognized by extracellular receptors present in neutrophils, macrophages and dendritic cells (DCs; such as Toll-like receptors [TLRs], for example)15–17 and trigger an intracellular signaling cascade led by the transcription factors Myd88 and NF-κB that culminate in the release of proinflammatory cytokines, such as IL-1β and IL-18 (a multiprotein complex responsible for activating caspase 1 and releasing mature forms of various cytokines),18 which, together with other cytokines (TNF-α, CXCL8, IL-6, among others), are responsible for the recruitment of leukocytes to the inflammation zone.
Once in the area to which they were recruited, leukocytes (mainly neutrophils and macrophages) release products stored in their granules, such as enzymes responsible for destroying the extracellular matrix (MMP-9, MMP-12, NE), which help their migration through the lung parenchyma but, in turn, generate damage in the lung, thus promoting the progressive reduction of its functionality.19
Adaptive immunity
Mostly, adaptive immunity appears due to the action of DCs that, in the first instance, pick up the signals of damage of diverse cell types or the antigens that it recognizes as strangers and takes them to the lymph nodes and it is there where it happens the antigen presentation by major histocompatibility complex (MHC) molecules either class I or class II, which results in the differentiation and polarization of T lymphocytes, which recognize the antigen presented by mediated T lymphocyte receptor (T cell receptor [TCR]) (Figure 2).20,21 In addition, the production of proinflammatory cytokines is required for this polarization to occur. For example, a microenvironment rich in IL-12 will lead to the activation and differentiation of natural killer (NK) lymphocytes.22
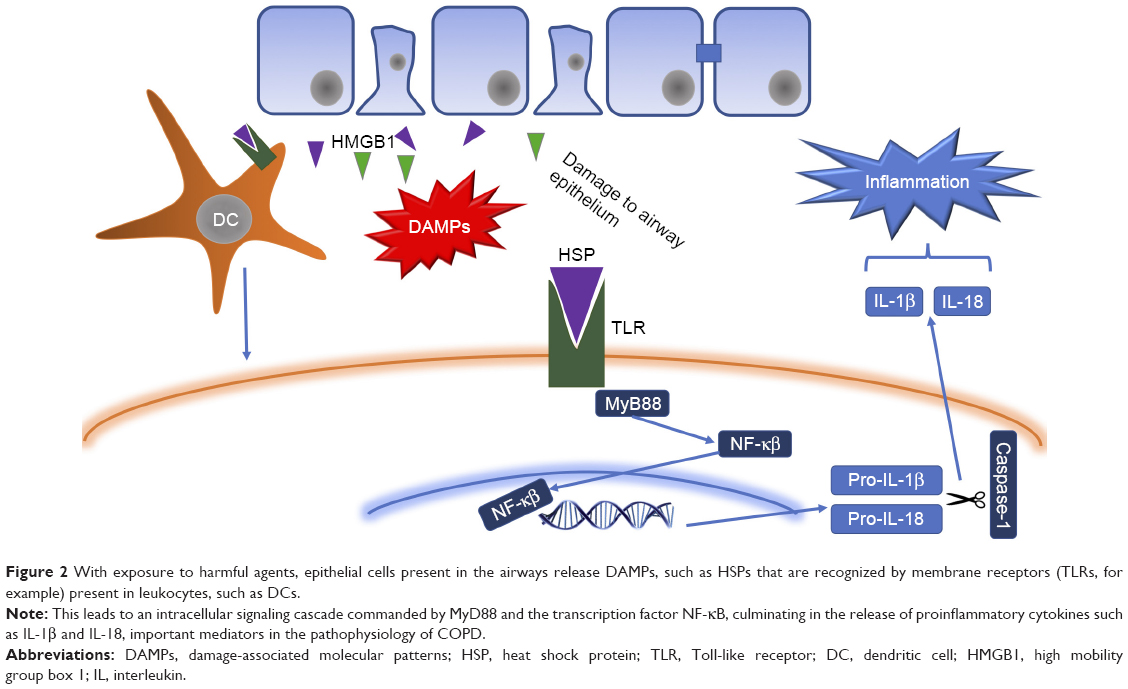 | Figure 2 With exposure to harmful agents, epithelial cells present in the airways release DAMPs, such as HSPs that are recognized by membrane receptors (TLRs, for example) present in leukocytes, such as DCs. |
It has recently been proposed that COPD may be an entity of autoimmune etiology, because autoantibodies have been found against peptides present in the extracellular matrix of lung tissue. Among them, the most important is elastin (anti-elastin antibodies are then formed). These autoantigens would be responsible, in large part, for mediating and perpetuating the inflammatory response present in the disease.2 In this context, B lymphocytes present in the peribronchial lymph nodes will be of special importance. Once sensitized by CD4+ T lymphocytes, they will be able to synthesize and secrete antibodies (which may be directed against host peptides).5
Th17 lymphocytes
