Back to Journals » OncoTargets and Therapy » Volume 12
TGFβ1 is essential for MSCs-CAFs differentiation and promotes HCT116 cells migration and invasion via JAK/STAT3 signaling
Authors Tan HX, Cao ZB, He TT, Huang T ![]() , Xiang CL
, Xiang CL ![]() , Liu Y
, Liu Y
Received 28 June 2018
Accepted for publication 17 April 2019
Published 5 July 2019 Volume 2019:12 Pages 5323—5334
DOI https://doi.org/10.2147/OTT.S178618
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr William C. Cho
Hao-Xiang Tan,1–3 Zhen-Bin Cao,4 Ting-Ting He,5 Tao Huang,1,3 Cai-Ling Xiang,1,3 Yu Liu1,3
1Department of Gastrointestinal Surgery, Hunan Provincial People’s Hospital, Changsha 410002, People’s Republic of China; 2Department of Gastrointestinal Surgery, Ruikang Hospital Affiliated to Guangxi University of Chinese Medicine, Nanning 530011, People’s Republic of China; 3Department of Gastrointestinal Surgery, First Affiliated Hospital of Hunan Normal University, Changsha 410002, People’s Republic of China; 4Department of Radiology, Ruikang Hospital Affiliated to Guangxi University of Chinese Medicine, Nanning 530011, People’s Republic of China; 5Department of Pathology, Ruikang Hospital Affiliated to Guangxi University of Chinese Medicine, Nanning 530011, People’s Republic of China
Background and purpose: Colorectal cancer (CRC) frequently metastasizes to the liver, which involves the participation of multiple cytokines. Tumor microenvironment (TME) composed of cancer-associated fibroblasts (CAFs) and tumor cells acts as an essential factor in cancer metastasis. Transforming growth factor β1 (TGFβ1) is a vital cytokine involved in migration and invasion of cancer cells. However, the underlying mechanisms remain unclear. In the present study, we aimed to investigate the role and molecular mechanisms of TGFβ1 in TME.
Methods: The conditioned medium prepared from colorectal cancer HCT116 and HT29 cells was used to culture mesenchymal stem cells (MSCs). The differentiation of MSCs to CAFs was detected by flow cytometry. The role of TGFβ1 in colorectal cancer cells metastasis was examined by wound-healing assay and transwell assay. And the activation of the Janus kinase/signal transducer and activator of transcription 3 (JAK/STAT3) signaling pathway was measured by Western blot assay.
Results: TGFβ1 induced the differentiation of MSCs to CAFs and improved HCT116 and HT29 cells migration and invasion. Meanwhile, TGFβ1 also upregulated the phosphorylation of STAT3 and enhanced the nuclear localization of p-STAT3, which activated JAK/STAT3 signaling pathway.
Conclusion: TGFβ1 induced the differentiation of MSCs into CAFs and promoted the migration and invasion of HCT116 and HT29 cells, which depended on the activation of JAK/STAT3 signaling pathway.
Keywords: TGFβ1, JAK/STAT3, MSCs-CAFs differentiation, migration and invasion
Introduction
Colorectal cancer (CRC) is one of the most common malignant tumor in the world.1 By the year 2030, 2.2 million patients will be diagnosed as colorectal cancer and the global burden of this disease will increase by 60%.1 The clinical data showed that about 14–18% of colorectal cancer patients suffered postoperative metastasis, and more than half of these metastases developed in liver, which had been considered as the main cause of death.2 Therefore, a better understanding of the underlying molecular mechanism involved in the metastasis of colorectal cancer is essential for preventing the progression and developing the more efficient therapies.
The “seed and soil theory” has revealed that the metastasis of malignant tumors is dependent on specific tumor microenvironment (TME), which has received more attention recently.3 The complex TME was composed of tumor cells, fibroblasts, immune and inflammatory cells, bone-marrow-derived inflammatory cells, as well as the extracellular matrix, microvessels and biomolecules.4 Studies have found that circulating tumor cells are dormant after reaching the corresponding target organs and require the corresponding TME to form metastases.5 Mesenchymal stem cells (MSCs) were involved in the formation of CRC primary lesion. And the differentiation of MSCs to cancer-associated fibroblast (CAFs) promoted the proliferation of cancer cells and the formation of tumor blood vessels.6,7 CAFs were heterogenous fibroblasts, and unlike the normal fibroblasts, CAFs could promote cancer stem cell renewal and atmosphere forming capacity and contribute to tumor genesis.8 Unfortunately, the mechanism of corresponding TME formation is still unclear.
The evidences showed multiple cytokines might be necessary in the differentiation of MSCs into CAFs. One of the key cytokines plays an important biological role in mediating tumor metastasis is TGFβ1.9 Studies have shown that TGFβ1 compels hematopoietic stem cell to differentiate toward myofibroblast via JAK/STAT3 signal pathway.10 TGFβ1 has also been reported to be secreted by tumor cells in a form of paracrine to stimulate the fibroblasts activation and MSCs differentiation.11,12 In addition, the exosome and cytokines secreted by tumor cells and CAFs may induce MSCs to synthesize α-SMA and tumor-promoting factors, which may promote the proliferation, migration and invasion of colorectal cancer cells.13 These evidences suggested that TGFβ1 might be involved as a significant active factor in the progression and development of CRC.
Thus, we hypothesize that the growth factor TGFβ1 induces differentiation of MSCs into CAFs and promotes the migration and invasion of HCT116 and HT29 cells via activating JAK/STAT3 signaling pathway, which ultimately accelerates the formation of TME for liver metastasis.
Materials and methods
Cell culture
The human colorectal carcinoma cell lines (HCT116, HT29) and bone-marrow-derived MSCs were obtained from ATCC. HCT116 and HT29 cells were cultured in McCoy’s 5a Medium (Gibco), supplemented with 2 mmol/L glutamine (Sigma, Spain) and 10% of FBS (fetal bovine serum, Gibco), at 37°C, 95% of air and 5% of carbon dioxide (CO2). MSCs were cultured in colorectal cancer cells’ conditioned medium (CM) at 37°C, 95% of air and 5% of CO2. colorectal cancer cells’ CM: HCT116 or HT29 cells were first cultured in general medium. When the cells were in the logarithmic growth phase, general medium was replaced with serum-free medium. The supernatant was collected as conditioned medium after 48 hrs. The TGFβ1-block peptide P17 were synthesized (AnaSpec, USA). P17 (100 mg/mL) was pre-treated with cells for blocking TGFβ1 effects. AG490 (10 µM), purchased from MedChemExpress, was subjected to block JAK/STAT3 signaling pathway.
ELISA assay
The supernatants of HCT116 and HT29 cells were collected after 48 hrs. The concentration of TGFβ1, TNFα, HGF and PDGF in the supernatants was determined by ELISA kit, purchased from R&D. The OD (optical density) value was detected by a microplate reader (Thermofisher, USA) at 450 nm.
Cell proliferation assay
Cell counting kit-8 (CCK8, Univbio, China) was utilized for cell proliferation assay. MSCs were planted in a 96-well plate (2.5×103 cells for each well), and cultured in colorectal cancer cells’ CM (HCT116-CM or HT29-CM group) or pre-treated with P17 conditioned medium (HCT116-CM+P17 or HT29-CM+P17 group). Serum-free medium were set as blank control group (Control-CM group). At the 24th/48th/72th hour after seeding, 100 µL of CCK-8 solution were added for staining. The absorbance at 450 nm was measured via a microplate reader (Thermofisher, USA) after a 2 hrs incubation at room temperature. Triplicates were performed for each condition.
