Back to Journals » Breast Cancer: Targets and Therapy » Volume 13
TGF-β-Induced TMEPAI Promotes Epithelial–Mesenchymal Transition in Doxorubicin-Treated Triple-Negative Breast Cancer Cells via SMAD3 and PI3K/AKT Pathway Alteration
Authors Wardhani BWK ![]() , Louisa M
, Louisa M ![]() , Watanabe Y, Setiabudy R, Kato M
, Watanabe Y, Setiabudy R, Kato M ![]()
Received 19 June 2021
Accepted for publication 3 September 2021
Published 21 September 2021 Volume 2021:13 Pages 529—538
DOI https://doi.org/10.2147/BCTT.S325429
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Pranela Rameshwar
Bantari WK Wardhani,1,2 Melva Louisa,3 Yukihide Watanabe,4 Rianto Setiabudy,3 Mitsuyasu Kato4
1Biomedical Sciences, Faculty of Medicine Universitas Indonesia, Jakarta, Indonesia; 2Department of Pharmacology, Faculty of Military Pharmacy, Indonesia Defense University, West Java, Indonesia; 3Department of Pharmacology and Therapeutics, Faculty of Medicine Universitas Indonesia, Jakarta, Indonesia; 4Department of Experimental Pathology, Graduate School of Comprehensive Human Sciences, Faculty of Medicine, University of Tsukuba, Tsukuba, Japan
Correspondence: Melva Louisa
Department of Pharmacology and Therapeutics, Faculty of Medicine Universitas Indonesia, Jl. Salemba Raya No. 6, Jakarta Pusat, Jakarta, 10430, Indonesia
Tel +62 21 3193-481
Email [email protected]
Introduction: Epithelial–mesenchymal transition (EMT) and overexpression of drug efflux transporters have been reported to cause doxorubicin resistance. Our previous study indicated that TMEPAI (transmembrane prostate androgen-induced protein) attenuated doxorubicin sensitivity in triple-negative breast cancer cells. However, how TMEPAI contributes to doxorubicin resistance in TNBC remains unclear. Thus, the present study aimed to elucidate the mechanism of TMEPAI in doxorubicin resistance in triple-negative breast cancer cells.
Methods: We used BT549, triple-negative cells wild type (WT), and BT549 TMEPAI knock-out. Both cells were treated with TGF-β 2 ng/mL for 24 hours, followed by TGF-β 2 ng/mL and doxorubicin 12.9 nM for another 24 hours. Afterward, the cells were harvested and counted. Cells were further lysed and used for RT-PCR and Western blot analysis. We determined the expression levels of proliferation, apoptosis, EMT markers, and drug efflux transporters. Additionally, we investigated the expressions of PI3K as well as SMAD3 and AKT phosphorylation.
Results: TNBC cells were shown to be less sensitive to doxorubicin in the presence of TMEPAI. TMEPAI was shown to alleviate the mRNA expressions of apoptosis markers: Bax, Bcl2, Caspase-3, and Caspase-9. Our results indicated that the presence of TMEPAI greatly amplifies EMT and increases drug efflux transporter expressions after doxorubicin treatment. Furthermore, our findings demonstrated that TMEPAI reduced the action of doxorubicin in inhibiting SMAD3 phosphorylation. TMEPAI was also shown to modify the effect of doxorubicin by reducing PI3K expressions and Akt phosphorylation in triple-negative breast cancer cells.
Conclusion: Our findings indicate that TMEPAI promotes EMT and drug efflux transporters at least in part by shifting doxorubicin action from SMAD3 phosphorylation reduction to PI3K/AKT inhibition in triple-negative breast cancer cells.