Migration of neutrophils to the inflammation region may be mediated largely by cytokines related to CD17+ Th17 lymphocytes (Figure 3), since it has been shown that IL-17A, IL-17F and IL-22 serve as inducers for the secretion of CXCL8, CXCL5, G-CSF, and GM-CSF by airway epithelial cells, which have as their mission the differentiation, proliferation and recruitment of these cells.23
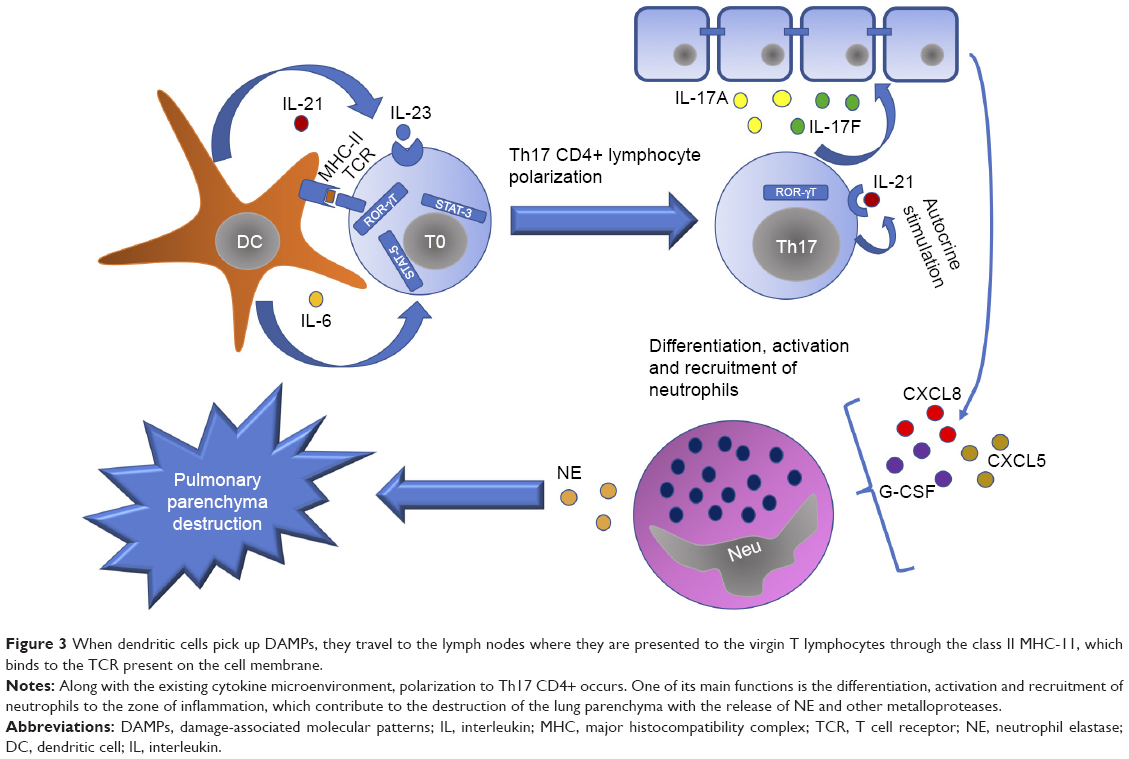 | Figure 3 When dendritic cells pick up DAMPs, they travel to the lymph nodes where they are presented to the virgin T lymphocytes through the class II MHC-11, which binds to the TCR present on the cell membrane. |
Th17 CD4+ lymphocytes are characterized by the production of IL-17 and are believed to be involved in protection against microorganisms for which Th1 and Th2 are not prepared to fight, such as certain extracellular bacteria and fungi. Its differentiation from naive T lymphocytes occurs from three signals: first, the binding of the TCR to the antigen; second, the action of costimulators (for example, B7 binding to CD28 present on the lymphocyte membrane); third, the environment of cytokines that exist in the medium, for example, IL-1β, IL-6, IL-21, IL-23 and TGF-β.24 In this way, naive T lymphocyte is polarized to Th17 lymphocyte from transcription factors characteristic of that cell line, such as RORγT, STAT-3 and STAT-5.25,26
Once polarization of the naive T lymphocyte occurs in Th17 lymphocyte, it synthesizes IL-21 to generate an autocrine stimulation and, thus, to activate the transcription factor RORγT for the synthesis of new cytokines, especially of the IL-17 family.27 In addition, IL-23 in combination with TGF-β also induces the activation of RORγT and subsequent expression of IL-17A. However, they can only do so after IL-6 or IL-21 has facilitated the expression of their receptors.28,29
It has also been proposed that, in addition to participating in the recruitment of leukocytes (mainly neutrophils), IL-17A influences most cells in the lung parenchyma, such as macrophages and DCs, which express receptors for IL-17A and synthesize proinflammatory cytokines such as IL-6 and TNF-α.30
On the other hand, IL-22 has been linked to multiple pathologies of an inflammatory nature, for example, psoriasis and rheumatoid arthritis, in addition to having an important role in the defense of the lungs against pathogens, especially extracellular bacteria, assisting production and release, for example, of defensins in the mucosa of the airways, in addition to maintaining the integrity of the epithelium.31 A study by Pichavant et al31 showed that, during Streptococcus pneumoniae infections in mice exposed to cigar smoke, cytokine levels related to Th17 lymphocytes are decreased, leading to exacerbations.33 In addition, IL-23 is associated with maintenance of the response generated by this group of cooperating T lymphocytes. Table 1 shows some of the main functions and characteristics of cytokines related to the Th17 profile.