Sphere formation assay
MSCs were seeded in 6-well Corning plates (2.5×103 cells for each well) with serum-free medium (Control-CM group), colorectal cancer cells’ CM (HCT116-CM or HT29-CM group) or pre-treated with P17 conditioned medium (HCT116-CM+P17 or HT29-CM+P17 group). The spheres in each well were numbered and photographed via an inverted microscope (Zeiss, Japan) after 15 days.14
Flow cytometry of CAFs biomarkers
CAFs biomarkers were recognized by a flow cytometer (BD Biosciences, USA). MSCs from various culture conditions were gathered. After resuspended with PBS in a 1.5 mL eppendorf tube, primary antibodies for CAFs specific protein: α-SMA, Vimentin or FSP1 (Mouse Anti-Human, Bioss, China) were added in eppendorf tubes, respectively (1:100). After a 1 hr incubation at room temperature, the supernatant was disposed. Cells were washed with PBS for 3 times. The FITC labeled secondary antibodies (Goat Anti-Mouse, Bioss, China) were added in each eppendorf tube (1:200) and incubated in dark for 1 hr at room temperature. The cells were washed for 3 times with PBS and then analyzed with BD FACS Calibur System (BD Biosciences, USA).
Quantitative real-time PCR
The qRT-PCR was performed according to the general protocol: RNA was extracted via TRIzol (Thermo Fisher, USA) and cDNA was synthesized via Prime Script RT Reagent kit (Takara, Japan). RT‑PCR was performed using ABI 7500 (Life Tech, USA). The relative expression levels of mRNA were determined using a SYBR-Green PCR master mix kit (Thermo Fisher, USA). Gene expression was evaluated using the 2−ΔΔCq method. GAPDH was used as control.
Wound-healing assay
The wound-healing assay was performed as common protocol: 3×105 of HCT116 or HT29 cells were seeded in each well of a 6-well plate (Corning, USA) in McCoy’s 5a Medium with 10% FBS, and cultured at 37°C, 5% of CO2 for 24 hrs to form a monolayer.15 The middle of the monolayer was scratched using a Cell Scraper (Sigma-Aldrich, USA). PBS was used to wash the well. Subsequently, serum-free medium (Control-CM group), serum-free medium with 10 μM TGFβ1 (Control-CM group + TGFβ1), colorectal cancer cells’ CM (HCT116-CM or HT29-CM group) or pre-treated with P17 conditioned medium (HCT116-CM+P17 or HT29-CM+P17 group) were added to the wells, respectively. The scratched monolayer was incubated at 37°C in an atmosphere of 5% CO2 for 48 hrs. Wound closure was measured in 3 random fields in each well using an inverted microscope (DMi1, Leica, Germany). Triplicates were performed for each group.
Transwell assay
The transwell assay for the ability of invasion was performed as the following protocol:15 the polycarbonate membranes with 8-μm pore (Corning, USA) were placed on 24-well Transwell plates (Corning, USA). Subsequently, the membranes were coated with matrigel (20 μg per well) and dried in air for 1 hr at 37°C. The other side of the membranes was coated with fibronectin (5 μg per well). 2×105 of HCT116 cells were placed on the Matrigel side of membranes. Serum-free medium (Control-CM group), serum-free medium with 10 μM TGFβ1 (Control-CM group + TGFβ1), colorectal cancer cell’s CM (HCT116-CM or HT29-CM group) or pre-treated with P17 conditioned medium (HCT116-CM+P17 or HT29-CM+P17 group) were added in the upper chamber. The concentration of FBS in upper chamber was 2.5% and the FBS in the lower chamber was 10%. The transwell plate was incubated for 8 hrs at 37°C and 5% CO2. The cells on the matrigel side of the membrane were scrubbed off with a cotton pad. The membranes were fixed in 95% ethanol and 5% acetic acid for 30 mins and stained with crystal violet. The number of cells in 3 random visual fields were calculated using an inverted microscope (DMi8, Leica, Germany). Triplicates were performed for each group.
Nuclear and cytoplasmic extraction and Western blot analysis
The Western blot assay was performed as the classic protocol.16 Cells cultured in different conditioned medium were collected and proteins were extracted by using RIPA buffer (Beotime Insitute of Biotechnology, China). The NE-PERTM Nuclear and Cytoplasmic Extraction Reagents Kit (Thermo Fisher, USA) was performed to measure p-STAT3 expression in cytoplasm or nuclei. The concentration of protein was measured using a Commassie Blue Staining Kit (Beyotime, China). Subsequently, 150 μL of lysate protein were mixed with 50 μL of 4× loading buffer and boiled for 5 mins. Equal amounts of total protein were separated by 10% SDS gel (sodium dodecyl sulphate-polyacrylamide) electrophoresis and transferred onto polyvinylidene difluoride membranes (Beyotime, China). The membranes were immunoblotted, respectively, with following primary antibodies: p-JAK1 (1:500), JAK1 (1:500), p-STAT3 (1:1000), STAT3 (1:1000), E-cadherin (1:500), N-cadherin (1:500) and vimentin (1:500), GAPDH (1:5000) and Lamin A (1:2000). All of the antibodies were obtained from Cell Signaling Technology, USA. After incubation overnight at 4°C, the antibodies were blocked with 0.5% of bovine serum albumin. The horseradish peroxidase linked secondary antibodies (Beyotime Institute of Biotechnology) were applied and DAB (3,3-diaminobenzidine tetrahydrochloride) (Beijing Liuyi Biotechnology Co., Ltd., Beijing, China) was used to visualize blot. The amount of total protein was semiquantified as ratio to GAPDH on each gel. Lamin A and GAPDH served as the normalized controls for nuclear and cytoplasmic fractions, respectively.
Statistical analysis
The data of this study were performed using SPSS 19.0 software (SPSS, USA) and GraphPad Prism 5.0 software (GraphPad, USA). Analysis of variance was calculated by the Student’s test (t-test) or one-way ANOVA analysis. The data were presented as mean ± standard deviation (SD). P<0.05 was considered to indicate a statistically significant result. All experiments were repeated at least three times.
Results
Colorectal cancer cells’ CM promotes the proliferation and differentiation of MSCs to CAFs
We first measured the expression level of growth factors in the conditioned medium of colorectal cancer cells (HCT116-CM and HT29-CM). In the conditioned medium, growth factors such as TGFβ1, TNFα, HGF and PDGF were examined via ELISA kit assay. The result showed that the concentration of TGFβ1, TNFα, HGF and PDGF were significantly upregulated both in HCT116-CM and HT29-CM (Figure 1A). Moreover, the number of MSCs cultured in serum-free medium (control-CM) was significantly less than that in colorectal cancer cells’ CM, and the cells were in poor condition with some cells floating in the medium (Figure 1B). The adherent cells in colorectal cancer cells’ CM group were spindle-like, which were different from the globoid cells in control-CM group. It indicated that these cells had differentiated to CAFs. The proliferation of MSCs was significantly increased after treated with colorectal cancer cells’ CM via CCK-8 assay (Figure 1C). And we found that the ability of sphere formation of colorectal cancer cells’ CM cultured MSCs was distinctly strengthened (Figure 1D). To ensure whether colorectal cancer cells’ CM could induce the differentiation of MSCs to CAFs, the CAFs-related biomarkers such as α-SMA, vimentin, FSP1 was detected by flow cytometry (Figure 1E). The results showed that colorectal cancer cells’ CM promoted the sphere formation of MSCs and increased the level of CAFs-related biomarkers, which suggested that colorectal cancer cells’ CM promoted the generation of CAFs.