Keywords: PMEPAI, TGF-β, SMAD3, vimentin, E-cadherin, drug efflux transporters
Introduction
Female breast cancer is still a global burden, being the predominant type of cancer and the leading cause of cancer-related mortality and morbidity.1 Triple-negative breast cancer (TNBC) is one of the types of breast cancer with the lack of molecular marker expressions: estrogen receptor (ER), progesterone receptors, and human epidermal growth factor 2 (HER2) molecular markers. Patients with the TNBC subtype are associated with a more aggressive disease state and poorer prognosis than non-TNBC.2–4 Currently, the backbone of treatments for TNBC patients is limited to cytotoxic agents such as anthracyclines, alkylators, and taxanes, still with evolving protocols.2,5–7 Despite the benefit of systemic chemotherapy in improving outcomes of TNBC patients, the problem with chemoresistance is significant, which accounts for about 90% of treatment failure cases.8–11
Various mechanisms have been suggested to be associated with cancer resistance to chemotherapy. Some of the proposed causes include the ABC transporter-mediated drug efflux, epithelial–mesenchymal transition (EMT), hypoxia, cancer stem cells and the regulation of several networks of signaling pathways.2,12–14 In TNBC, TGF-β1 was found to play a critical role in treatment-resistant development. In a recent study, Xu et al revealed that endogenous TGF-β1 affects cancer cell stemness and causes an increase in anti-apoptotic markers as well as EMT after low-dose epirubicin treatments.15
Our previous study showed that TGF-β-induced TMEPAI (transmembrane prostate androgen-induced protein) caused the decrease anticancer response of TNBC to doxorubicin and paclitaxel.16 Overexpression of TMEPAI was found in many types of cancer, including breast cancer. Singha et al reported that more patients with invasive type have high expressions of TMEPAI compared with those in non-invasive cells such as luminal A type of breast cancer cells,17 while Nie et al reported that TMEPAI expressions were higher in HER2+ and TNBC types vs luminal A types of breast cancer cells.18
High expression of TMEPAI in TNBC patients and is associated with poor prognosis.17 Various studies indicated that TMEPAI is induced by TGF-β signaling and involved in multiple signaling pathways.19,20 Furthermore, Singha et al suggested that TGF-β-induced TMEPAI inhibited Smad signaling increased PI3K/Akt signaling by downregulating PTEN, which led to tumor progression in TNBC.21 However, the mechanism on how TMEPAI may alter the cytotoxic effect of doxorubicin in TNBC is not fully understood. Therefore, we aimed to investigate the role of TMEPAI in TNBC response to doxorubicin, mainly via Smad and PI3K/Akt pathways.
Materials and Methods
Cell Culture and Drug Treatment
Triple-negative breast cancer cells, BT549 wild type (WT) cells were purchased from the ATCC by the Laboratory of Experimental Pathology, University of Tsukuba. BT549 TMEPAI-knocked-out (KO) cells were established previously using the CRISPR-Cas9 technique.
Cells were cultured and maintained using culture medium described previously.16 Experiments were done when the cells reached 70% confluency under cell starvation in 1% Fetal Bovine Serum (Gibco) in a 10-cm tissue-culture dish (Corning). Cells were then treated with recombinant human TGF-β (Wako) 2 ng/mL for 24 hours, followed by TGF-β 2 ng/mL and doxorubicin (Wako) 12.9 nM for another 24 hours. The dose of doxorubicin was based on our previous experiment that showed the IC50 of doxorubicin in BT549 cells was 12.9 nM.16 Afterward, cells were harvested and counted. Cell viability was calculated using the trypan blue exclusion method. Then, these cells were lysed and isolated for RNA and protein for further analysis. All the treatments were done in four separate experiments in duplicate.
RT-PCR
Total RNA from cell lysates was extracted using Total Mini Kit Cultured Cells (Geneaid). RNA concentrations were measured using Nanodrop 200 UV-Vis Spectrophotometer. We did a synthesis of cDNA from 100 ng of total RNA using Revertra Ace (Toyobo). Then, we used equal amounts of cDNA (100 ng) to perform quantitative reverse-transcriptase polymerase chain reaction (RT-PCR) on a LightCycler®480 Roche using Thunderbird® SYBR qPCR Mix (Toyobo). The mRNA expressions were quantified using the 2−ΔΔCT method, with β-actin as the reference gene. The primer sequences used in the study are explained in Table 1.
 |
Table 1 Primer Sequence Used in the Study |
Western Blotting
Cell lysates were subjected to SDS-page electrophoresis gel to determine protein expressions of β-actin, phosphorylated SMAD3, total SMAD3, phosphorylated AKT, AKT, PI3K, Vimentin, E-Cadherin, and MRP-1 (Cell Signaling). Equal amounts of protein (50 µg) were boiled with Laemmli Buffer for 5 minutes, continued with sodium dodecyl sulfate-polyacrylamide (SDS-PAGE) electrophoresis using 8–12.5% gel. Separated proteins were transferred to nitrocellulose membrane (Biorad) and blocked with 5% skim milk in Tris-buffered saline (TBS) containing 0.01% Tween-20 (TBST) for 1 hour and 2 hours for phosphorylated protein. Membranes were washed three times for 5 minutes in TBST. Primary antibodies were incubated in 1:1000 dilution of TBST buffer at 4°C overnight. Anti-rabbit secondary antibody was incubated in 1:4000 dilution of TBST buffer for 1 hour at room temperature. Detection of protein expression was done on Gel Documentation System (UvTech) using additionally enhanced chemiluminescence (ImmunoStar, Wako).