 | Table 1 Principal characteristics of cytokines related with Th17 lymphocytes |
Characteristics of the genes-related to Th17 lymphocytes
Generally, genes have characteristics common to each other but differ from one another at different levels, ranging from the locus, number of introns and exons, number of base pairs and the position in which they are within the genome (forward/reverse strand). In this case, the genes related to the Th17 profile have similar characteristics, such as being on the same chromosome and similar size in terms of base pairs. Table 2 shows the main characteristics of these genes.
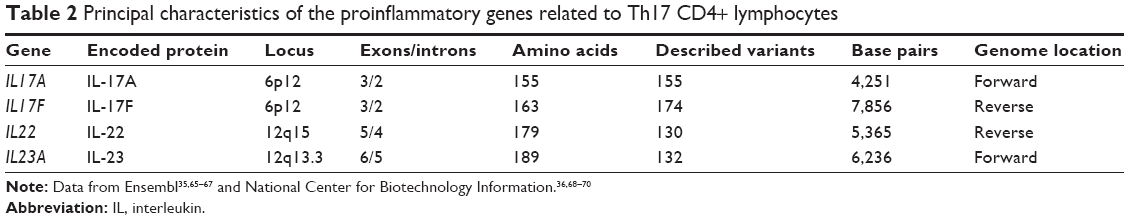 | Table 2 Principal characteristics of the proinflammatory genes related to Th17 CD4+ lymphocytes |
COPD infectious exacerbations
In general, COPD patients tend to experience frequent exacerbations. Of these, the most frequent are those of infectious origin, with bacteria and viruses being the most prevalent etiological agents, with 50%–70% and 30%, respectively.37 The clinical manifestations present during exacerbations are a consequence of the action of the virus/bacteria and of the immune response against the pathogen that assembles the host.38
Bacteria
Previously, the lungs were believed to be a sterile area, free of microorganisms.39 However, recent studies have shown that this is not entirely true and that the lungs contain, depending on the disease or stage in which they are found, a variable content of microbiomas.40 In the upper airways, there are also colonizing microorganisms; nose, for example, is colonized by bacteria such as Staphylococcus aureus, S. epidermidis and Corynebacteria.41 The nasopharynx is colonized under normal conditions by non-hemolytic, α-hemolytic Streptococcus and some Neisseria species, in addition to S. pneumoniae and Haemophilus influenzae.42
The presence of bacteria in patients who have had an exacerbation does not prove that these alone are sufficient to trigger the exacerbation of the symptomatology of patients with COPD, because they can also be isolated in individuals who do not have this pathology.39
In healthy individuals, different species of bacteria are constantly inhaled. However, due of the responsiveness of their immune system that is not compromised, they are not capable of infecting the host. This response is generated mainly by the presence of antimicrobial molecules present in the respiratory mucosa such as defensins, pentraxin-3, lactoferrin, lysozyme and cathelicidin, as well as immunoglobulin A (IgA) and tissue resident cells.39
An important factor associated with the predisposition of COPD patients to present exacerbations is the metaplasia of squamous epithelial cells that appears as a consequence of the damage generated to the airway epithelium by the chemical compounds of the cigarette. This event, together with the loss of ciliated epithelial cells, promotes the accumulation of infectious agents, generating an imbalance in the pulmonary microbioma and, therefore, a greater probability of producing an infection triggering exacerbations.41 In addition, the loss of the tight junctions between epithelial cells acting as an impermeable barrier is a factor that helps colonization by potentially pathogenic bacteria. It has also been shown that patients with COPD who are vaccinated against some pneumococcal strains are less prone to exacerbations due to this infectious agent. Table 3 shows the main bacteria causing exacerbations in patients with COPD.