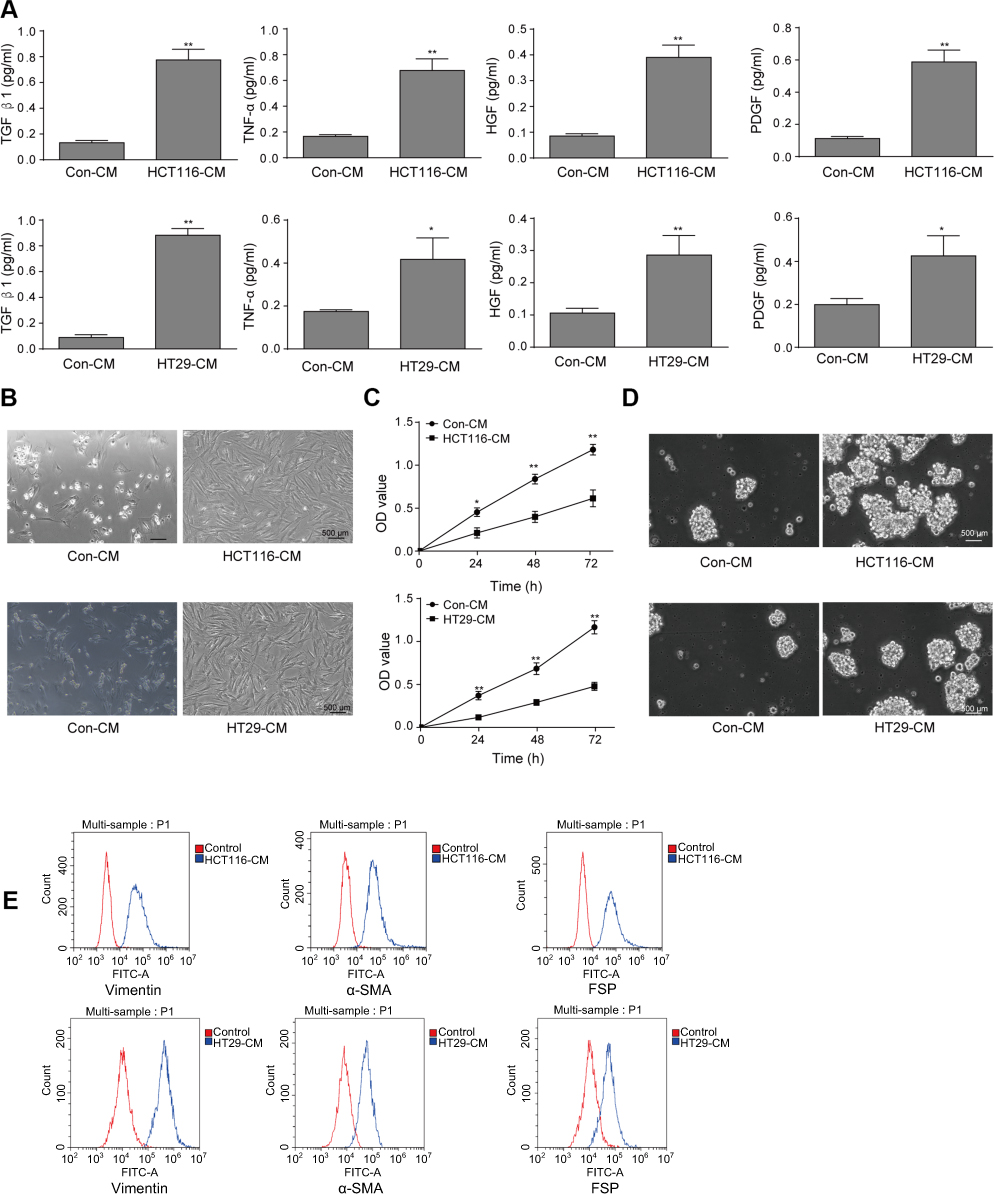 |
Figure 1 The effects of conditioned medium of colorectal cancer cells on the differentiation of MSCs to CAFs. (A) After HCT116 and HT29cells were cultured with serum-free medium for 48 hrs, the media was saved to perform TGFβ1, TNFα, HGF and PDGF levels via Enzyme-linked immunosorbent assay (ELISA). *P<0.05 and **P<0.01 vs Con-CM. (B) The condition of MSCs cultured under serum-free medium (Con-CM) or HCT116 or HT29 conditioned medium (HCT116-CM or HT29-CM) for 48 hrs. Scale bar, 500 µm. (C) CCK-8 assay to detect the proliferation of MSCs for indicated times. *P<0.05 and **P<0.01 vs Con-CM. (D) Sphere formation assay of MSCs incubated with Con-CM and HCT116-CM or HT29-CM. Scale bar, 500 µm. (E) For flow cytometry, the CAFs-related biomarkers such as α-SMA, vimentin, FSP1 were detected. The experiments were repeated at least three times.Abbreviations: Con, control; MSCs, mesenchymal stem cells; CAFs, cancer-associated fibroblasts. |
TGFβ1 induces the proliferation and differentiation of MSCs to CAFs
To further verify our hypothesis that TGFβ1 is the key factor to mediate the metastasis, we applied TGFβ1 blocking peptide P17 for pre-treating with colorectal cancer cells. As shown in Figure 2A, the number of MSCs in the control-CM group and the HCT116-CM+P17 or HT29-CM+P17 group was significantly less than that in the HCT116-CM or HT29-CM group, with some cells floating in the medium. And the result of CCK-8 assay also showed that the increasing proliferation of MSCs incubated with HCT116-CM or HT29-CM group was inhibited by exposure to P17 treatment (Figure 2B). In addition, the sphere formation analysis was similar to CCK-8 assay (Figure 2C). Moreover, flow cytometry revealed that the CAFs-related biomarkers such as α-SMA, vimentin, FSP1 were downregulated in HCT116-CM+P17 or HT29-CM+P17 group and control-CM group compared to those in HCT116-CM or HT29-CM group (Figure 2D), which suggested that the differentiation of MSCs cells was prohibited with TGFβ1-blocking peptide P17. These findings suggested that TGFβ1 acted as a key factor modulating the differentiation of MSCs to CAFs.