Statistical Analysis
The difference between the two groups (TMEPAI wild type versus knock-out) was determined using Student’s t-test. Comparison between four treatment groups was examined using one-way ANOVA followed by post-hoc Tukey or Games Howell test.
Results
TMEPAI Impaired the Cytotoxic Effect of Doxorubicin
Doxorubicin treatment resulted in reduced cell viability in TNBC, both in the parental and TMEPAI knock-out cells. However, the magnitude of the cytotoxic effect was more pronounced in the absence of TMEPAI (Figure 1). A higher percentage of cell viability in WT cells suggested that TMEPAI positive TNBC was less sensitive to doxorubicin.
 |
Figure 1 Reduction of cell viability after doxorubicin treatment was more pronounced in TMEPAI knock-out cells. Cells were treated with TGF-β and doxorubicin. We did all experiments four times in duplicate. Analyses were done using an independent t-test. Data are presented in mean ± SD. *p<0.05 vs WT (wild-type cells/TMEPAI positive cells). |
TMEPAI Caused a Modest Change in Ki-67 mRNA Expressions in Doxorubicin-Treated Cells
We investigated the effect of TMEPAI on the expressions of cell proliferation markers Ki-67 after treatment with doxorubicin. Doxorubicin treatment significantly decreased cell proliferation markers. Nonetheless, the absence of TMEPAI resulted in a slight change in mRNA Ki-67 expressions as observed in the knock-out cells after treatment with doxorubicin (Figure 2).
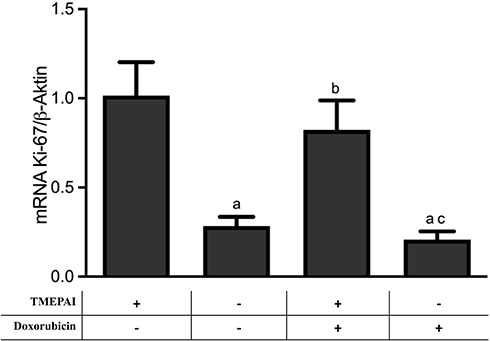 |
Figure 2 TMEPAI was modestly affecting mRNA expressions of Ki-67 in doxorubicin-treated cells. Cells were treated with TGF-β and doxorubicin. Data are presented as mean ± SD. We did all experiments four times in duplicate. ap <0.05 vs TMEPAI (+) cells without doxorubicin; bp<0.05 vs TMEPAI (-) cells without doxorubicin cp<0.05 vs (+) TMEPAI (+) cells with doxorubicin after analysis using one-way ANOVA followed by Games-Howell test. |
TMEPAI Ameliorates the Pro-Apoptotic Effect of Doxorubicin
We found that the presence of TMEPAI alleviated the apoptotic effect of doxorubicin as shown by the decreased mRNA expressions of all apoptotic markers investigated, Bax, BCl2, Caspase-3, and Caspase-9 (Figure 3).
 |
Figure 3 TMEPAI ameliorated mRNA expressions of apoptosis markers in doxorubicin-treated cells. (A) Bax; (B) Bcl2; (C) Caspase-3; and (D) Caspase-9. Cells were treated with TGF-β and doxorubicin. Data are presented in mean ± SD. We did the experiments four times in duplicate. ap <0.05 vs TMEPAI (+) cells without doxorubicin; bp<0.05 vs TMEPAI (-) cells without doxorubicin. cp<0.05 vs TMEPAI (+) cells with doxorubicin after analysis using one-way ANOVA followed by Tukey or Games-Howell test. |
TMEPAI Altered the Effect of Doxorubicin on Smad3 and PI3K/AKT Pathway
We investigated the TGF-β signaling pathway downstream: the canonical pathway Smad3 and non-canonical PI3K/AKT. Our results suggested that doxorubicin had a more potent effect in reducing Smad3 phosphorylation in the absence of TMEPAI. In contrast, doxorubicin showed a more substantial inhibitory effect on PI3K/AKT activation in the presence of TMEPAI. Our finding was supported by TMEPAI inducing PI3K/AKT activation (Figure 4A–C).