 | Table 3 Principal causative bacteria of COPD exacerbations |
Viruses
In the past, it was believed that only bacteria were capable of generating exacerbations in patients with COPD.48,52 However, the occurrence of these in the winter and symptoms similar to cold led to the belief that viruses were also associated with the complications of this obstructive pathology of the airways.53 On the other hand, viral infections of the respiratory tract have been shown to influence the lung microbiome in patients with COPD, which indicates that both microorganisms can coexist and predispose to exacerbations.54 The most prevalent viruses during acute exacerbations of COPD are rhinovirus, coronavirus, influenza, respiratory syncytial virus and parainfluenza, among others.51,55
There are many factors that influence virus infection in patients with COPD. Among them, ICAM-1 overexpression stands out, where, for example, rhinoviruses are anchored and more easily enter epithelial cells, favoring infection.41 It has been described in several studies that the functionality of NK lymphocytes in patients with COPD is severely diminished, which leads to a greater propensity for virus infections (rhinovirus, respiratory syncytial virus, influenza, etc.), causal agents of a high percentage of exacerbations in this type of patients. The low antiviral activity is due, in large part, to the inhibition of the secretion of cytokines such as TNF-α, IFN-γ and IL-12, in addition to the low production of enzymes such as perforins, so they cannot eliminate the cells already infected by virus, so that the infection continues its course without having a response that attenuates it.56
It has been proposed that vitamin D (1,25-dihydroxyvitamin D3 [1,25D]) plays a key role on lung immunity. During a viral infection, vitamin D induces IκBα (a potent inhibitor of NF-κB) expression in airway epithelial cells, favoring a reduction of proinflammatory cytokines and no change in viral clearance.57 This increase of 1,25D in airways will contribute to control tissue damage, while maintaining viral clearance. In COPD, vitamin D deficiency is highly prevalent and correlates with severity of COPD.58 An important mechanism through which 1,25D can predispose to present an exacerbation is by suppressing Th1 (IL-2, IFN-γ, TNF-α) and Th17 (IL-17A, IL-17F) cytokines and promoting regulatory T lymphocytes (Treg) (IL-10, TGF-β) function.59
Lower levels of 1,25D in COPD may be explained by different mechanisms, for example, the reduction of cutaneous vitamin D3 production caused by smoking and limited sunlight exposure. Other mechanisms of vitamin D deficiency could be reduced vitamin D activation in liver and kidneys, increased vitamin D sequestration in adipose tissue and decreased intestinal absorption.60
As with pneumococcus, it is recommended that COPD patients be immunized annually with the influenza vaccine, since it is also one of the main etiological agents of exacerbations. In addition, several studies suggest that epidemiologically speaking, influenza vaccine is more effective than pneumococcus.42
Th17 CD4+ lymphocytes in infectious exacerbations
In COPD exacerbations, there is an increase in the lungs and respiratory tracts cellularity. Therefore, an increase in the number of leukocytes, such as neutrophils and macrophages in the airway, and the increase in proinflammatory cytokines are directly related to the severity of the exacerbations.39 Th17 CD4+ lymphocytes synthesize IL-17A that promotes the activation of bronchial fibroblasts, epithelial cells and smooth muscle cells, inducing them to produce proinflammatory cytokines responsible for the recruitment of neutrophils and their local infiltration, thus aggravating the COPD symptomatology.55 IL-17A promotes inflammation by coordinating granulopoiesis and neutrophil mobilization. This cytokine is particularly central to lung immunity because it has been demonstrated that innate host defenses to respiratory pathogens are compromised in mice lacking this proinflammatory cytokine or its receptor (IL-17RA), leading to reduced neutrophil activation, differentiation and recruitment and increased bacterial proliferation.61
Le Rouzic et al62 in their study showed that chronic exposure to cigarette smoke extract (CSE) inhibits S. pneumoniae-induced monocyte-derived dendritic cells (MDDC) maturation and secretion of cytokines involved in Th1 and Th17 T cell differentiation, among which are IL-1β, IL-6, IL-12 and IL-23 (which is necessary to Th17 polarization). These events can contribute to colonization of pathogens capable of generating exacerbations.
Qiu et al63 found that IL-27 significantly augmented T-bet expression by naive CD4+ T cells in Th1 conditions. In contrast, the expression of ROR-γT was dramatically suppressed by the addition of IL-27. Both in COPD patients and in mice, cigarette smoke exposure induced the upregulation of IL-27R (WSX-1) by naive CD4+ T cells. IL-27 promotes the expression of Th1 cells but inhibits the expression of Th17 cells in vitro. Also, IL-27R expression is increased during bacterial and parasitic infections, bringing on an exacerbation.