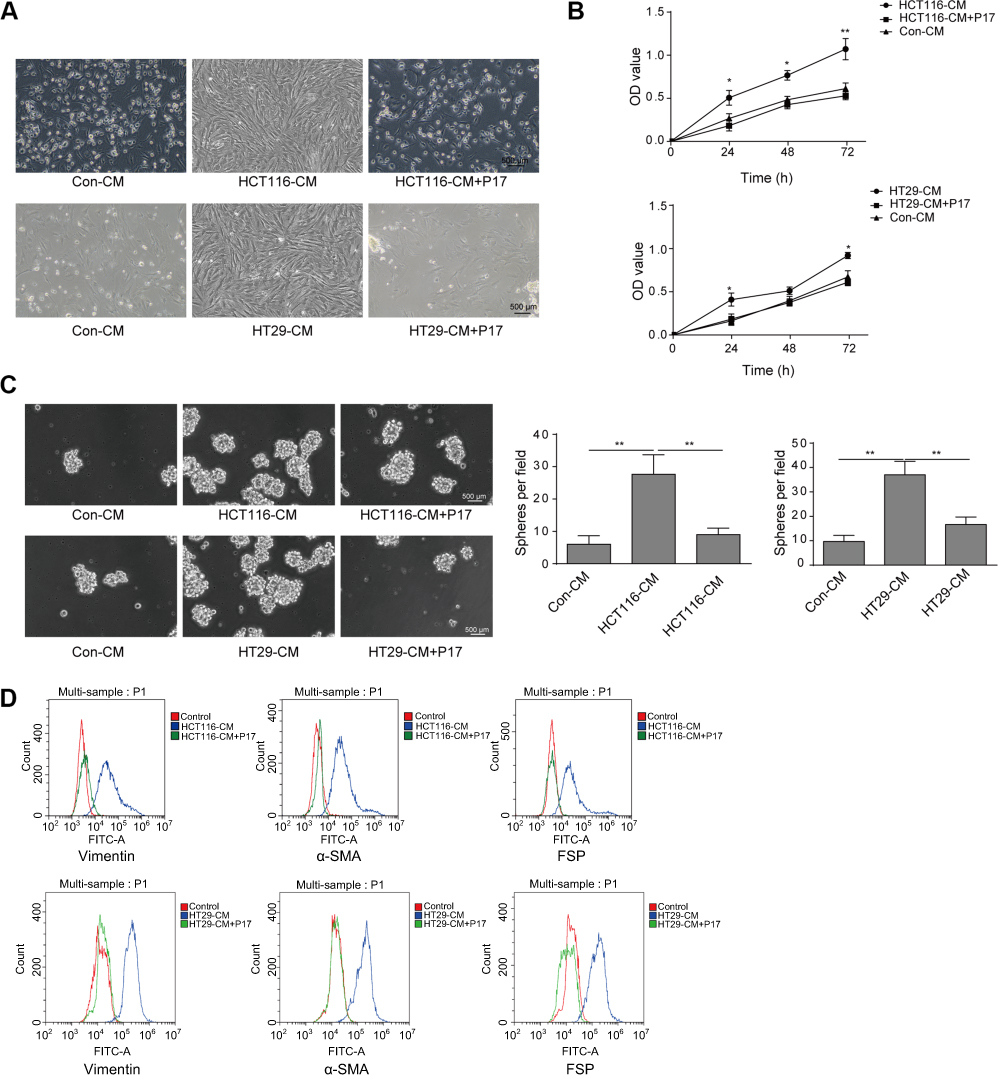 |
Figure 2 TGFβ1-blocking peptide P17 suppresses the proliferation and differentiation of MSCs to CAFs. (A) P17 was pre-treated colorectal cancer cells and then the condition of MSCs incubated with various conditioned medium was detected. Scale bar, 500 µm. (B) Proliferation of MSCs was detected by CCK8 assay. (C) The ability of MSCs to form spheres in Con-CM group and HCT116-CM+P17 or HT29+P17 group was inhibited compared with HCT116-CM or HT29-CM group. **P<0.01 vs Con-CM, **P<0.01 vs HCT116-CM and **P<0.01 vs HT29-CM group. (D) The expression of CAFs-related proteins such as α-SMA, vimentin, FSP was measured by flow cytometry. All experiments were at least repeated three times. Abbreviations: Con, control; MSCs, mesenchymal stem cells; CAFs, cancer-associated fibroblasts; CM, conditioned medium. |
TGFβ1 enhances the migration and invasion of HCT116 and HT29 cells
To determine the role of TGFβ1 on metastasis, the migration and invasion of HCT116 and HT29 cells were assessed by wound-healing assay and transwell assay at indicated times. As shown in Figure 3A and B, HCT116 and HT29 cells migration and invasion significantly increased in the control-CM+TGFβ1 group and HCT116-CM or HT29-CM group, while this effect was abolished by TGFβ1-blocking peptide P17. Epithelial-mesenchymal transition (EMT) is a vital mechanism of cancer metastasis. To further investigate, the relative expression of E-cadherin, N-cadherin and vimentin were measured by qRT-PCR and Western blot assay. As shown in Figure 3C and D, the expression of N-cadherin and vimentin were upregulated, whereas the expression of E-cadherin was downregulated after TGFβ1 treatment. These molecules showed an opposite expression following exposure to P17. Therefore, these results demonstrated that TGFβ1 enhances the migration and invasion of HCT116 and HT29 cells.
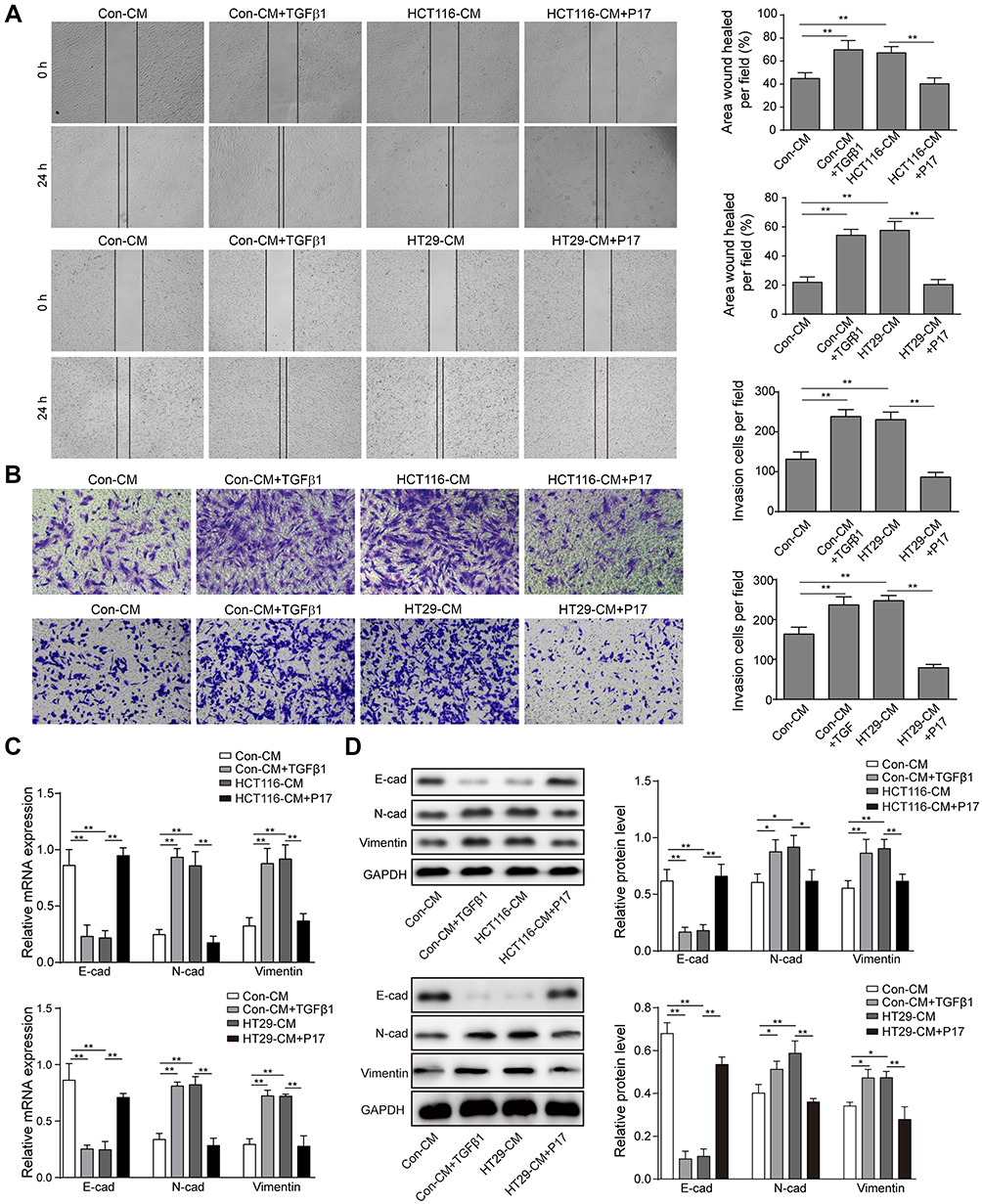 |
Figure 3 Blocking TGFβ1 inhibits the migration and invasion of HCT116 and HT29 cells. (A) In wound-healing assay, HCT116 cells were pre-treated with TGFβ1 or P17 before incubating with different CM and the cells motility was measured. **P<0.01. (B) Transwell invasion assay of TGFβ1 or P17 pre-treated HCT116 cells following exposure to different CM for 48 hrs. **P<0.01. (C) qRT-PCR analysis of epithelial-mesenchymal transition (EMT) markers such as N-cadherin, E-cadherin and vimentin. **P<0.01. (D) Western blot analysis of EMT markers such as N-cadherin, E-cadherin and vimentin. *P<0.05 and **P<0.01 vs Con-CM. *P<0.05 and **P<0.01 vs HCT116-CM. **P<0.01 vs HT29-CM. Data are presented as mean ± standard deviation of three independent experiments. Abbreviations: Con, control; CM, conditioned medium. |
JAK/STAT3 signaling is involved in TGFβ1-induced migration and invasion of HCT116 and HT29 cells