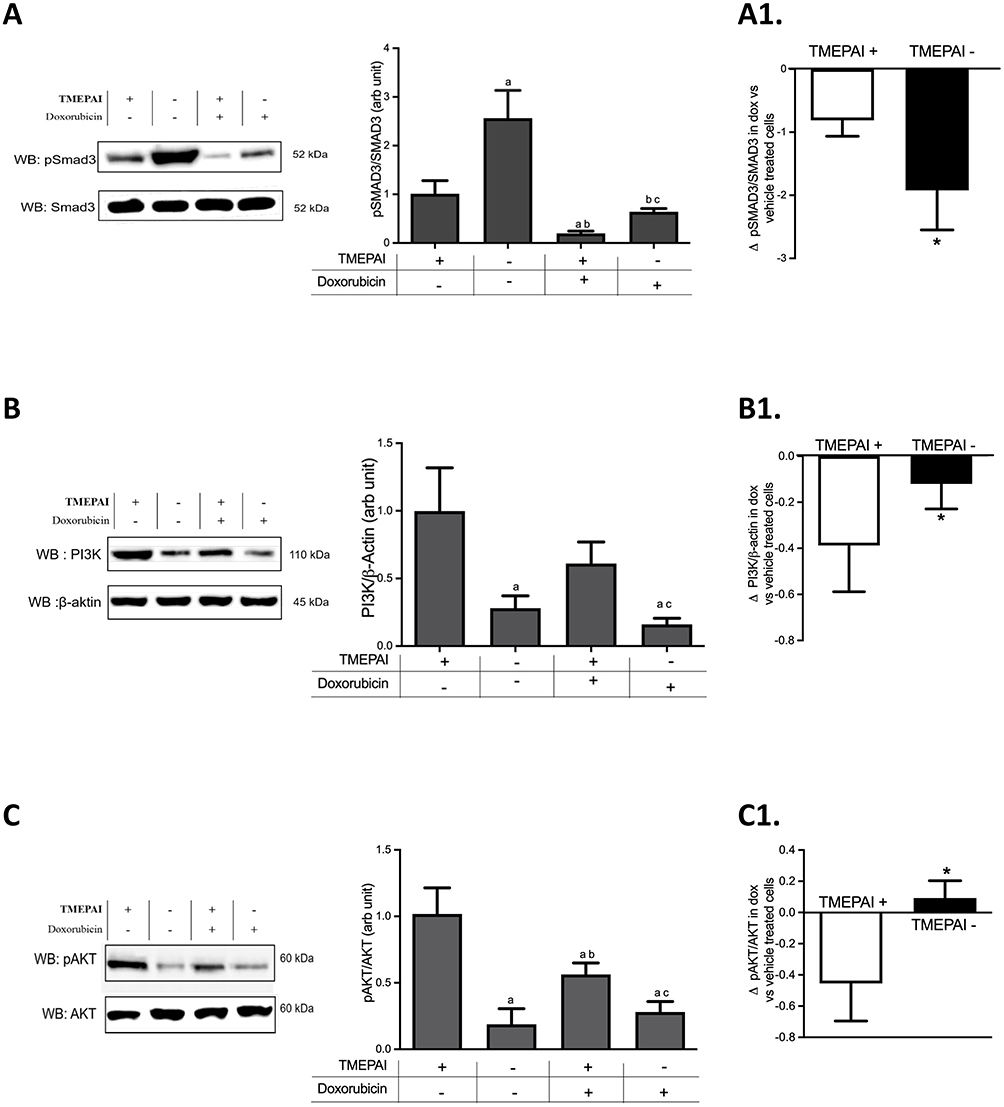 |
Figure 4 TMEPAI modified the effect of doxorubicin on SMAD3 and PI3K/AKT expressions (A) pSMAD3/SMAD3; (B) PI3K/β-actin; (C) pAKT/AKT; and the differences the differences of expressions in doxorubicin vs vehicle treated cells; (A1) Δ pSMAD3/SMAD3; (B1) Δ PI3K/β-actin (C1) Δ pAKT/AKT. Cells were treated with TGF-β and doxorubicin. Data are presented in mean ± SD from four separate experiments in duplicate. ap <0.05 vs (+) TMEPAI (+) cells without doxorubicin, bp<0.05 vs TMEPAI (-) cells without doxorubicin, cp<0.05 vs TMEPAI (+) cells with doxorubicin and *p<0.05 vs TMEPAI (+) cells after analysis using one-way ANOVA followed by Tukey or Games-Howell test. |
TMEPAI Enhanced EMT in Doxorubicin-Treated Cells
Epithelial–mesenchymal transition (EMT) is a related mechanism of TNBC resistance to anticancer drugs, including doxorubicin. After doxorubicin treatment, we analyzed several EMT markers: mRNA expression levels of snail, zeb-1, and twist and protein expressions of vimentin and E-Cadherin (Figure 5). Our results indicated that the presence of TMEPAI potently amplified doxorubicin-induced EMT.