Receptors on the cell membrane of antigen-presenting phagocytes (APCs) such as TLRs recognize the presence of lipopolysaccharide in the membrane of bacteria such as H. influenzae and M. catharralis that colonize in a large part of the respiratory tract under normal conditions and that imbalance in the pulmonary microbiome exerts a stimulus that allows phagocytes to synthesize and release proinflammatory cytokines, in addition to phagocytosing the pathogen to subsequently present the antigen to T lymphocytes naive and allow their polarization together with the cytokine microenvironment present at that time. These data suggest that CD17+ Th17 lymphocytes play an important role in the immune response to bacteria such as S. pneumoniae, S. aureus and M. catharralis.56
DCs are professional APCs whose principal function is linking the innate and adaptive immune responses, a central mechanism in COPD. A chronic exposure to CSE has been related to a deficiency in the phagolysosome trafficking in DC, secondary to low expression of maturation markers such as CD83, which leads to a deficient antigenic presentation and, consequently, an inadequate response against bacteria such as S. pneumoniae.62
van der Sluijs et al64 reported in their study that inhibiting the production of IL-10 (inhibitory cytokine) by Tregs decreases the likelihood of secondary S. pneumoniae infection.
In the case of viruses, pathogen-associated molecular patterns are recognized by TLRs present in endolysosomes or by RIG1 receptors (RLRs) in the cytoplasm. When this happens, the signaling pathway led by NF-κB and IRF3 culminates in the synthesis and release of proinflammatory cytokines, in addition to type I IFNs, such as IFN-α and IFN-β, creating an antiviral state that rests the infection. With the production of these IFNs, inhibition of the response of this strain of cooperating T lymphocytes, such as Th17 CD4+, is generated, causing greater susceptibility to secondary bacterial infections, thus aggravating the health status of patients with COPD susceptible to this type of infections.56
Since cytokines such as IL-17A and IL-17F do not exist in the airway microenvironment, the production of neutrophil chemoattractants by airway epithelial cells such as CXCL8 and CXCL5 cannot be induced and the action required attracting neutrophils, and therefore, there is not adequate containment of extracellular bacteria.34
Another way that has been recently described as an important mediator of Th17 CD4+ inducers is a serum amyloid A (SAA), which promotes expression of inflammatory mediators and neutrophil chemotaxis and survival by the ALX-FPR2 receptor. SAA is also a potent endogenous ligand that stimulates expression of Th17 polarizing mediators and influences in vitro Th17 differentiation of CD4+ T cells.61
Conclusion
Mechanisms involved in infectious COPD exacerbations are not entirely clear. Various cellular types and molecules are involved in its pathogenesis. Importantly, the Th17 profile has been linked as one of the main participants, which involves a complex system of proinflammatory cells and molecules. For these reasons, it is essential to conduct clinical and basic studies to have a broader perspective of the molecular and cellular mechanisms that take place during COPD exacerbations, which can compromise the life of patients.
Acknowledgment
The authors wish to thank the Programa Interinstitucional para el Fortalecimiento de la Investigación y el Posgrado del Pacífico (DELFIN) for the scholarship obtained to carry out the initial Marco Antonio Ponce-Gallegos research for this article in the HLA laboratory.
Disclosure
The authors report no conflicts of interest in this work.