STAT3 is a key transcription factor, which is involved in cell proliferation, survival and stress. It was reported that the activation of JAK/STAT3 induced by TGFβ1 promoted the tumorgenesis of lung carcinoma cells. To verify our hypothesis, the STAT3 inhibitor AG490 was pre-treated with HCT116 cells. First, the JAK/STAT3 protein expression was measured by Western blot. The result showed that TGFβ1 activated the phosphorylation of JAK1 and STAT3 (Figure 4A). And then, in order to further determine JAK/STAT3 signaling may be involved in regulating TGFβ1-induced EMT, the agent AG490, a STAT3 inhibitor, was used. As shown in Figure 4B and C, the TGFβ1 increased the level of N-cadherin and vimentin expression and decreased the level of E-cadherin expression, whereas this effect were reversed by AG490. Meanwhile, the invasive and migratory cells number was also markedly decreased while adding AG490 (Figure 5A–D), which is similar to the effect of P17. To further explore the correlation of TGFβ1 and JAK/STAT pathway, we detected the expression of p-STAT3 in cytoplasm and nucleus. We found a considerable increase in p-STAT3 expression in nuclues versus the cytosol (Figure 5E). GAPDH was used as a cytosol marker and Lamin A was used as a nuclues marker. Thus, the results confirmed that TGFβ1 enhanced p-STAT3 expression and nuclear location, while this effect was blocked by AG490 or P17, which verified that the function of TGFβ1 in metastasis was in a STAT3-dependent manner.
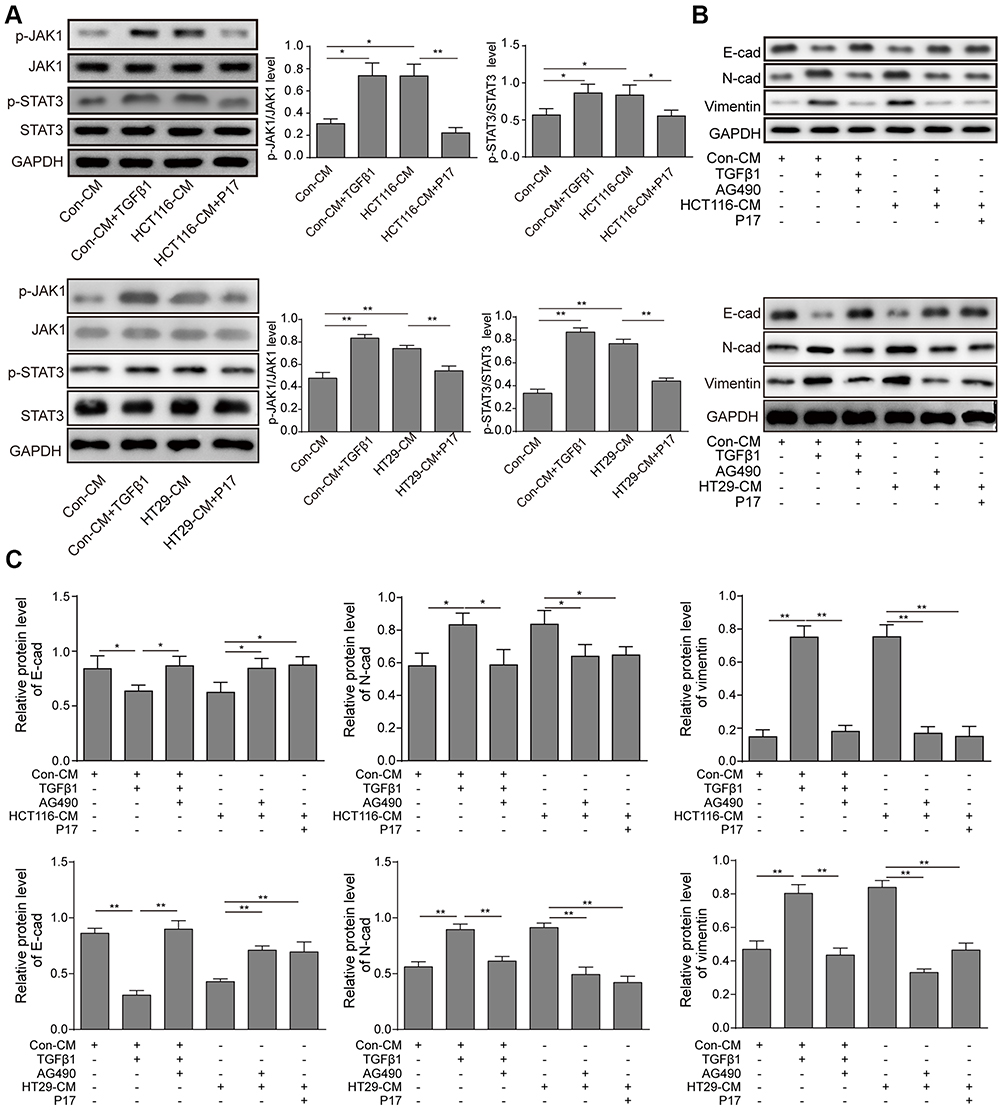 |
Figure 4 TGFβ1 induces HCT116 and HT29 cells migration and invasion via activating JAK/STAT3 signaling. (A) The expression of p-JAK1, JAK1, p-STAT3, STAT3 was measured by Western blot analysis. *P<0.05 and **P<0.01 vs Con-CM; *P<0.05 and **P<0.01 vs HCT116-CM. **P<0.01 vs.HT29-CM. (B and C) Western blot assay of EMT markers., *P<0.05 and **P<0.01 vs Con-CM, *P<0.05 and **P<0.01 vs Con-CM+TGFβ1, *P<0.05 and **P<0.01 vs HCT116-CM, **P<0.01 vs HT29-CM. (D and E) All data were presented as the mean ± standard among three independent experiments. Abbreviations: Con, control; CM, conditioned medium. |
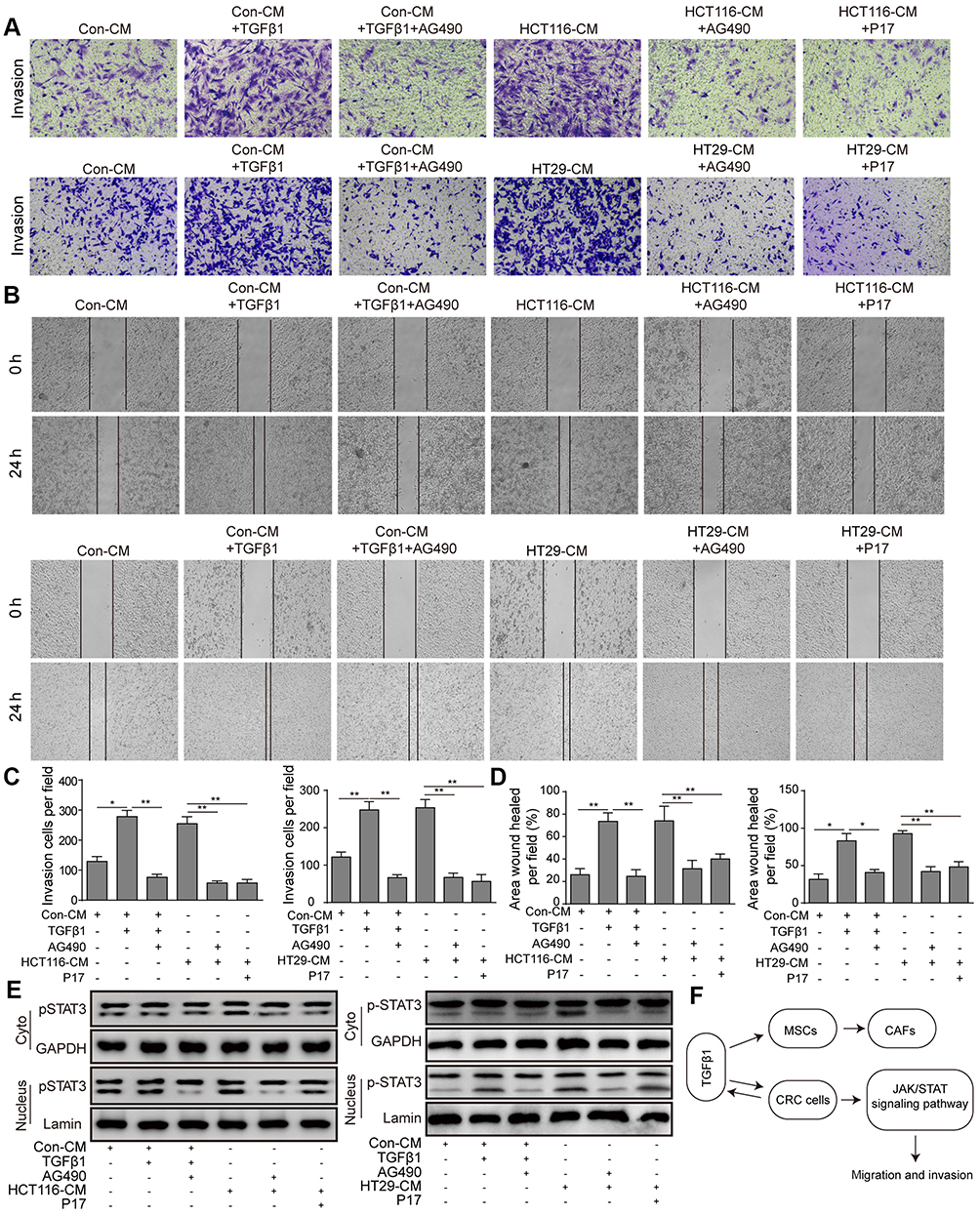 |
Figure 5 TGFβ1 induces HCT116 and HT29 cells migration and invasion via activating JAK/STAT3 signaling. (A–D) The migration and invasion of HCT116 cells was determined by wound-healing and transwell assays. *P<0.05 and **P<0.01 vs Con-CM, *P<0.05 and **P<0.01 vs Con-CM+TGFβ1, **P<0.01 vs HCT116-CM or HT29-CM. (E) p-STAT3 protein levels in HCT116 cells subcellular fractions with TGFβ1, P17 or AG490. GAPDH and Lamin A were used to act as cytoplasmic and nuclear markers, respectively. (F) The molecular pathway of TGFβ1 inducing MSCs-CAFs' differentiation and CRC metastasis. Abbreviations: Con, control; CM, conditioned medium; MSCs; mesenchymal stem cells; CAFs, cancer-associated fibroblasts; CRC, colorectal cancer. |
Discussion