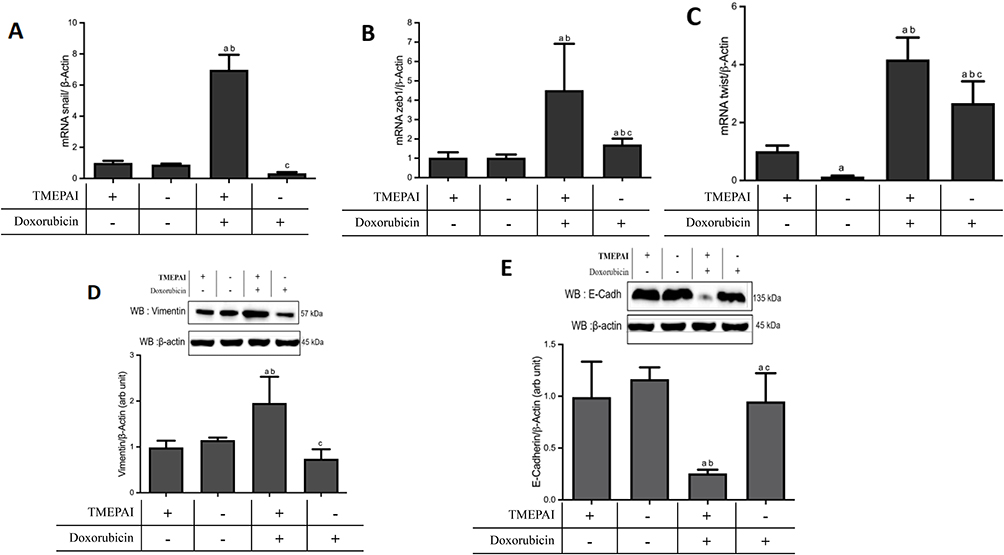 |
Figure 5 Marked enhancement of EMT in TMEPAI positive cells, as shown by EMT markers. (A) mRNA expressions of snail/β-actin, (B) mRNA expressions of zeb1/β-actin, (C) mRNA expressions of twist/β-actin, (D) protein expressions of Vimentin/β-actin; (E) protein expressions of E-Cadherin/β-actin. Cells were treated with TGF-β and doxorubicin. Data are presented in mean ± SD from four separate experiments in duplicate. ap<0.05 vs TMEPAI (+) cells without doxorubicin, bp<0.05 vs TMEPAI (-) cells without doxorubicin, cp<0.05 vs TMEPAI (+) cells with doxorubicin; after analysis using one-way ANOVA followed by Tukey or Games-Howell test. |
TMEPAI Increased Drug Efflux Transporters Expressions in Doxorubicin-Treated TNBC
One anticancer resistance mechanism is drug efflux transporter overexpression, including P-glycoprotein, BCRP, and MRP-1. The presence of TMEPAI caused an alleviation in drug efflux transporters expressions after treatment with doxorubicin, particularly P-glycoprotein and BCRP (Figure 6).