References
Daldegan MB, Teixeira MM, Talvani A. Concentration of CCL11, CXCL8 and TNF-alpha in sputum and plasma of patients undergoing asthma or chronic obstructive pulmonary disease exacerbation. Braz J Med Biol Res. 2005;38(9):1359–1365. | ||
Hong SC, Lee S-H. Role of th17 cell and autoimmunity in chronic obstructive pulmonary disease. Immune Netw. 2010;10(4):109–114. | ||
Bergin DA, Reeves EP, Hurley K, et al. The circulating proteinase inhibitor α-1 antitrypsin regulates neutrophil degranulation and autoimmunity. Sci Transl Med. 2014;6(217):217ra1. | ||
Menezes AM, Montes de Oca M, Pérez-Padilla R, et al. Increased risk of exacerbation and hospitalization in subjects with an overlap phenotype: COPD-asthma. Chest. 2014;145(2):297–304. | ||
Rovina N, Koutsoukou A, Koulouris NG. Inflammation and immune response in COPD: where do we stand? Mediators Inflamm. 2013;2013:1–9. | ||
Vestbo J, Hurd SS, Agustí AG, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease GOLD executive summary. Am J Respir Crit Care Med. 2013;187(4):347–365. | ||
Secretaría de Salud; Instituto Nacional de Enfermedades Respiratorias [National Institute of Respiratory Diseases]. Clínica de EPOC [COPD Clinic]; [cited August, 2016]. Available from: http://www.iner.salud.gob.mx/principales/investigaci%C3%B3n/por-departamento/investigaci%C3%B3n-en-tabaquismo-y-epoc/investigaci%C3%B3n-en-tabaquismo-y-epoc/cl%C3%ADnica-de-epoc.aspx. Accessed May 27, 2017. Spanish. | ||
World Health Organization. Chronic obstructive pulmonary disease (COPD); [cited August 2, 2016]. Available from: http://www.who.int/respiratory/copd/en/. Accessed May 27, 2017. | ||
De Boer WI, Alagappan VKT, Sharma HS. Molecular mechanisms in chronic obstructive pulmonary disease. Cell Biochem Biophys. 2007;47(9):131–147. | ||
Global Initiative for Chronic Obstructive Lung Disease. Global Strategy for the Diagnosis, Management and Prevention of COPD – 2016. Available from: http://goldcopd.org/global-strategy-diagnosis-management-prevention-copd-2016/. Accessed May 27, 2017. | ||
Suissa S, Dell’Aniello S, Ernst P. Long-term natural history of chronic obstructive pulmonary disease: severe exacerbations and mortality. Thorax. 2012;67(11):957–963. | ||
Dye JA, Adler KB. Effects of cigarette smoke on epithelial cells of the respiratory tract. Thorax. 1994;49:825–834. | ||
Shaykhiev R, Crystal RG. Early events in the pathogenesis of chronic obstructive pulmonary disease: smoking-induced reprogramming of airway epithelial basal progenitor cells. Ann Am Thorac Soc. 2014;11(suppl 5):S252–S258. | ||
Pouwels SD, Heijink IH, ten Hacken NHT, et al. DAMPs activating innate and adaptive immune responses in COPD. Mucosal Immunol. 2014;7(2):215–226. | ||
Maes T, Bracke KR, Vermaelen KY, et al. Murine TLR4 is implicated in cigarette smoke-induced pulmonary inflammation. Int Arch Allergy Immunol. 2006;141(4):354–368. | ||
Doz E, Noulin N, Boichot E, et al. Cigarette smoke-induced pulmonary inflammation is TLR4/MyD88 and IL-1R1/MyD88 signaling dependent. J Immunol. 2008;180(2):1169–1178. | ||
Freeman CM, Martinez MJ, Han MK, et al. Lung CD8+ T cells in COPD have increased expression of bacterial TLRs. Respir Res. 2013;1(14):1–13. | ||
Birrell MA, Eltom S. The role of the NLRP3 Inflammasome in the pathogenesis of airway disease. Pharmacol Ther. 2011;130(3):364–370. | ||
Hoenderdos K, Condliffe A. The neutrophil in COPD: too little too late, or too much too soon? Am J Respir Cell Mol Biol. 2013;48(5):531–539. | ||
Collin M, Bigley V, Haniffa M, Hambleton S. Human dendritic cell deficiency: the missing ID? Nat Rev Immunol. 2011;11(9):575–583. | ||
Bar-On L, Jung S. Defining dendritic cells by conditional and constitutive cell ablation. Immunol Rev. 2010;234(1):76–89. | ||
Cook PC, MacDonald AS. Dendritic cells in lung immunopathology. Semin Immunopathol. 2016;38(4):449–460. | ||
Aujla SJ, Dubin PJ, Kolls JK. Interleukin-17 in pulmonary host defense. Exp Lung Res. 2007;33(10):507–518. | ||
Tesmer LA, Lundy SK, Sarkar S, Fox DA. Th17 cells in human disease. Immunol Rev. 2008;223:87–113. | ||
Wei L, Laurence A, Elias KM, O’Shea JJ. IL-17s produced by Th17 cells and drives IL-17 production in a STAT3-dependent manner. J Biol Chem. 2007;282:34605–34610. | ||