It was realized that the TME played a key part in tumor progression.17–19 One of the fundamental part of TME is CAFs.20,21 CAFs were involved in tumor-associated inflammation through NF-κB pathway.22,23 The activated NF-κB induced the overexpression of chemokine CCL2 and pro-inflammatory cytokine, cyclooxygenase 2 (COX-2), which produced in CAFs.24 The chemokine CCL2 could recruit the blood mononuclear cells to tumor site, which is helpful for the generation of tumor-related macrophages.24 Meanwhile, COX-2 induces the expression of prostaglandin E2, which promotes the proliferation of malignant colonic epithelial cells.25,26 On the other hand, the CAFs secreted ECM (extracellular matrix), collagen and lysyl oxidase (LOX).27,28 ECM usually served as a pool of tumor growth factors, and LOX cross-linked to type I collagen to form a scaffold for tumorigenesis.28 Interestingly, the mechanical stress from the firm collagen scaffold induced CAFs to express MMPs (matrix metalloproteinase).27 The CAFs-induced MMPs could mediate the degradation of ECM, which caused cancer cell invasion.29 The origin of CAFs was complexed. Stellate cell, smooth muscle cell, endothelial cell, epithelial cell or resident fibroblast could be transformed into CAFs via different mechanisms.24 It is difficult to identify the source of CAFs and conduct the appropriate treatment of colorectal cancer. Here we found that MSCs, as the essential part of TME, differentiated into CAFs within specific microenvironment, which participated in the cancer metastasis.
The evidences showed that the TGFβ1 participated in tumorigenesis, development and metastasis.30 The high level of TGFβ1 was associated with poor postoperative metastasis in colorectal cancer.31 In the present study, we observed that TGFβ1 promoted the differentiation of MSCs into CAFs via upregulating the expression of α-SMA, vimentin and FSP1, which were considered as the CAFs biomarkers. The α-SMA (α-smooth muscle actin) was the fibrotic specific protein which played an important role in fibrogenesis. It had been widely used in the evaluation of CRC progression in recent years.26,32,33 Vimentin was an intermediate filament protein, usually expressed in mesenchymal cells. Vimentin composed the cytoskeleton with microtubules and microfilaments; thus, it was known as the biomarker of mesenchymal-derived cells. The level of vimentin might indicate the progress of CAFs.34–37 FSP1, also known as CXCL1 (C-X-C motif chemokine ligand 1), was a growth factor combining with G-protein coupled receptor. FSP1 could be secreted by CAFs and enhanced the progress of cancers.38,39 In this study, we found the aberrant expression of these CAFs-related biomarkers. One possible mechanism might be: TGFβ1 bound to TGFβ1 receptors on MSCs and activated Smad2/3 and P38,40 mediate the activation of CXCR4 (C-X-C chemokine receptor type 4)/SDF1 (stromal cell-derived factor-1) pathway, and upregulated the expression of fibroblast-specific proteins such as α-SMA, promoted the differentiation of MSCs into CAFs.41 We used the synthetic peptide P17 to block TGFβ1 signal pathway, and found the levels of α-SMA, vimentin and FSP1 were downregulated. It demonstrated that TGFβ1 played a critical role in the differentiation of MSCs into CAFs.