Fantini MC, Rizzo A, Fina D, et al. IL-21 regulates experimental colitis by modulating the balance between Th1 and Th17 cells. Eur J Immunol. 2007;37:3155–3163. | ||
Zhou L, Ivanov II, Spolski R, et al. IL-6 programs TH-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8:967–974. | ||
Korn T, Bettelli E, Gao W, et al. IL-21 initiates an alternative pathway to induce proinflammatory TH17 cells. Nature. 2007;448:484–487. | ||
Aujla SJ, Chan YR, Zheng M, et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med. 2008;14(3):275–281. | ||
Doe C, Bafadhel M, Siddiqui S, et al. Expression of the T helper 17-associated cytokines IL-17A and IL-17F in asthma and COPD. Chest. 2010;138(5):1140–1147. | ||
Pichavant M, Sharan R, Le Rouzic O, et al. IL-22 defect during Streptococcus pneumoniae infection triggers exacerbation of chronic obstructive pulmonary disease. EBioMedicine. 2015;2(11):1686–1696. | ||
Sonnenberg GF, Nair MG, Kirn TJ, Zaph C, Fouser LA, Artis D. Pathological versus protective functions of IL-22 in airway inflammation are regulated by IL-17A. J Exp Med. 2010;207(6):1293–1305. | ||
Doisne JM, Soulard V, Becourt C, et al. Cutting edge: crucial role of IL-1 and IL-23 in the innate IL-17 response of peripheral lymph node NK1.1 - invariant NKT cells to bacteria. J Immunol. 2011;186(2):662–666. | ||
Iwakura Y, Ishigame H. The IL-23/IL-17 axis in inflammation. J Clin Invest. 2006;116(5):1218–1222. | ||
Ensembl. IL17A; [cited August 2, 2016]. Available from: http://www.ensembl.org/Homo_sapiens/Gene/Summary?db=core;g=ENSG00000112115;r=6:52186387-52190638;t=ENST00000340057. Accessed May 27, 2017. | ||
National Center for Biotechnology Information; US National Library of Medicine. IL17A; [cited August 2, 2016]. Available from: https://www.ncbi.nlm.nih.gov/gene/3605. Accessed May 27, 2017. | ||
Huang YJ, Charlson ES, Collman RG, Colombini-Hatch S, Martinez FD, Senior RM. The role of the lung microbiome in health and disease. A National Heart, Lung, and Blood Institute workshop report. Am J Respir Crit Care Med. 2013;187(12):1382–1387. | ||
Kiley JP. Advancing respiratory research. Chest. 2011;140(2):497–501. | ||
Beasley V, Joshi PV, Singanayagam A, Molyneaux PL, Johnston SL, Mallia P. Lung microbiology and exacerbations in COPD. Int J Chron Obstruct Pulmon Dis. 2012;7:555–569. | ||
Santos S, Marín A, Serra-Batlles J, et al. Treatment of patients with COPD and recurrent exacerbations: the role of infection and inflammation. Int J Chron Obstruct Pulmon Dis. 2016;11:515. | ||
Austrian R. The bacterial flora of the respiratory tract. Some knowns and unknowns. Yale J Biol Med. 1968;40:400–413. | ||
Hui DS, Ip M, Ling T, et al. A multicentre surveillance study on the characteristics, bacterial aetiologies and in vitro antibiotic susceptibilities in patients with acute exacerbations of chronic bronchitis. Respirology. 2011;16(3):532–539. | ||
Choi KJ, Cha SI, Shin KM, et al. Prevalence and predictors of pulmonary embolism in Korean patients with exacerbation of chronic obstructive pulmonary disease. Respiration. 2013;85(3):203–209. | ||
Desai H, Eschberger K, Wrona C, et al. Bacterial colonization increases daily symptoms in patients with chronic obstructive pulmonary disease. Ann Am Thorac Soc. 2014;11(3):303–309. | ||
Parameswaran GI, Wrona CT, Murphy TF, Sethi S. Moraxella catarrhalis acquisition, airway inflammation and protease-antiprotease balance in chronic obstructive pulmonary disease. BMC Infect Dis. 2009;9(1):178. | ||
Miravitlles M, Espinosa C, Fernandez-Laso E, Martos JE, Maldonado JA, Gallego M. Relationship between bacterial flora in sputum and functional impairment in patients with acute exacerbations of COPD. Chest. 1999;116:40–46. | ||
Sethi S. Infection as a comorbidity of COPD. Eur Respir J. 2010;35(6):1209–1215. | ||
Ramírez-Venegas A, Sansores RH. Consenso Mexicano de EPOC: Manejo de un paciente con EPOC inestable. Exacerbación leve, moderada y grave [Mexican consensus of COPD: management of a patient with unstable COPD. Mild, moderate and severe exacerbation]. Neumol Cir Torax. 2007;66(S2):54–70. Spanish. | ||
Jenkins CR, Celli B, Anderson JA, et al. Seasonality and determinants of moderate and severe COPD exacerbations in the TORCH study. Eur Respir J. 2012;39(1):38–45. | ||
Molyneaux P, Mallia P, Cox MJ, et al. Outgrowth of the bacterial airway microbiome after rhinovirus exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;188(10):1224–1231. | ||
Mallia P, Footitt J, Sotero R, et al. Rhinovirus infection induces degradation of antimicrobial peptides and secondary bacterial infection in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;186(11):1117–1124. | ||
Greenberg SB, Allen M, Wilson J, Atmar RL. Respiratory viral infections in adults with and without chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2000;162(1):167–173. | ||
Mian MF, Lauzon NM, Stampfli MR, Mossman KL, Ashkar AA. Impairment of human NK cell cytotoxic activity and cytokine release by cigarette smoke. J Leukoc Biol. 2008;83(3):774–784. | ||
Bhowmik A, Seemungal TA, Sapsford RJ, Wedzicha JA. Relation of sputum inflammatory markers to symptoms and lung function changes in COPD exacerbations. Thorax. 2000;55(2):114–120. | ||
D’Anna SE, Balbi B, Cappello F, Carone M, di Stefano A. Bacterial–viral load and the immune response in stable and exacerbated COPD: significance and therapeutic prospects. Int J Chron Obstruct Pulmon Dis. 2016;11(1):445–453. | ||
Lester SN, Li K. Toll-like receptors in antiviral innate immunity. J Mol Biol. 2014;426(6):1246–1264. | ||
Hansdottir S, Monick MM, Lovan N, Powers L, Gerke A, Hunninghake GW. Vitamin D decreases respiratory syncytial virus induction of NFkappaB-linked chemokines and cytokines in airway epithelium while maintaining the antiviral state. J Immunol. 2010;184(2):965–974. | ||
Janssens W, Bouillon R, Claes B, et al. Vitamin D deficiency is highly prevalent in COPD and correlates with variants in the vitamin D-binding gene. Thorax. 2010;65(3):215–220. | ||
Daniel C, Sartory NA, Zahn N, Radeke HH, Stein JM. Immune modulatory treatment of trinitrobenzene sulfonic acid colitis with calcitriol is associated with a change of a T helper (Th) 1/Th17 to a Th2 and regulatory T cell profile. J Pharmacol Exp Ther. 2008;324(1):23–33. | ||
Persson LJ, Aanerud M, Hiemstra PS, Hardie JA, Bakke PS, Eagan TM. Chronic obstructive pulmonary disease is associated with low levels of Vitamin D. PLoS One. 2012;7(6):1–8. | ||
Anthony D, Seow HJ, Uddin M, et al. Serum amyloid a promotes lung neutrophilia by increasing IL-17A levels in the mucosa and γδ T cells. Am J Respir Crit Care Med. 2013;188(2):179–186. | ||
Le Rouzic O, Koné B, Kluza J, et al. Cigarette smoke alters the ability of human dendritic cells to promote anti-Streptococcus pneumoniae Th17 response. Respir Res. 2016;17(1):94. | ||
Qiu S-L, Duan M-C, Liang Y, et al. Cigarette smoke induction of interleukin-27/WSX-1 regulates the differentiation of Th1 and Th17 cells in a smoking mouse model of emphysema. Front Immunol. 2016;7:553. | ||
van der Sluijs KF, van Elden LJ, Nijhuis M, et al. IL-10 is an important mediator of the enhanced susceptibility to pneumococcal pneumonia after influenza infection. J Immunol. 2004;172(12):7603–7609. | ||
Ensembl. IL17F; [cited August 2, 2016]. Available from: http://www.ensembl.org/Homo_sapiens/Gene/Summary?db=core;g=ENSG00000112116;r=6:52236681-52244537. Accessed May 27, 2017. | ||
Ensembl. IL23A; [cited August 2, 2016]. Available from: http://www.ensembl.org/Homo_sapiens/Gene/Summary?db=core;g=ENSG00000110944;r=12:56334174-56340410. Accessed May 27, 2017. | ||
Ensembl. IL22; [cited in August 2 of 2016]. Available from: http://www.ensembl.org/Homo_sapiens/Gene/Summary?db=core;g=ENSG00000127318;r=12:68248242-68253607. Accessed May 27, 2017. | ||
National Center for Biotechnology Information; US National Library of Medicine. IL17F; [cited August 2, 2016]. Available from: https://www.ncbi.nlm.nih.gov/gene/112744. Accessed May 27, 2017. | ||
National Center for Biotechnology Information; US National Library of Medicine. IL23A; [cited August 2, 2016]. Available from: https://www.ncbi.nlm.nih.gov/gene/51561. Accessed May 27, 2017. | ||
National Center for Biotechnology Information; US National Library of Medicine. IL22; [cited August 2, 2016]. Available from: https://www.ncbi.nlm.nih.gov/gene/50616. Accessed May 27, 2017. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.