On the other hand, we found TGFβ1 could activate JAK/STAT3 pathway. The activation of the STAT3 signaling pathway has been proved to associate with cancer cell invasion and metastasis.42 And the aberrantly activation STAT3 was found in various tumor tissues.43,44 After JAK was phosphorylated, it recruited STAT3 monomers to produce homologous or heterogonous dimers.45,46 Subsequently, STAT3 enters the nucleus and binds to DNA to mediate the synthesis of downstream protein, which involved in cell proliferation, differentiation, apoptosis, angiogenesis, immune response and metastasis.45,46 The previous study also showed the important role of TGFβ1 in the TME. TGF-β1 could directly regulate the expression of MMP9, VEGF and SDF-1. In the TME, tumor cells could remodel the surrounding tissues by MMP to achieve invasion and metastasis; VEGF and SDF-1 secreted by cancer cells could promote endothelial cell proliferation. These chemokines in TME and promotes cell proliferation, migration and mediates metastasis and/or homing to the secondary site.47–49 In the present study, the invasion and migration of colorectal cancer cells were suppressed after STAT3 signaling pathway was blocked by AG490. Combining the results above, we deduced TGFβ1 activated the JAK/STAT3 pathway, thereby promoting the migration and invasion of colorectal cancer cells.
Conclusions
In conclusion, the findings of the present study verified the biological functions of TGFβ1 in the differentiation of MSCs into CAFs, and provided evidence that the TGFβ1 promoted the invasion and metastasis of HCT116 and HT29 cells. In conclusion, TGFβ1 is an essential growth factor to advance the formation of TME through the activation of JAK/STAT3 signaling pathway, which promotes the tumor metastasis (Figure 5F). This mechanism represents potential molecular targets for the treatment of this disease. However, due to multiple growth factors secreted by cancer cells, we need to further explore the effects of other growth factors such as PDGF, HGF on cancer metastasis and the present study of TGFβ1 in TME is desired to confirm in vivo.
Acknowledgment
We would like to give our sincere gratitude to the reviewers for their constructive comments.
Disclosure
The authors report no conflicts of interest in regard to this work.
References
1. Arnold M, Sierra MS, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global patterns and trends in colorectal cancer incidence and mortality. Gut. 2017;66(4):683–691. doi:10.1136/gutjnl-2015-310912
2. Valderrama-Treviño AI, Barrera-Mera B, Ceballos-Villalva JC, Montalvo-Javé EE. Hepatic metastasis from colorectal cancer. Euroasian J Hepatogastroenterol. 2017;7(2):166–175. doi:10.5005/jp-journals-10018-1241
3. Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989;8(2):98–101.
4. Korneev KV, Atretkhany K-SN, Drutskaya MS, Grivennikov SI, Kuprash DV, Nedospasov SA. TLR-signaling and proinflammatory cytokines as drivers of tumorigenesis. Cytokine. 2017;89:127–135. doi:10.1016/j.cyto.2016.01.021
5. Liu Y, Cao X. Characteristics and significance of the pre-metastatic niche. Cancer Cell. 2016;30(5):668–681. doi:10.1016/j.ccell.2016.09.011
6. Anderberg C, Pietras K. On the origin of cancer-associated fibroblasts. Cell Cycle. 2009;8(10):1461–1462. doi:10.4161/cc.8.10.8557
7. Navab R, Strumpf D, Bandarchi B, et al. Prognostic gene-expression signature of carcinoma-associated fibroblasts in non-small cell lung cancer. Proc Natl Acad Sci U S A. 2011;108(17):7160–7165. doi:10.1073/pnas.1014506108
8. Tsuyada A, Chow A, Wu J, et al. CCL2 mediates cross-talk between cancer cells and stromal fibroblasts that regulates breast cancer stem cells. Cancer Res. 2012;72(11):2768–2779. doi:10.1158/0008-5472.CAN-11-3567
9. Tauriello DVF, Palomo-Ponce S, Stork D, et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. 2018;554(7693):538–543. doi:10.1038/nature25492
10. Tang LY, Heller M, Meng Z, et al. Transforming growth factor-beta (TGF-beta) directly activates the JAK1-STAT3 axis to induce hepatic fibrosis in coordination with the SMAD pathway. J Biol Chem. 2017;292(10):4302–4312. doi:10.1074/jbc.M116.773085
11. Cirri P, Chiarugi P. Cancer associated fibroblasts: the dark side of the coin. Am J Cancer Res. 2011;1(4):482–497.
12. Mishra PJ, Banerjee D. Activation and differentiation of mesenchymal stem cells. Methods Mol Biol. 2017;1554:201–209. doi:10.1007/978-1-4939-6759-9_13
13. Guan J, Chen J. Mesenchymal stem cells in the tumor microenvironment. Biomed Rep. 2013;1(4):517–521. doi:10.3892/br.2013.103
14. Zhuang J, Lu Q, Shen B, et al. TGFbeta1 secreted by cancer-associated fibroblasts induces epithelial-mesenchymal transition of bladder cancer cells through lncRNA-ZEB2NAT. Sci Rep. 2015;5:11924. doi:10.1038/srep11924
15. Bartosh TJ, Ylostalo JH. Preparation of anti-inflammatory mesenchymal stem/precursor cells (MSCs) through sphere formation using hanging-drop culture technique. Curr Protoc Stem Cell Biol. 2014;28:Unit 2B 6. doi:10.1002/9780470151808.sc02b06s28
16. Justus CR, Leffler N, Ruiz-Echevarria M, Yang LV. In vitro cell migration and invasion assays. J Vis Exp. 2014;(88). doi:10.3791/51046
17. Geng B, Zhang C, Wang C, et al. IkappaB-kinase-epsilon in the tumor microenvironment is essential for the progression of gastric cancer. Oncotarget. 2017;8(43):75298–75307. doi:10.18632/oncotarget.20778
18. Huang A, Cao S, Tang L. The tumor microenvironment and inflammatory breast cancer. J Cancer. 2017;8(10):1884–1891. doi:10.7150/jca.17595
19. Novikova MV, Khromova NV, Kopnin PB. Components of the hepatocellular carcinoma microenvironment and their role in tumor progression. Biochemistry (Mosc). 2017;82(8):861–873. doi:10.1134/S0006297917080016
20. Belli C, Trapani D, Viale G, et al. Targeting the microenvironment in solid tumors. Cancer Treat Rev. 2018;65:22–32. doi:10.1016/j.ctrv.2018.02.004
21. Liao Z, Tan ZW, Zhu P, Tan NS. Cancer-associated fibroblasts in tumor microenvironment - accomplices in tumor malignancy. Cell Immunol. 2018. doi:10.1016/j.cellimm.2017.12.003
22. Bohonowych JE, Hance MW, Nolan KD, Defee M, Parsons CH, Isaacs JS. Extracellular Hsp90 mediates an NF-kappaB dependent inflammatory stromal program: implications for the prostate tumor microenvironment. Prostate. 2014;74(4):395–407. doi:10.1002/pros.22761
23. Yeung TL, Leung CS, Wong -K-K, et al. TGF-beta modulates ovarian cancer invasion by upregulating CAF-derived versican in the tumor microenvironment. Cancer Res. 2013;73(16):5016–5028. doi:10.1158/0008-5472.CAN-13-0023
24. Mukaida N, Sasaki S. Fibroblasts, an inconspicuous but essential player in colon cancer development and progression. World J Gastroenterol. 2016;22(23):5301–5316. doi:10.3748/wjg.v22.i23.5301
25. Roulis M, Nikolaou C, Kotsaki E, et al. Intestinal myofibroblast-specific Tpl2-Cox-2-PGE2 pathway links innate sensing to epithelial homeostasis. Proc Natl Acad Sci U S A. 2014;111(43):E4658–E4667. doi:10.1073/pnas.1415762111
26. Sasaki Y, Nakatani Y, Hara S. Role of microsomal prostaglandin E synthase-1 (mPGES-1)-derived prostaglandin E2 in colon carcinogenesis. Prostaglandins Other Lipid Mediat. 2015;121(Pt A):42–45. doi:10.1016/j.prostaglandins.2015.06.006
27. Levental KR, Yu H, Kass L, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139(5):891–906. doi:10.1016/j.cell.2009.10.027
28. Malik R, Lelkes PI, Cukierman E. Biomechanical and biochemical remodeling of stromal extracellular matrix in cancer. Trends Biotechnol. 2015;33(4):230–236. doi:10.1016/j.tibtech.2015.01.004
29. Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141(1):52–67. doi:10.1016/j.cell.2010.03.015
30. Liu H, Wang S, Ma W, Lu Y. Transforming growth factor beta1 promotes migration and invasion of human hepatocellular carcinoma cells via up-regulation of connective tissue growth factor. Cell Biochem Biophys. 2015;73(3):775–781. doi:10.1007/s12013-015-0693-6
31. Makrodouli E, Oikonomou E, Koc M, et al. BRAF and RAS oncogenes regulate Rho GTPase pathways to mediate migration and invasion properties in human colon cancer cells: a comparative study. Mol Cancer. 2011;10:118. doi:10.1186/1476-4598-10-93
32. Arena S, Salati M, Sorgentoni G, Barbisan F, Orciani M. Characterization of tumor-derived mesenchymal stem cells potentially differentiating into cancer-associated fibroblasts in lung cancer. Clin Transl Oncol. 2018;20:1582–1591. doi:10.1007/s12094-018-1894-4
33. Nishishita R, Morohashi S, Seino H, et al. Expression of cancer-associated fibroblast markers in advanced colorectal cancer. Oncol Lett. 2018;15(5):6195–6202. doi:10.3892/ol.2018.8097
34. Ding L, Ren J, Zhang D, et al. A novel stromal lncRNA signature reprograms fibroblasts to promote the growth of oral squamous cell carcinoma via LncRNA-CAF/interleukin-33. Carcinogenesis. 2018;39(3):397–406. doi:10.1093/carcin/bgy006
35. Kilvaer TK, Rakaee M, Hellevik T, et al. Tissue analyses reveal a potential immune-adjuvant function of FAP-1 positive fibroblasts in non-small cell lung cancer. PLoS One. 2018;13(2):e0192157. doi:10.1371/journal.pone.0192157
36. Hogervorst M, Rietveld M, de Gruijl F, El Ghalbzouri A. A shift from papillary to reticular fibroblasts enables tumour-stroma interaction and invasion. Br J Cancer. 2018;118(8):1089–1097. doi:10.1038/s41416-018-0024-y
37. Strong AL, Pei DT, Hurst CG, Gimble JM, Burow ME, Bunnell BA. Obesity enhances the conversion of adipose-derived stromal/stem cells into carcinoma-associated fibroblast leading to cancer cell proliferation and progression to an invasive phenotype. Stem Cells Int. 2017;2017:1–11. doi:10.1155/2017/9216502
38. Zhang H, Yue J, Jiang Z, et al. CAF-secreted CXCL1 conferred radioresistance by regulating DNA damage response in a ROS-dependent manner in esophageal squamous cell carcinoma. Cell Death Dis. 2017;8(5):e2790. doi:10.1038/cddis.2017.518
39. Miyake M, Hori S, Morizawa Y, et al. CXCL1-mediated interaction of cancer cells with tumor-associated macrophages and cancer-associated fibroblasts promotes tumor progression in human bladder cancer. Neoplasia. 2016;18(10):636–646. doi:10.1016/j.neo.2016.08.002
40. Hou J, Wang L, Hou J, et al. Peroxisome proliferator-activated receptor gamma promotes mesenchymal stem cells to express connexin43 via the inhibition of TGF-beta1/smads signaling in a rat model of myocardial infarction. Stem Cell Rev. 2015;11(6):885–899. doi:10.1007/s12015-015-9615-7
41. Quante M, Tu SP, Tomita H, et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell. 2011;19(2):257–272. doi:10.1016/j.ccr.2011.01.020
42. Pinciroli P, Pereira DM, Amaral JD, et al. An IL6-correlated signature in serous epithelial ovarian cancer associates with growth factor response. BMC Genomics. 2013;14:508. doi:10.1186/1471-2164-14-181
43. Cheng GZ, Zhang WZ, Sun M, et al. Twist is transcriptionally induced by activation of STAT3 and mediates STAT3 oncogenic function. J Biol Chem. 2008;283(21):14665–14673. doi:10.1074/jbc.M707429200
44. Colomiere M, Ward AC, Riley C, et al. Cross talk of signals between EGFR and IL-6R through JAK2/STAT3 mediate epithelial-mesenchymal transition in ovarian carcinomas. Br J Cancer. 2009;100(1):134–144. doi:10.1038/sj.bjc.6604794
45. Groner B, von Manstein V. Jak Stat signaling and cancer: opportunities, benefits and side effects of targeted inhibition. Mol Cell Endocrinol. 2017;451:1–14. doi:10.1016/j.mce.2017.05.033
46. Lu R, Zhang YG, Sun J. STAT3 activation in infection and infection-associated cancer. Mol Cell Endocrinol. 2017;451:80–87. doi:10.1016/j.mce.2017.02.023
47. Moore-Smith LD, Isayeva T, Lee JH, Frost A, Ponnazhagan S. Silencing of TGF-beta1 in tumor cells impacts MMP-9 in tumor microenvironment. Sci Rep. 2017;7(1):8678. doi:10.1038/s41598-017-09062-y
48. Park BV, Freeman ZT, Ghasemzadeh A, et al. TGFbeta1-mediated SMAD3 enhances PD-1 expression on antigen-specific T cells in cancer. Cancer Discov. 2016;6(12):1366–1381. doi:10.1158/2159-8290.CD-15-1347
49. Lamprecht S, Sigal-Batikoff I, Shany S, et al. Teaming up for trouble: cancer cells, transforming growth factor-beta1 signaling and the epigenetic corruption of stromal naive fibroblasts. Cancers (Basel). 2018;10(3). doi:10.3390/cancers10110400
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.