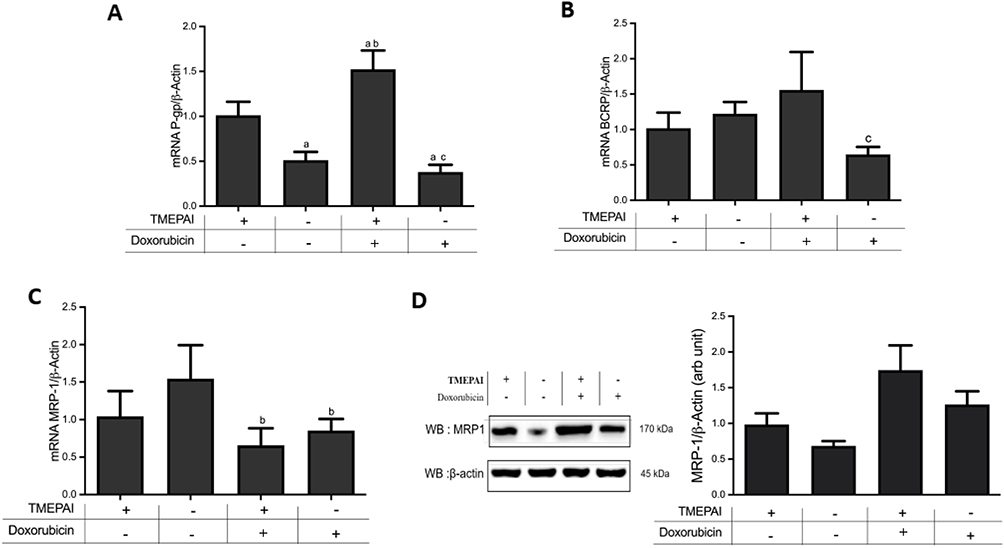 |
Figure 6 TMEPAI induced drug efflux transporters expressions in doxorubicin-treated cells. (A) mRNA expressions of P-glycoprotein/β-actin, (B) mRNA expressions of BCRP/β-actin, (C) mRNA expressions of MRP1/β-actin, (D) protein expressions of MRP1/β-actin. Data are presented in mean ± SD from four separate experiments in duplicate. ap <0.05 vs TMEPAI (+) cells without doxorubicin, bp<0.05 vs TMEPAI (-) cells without doxorubicin, cp<0.05 vs TMEPAI (+) cells with doxorubicin after analysis using one-way ANOVA followed by Tukey’s test. |
Discussion
Previously, we reported that TGF-β-induced TMEPAI was involved in altering the sensitivity of TNBC to doxorubicin.16 In our present study, we confirmed that TMEPAI participated in reducing the response of TNBC to doxorubicin, mainly by promoting EMT and drug-efflux transporters.
To investigate the role of TMEPAI in TNBC resistance to doxorubicin, we used BT549 cells that displayed high expression of TMEPAI and BT549 TMEPAI knock-out, which was previously established with the CRISPR Cas9 technique.16,29 TMEPAI was known to be induced by TGF-β in TNBC and involved in tumor progression through its interaction with Smads in canonical pathways, as well as regulating Smad-independent pathway by activating MAPK and PI3K/AKT.17,30 Studies had revealed the critical role of TGF-β in the development of anticancer resistance, by causing EMT, increasing tumor heterogeneity, as well as generating stemness and metastatic markers.15,31,32 However, how TGF-β-induced TMEPAI might play roles in anticancer resistance, doxorubicin in particular, remains unclear.
Prior treatment with TGF-β in TMEPAI positive cells is essential for TNBC response to doxorubicin. Studies from clinical samples from patients have confirmed that most TNBC patients have a high expression of TGF-β1.33,34 A previous study by Wardhani et al reported that without TGF-β stimulation, the response of TNBC Hs578T to cytotoxic drugs would be reversed.35 In our study, doxorubicin retained its cytotoxic effect result in both TMEPAI positive and negative cells. Nonetheless, TMEPAI knock-out cells showed a more potent result. In TMEPAI knock-out BT549, we observe only a slight decrease in Ki-67 mRNA expressions after doxorubicin treatment. Previous studies had suggested that TMEPAI might play a paradoxical role in cancer progression.17,21,36 TMEPAI potentiates the non-canonical pathway in the presence of high TGF-β and supports cell proliferation,17,21 it also inhibits Wnt/β-catenin signaling that reduces breast cancer metastasis.36 A recent study by Puteri et al might explain the phenomenon that double PY motifs and a SIM of TMEPAI isoforms are essential for colony and sphere formation, however not for monolayer cell formation.29 It was known that BT549 is a triple-negative breast cancer cell with a high expression of Ki-67,37–39 and doxorubicin can still exert anticancer activity in cells that proliferate actively.40,41
One of the mechanisms of doxorubicin-induced cytotoxicity is activating caspases, modifying Bax and Bcl2 expressions, and altering the actin cytoskeleton of tumor cells. Reduced sensitivity of cancer cells to doxorubicin can be shown by dysregulation of apoptotic marker expressions.8,42,43 Our RT-PCR analysis suggested that doxorubicin might have a weaker pro-apoptotic activity in TMEPAI positive cells than knock-out cells. However, we acknowledge that a better correlation to apoptotic markers will be achieved if we analyze using Western blotting. A different result was found in hematological cancer such as multiple myeloma, where TMEPAI expression is relatively low. TMEPAI was shown to enhance apoptosis in multiple myeloma cells by decreasing c-Maf stability that resulted in protein degradation.44
Previous studies had reported the role of TMEPAI in canonical (Smad-dependent) and non-canonical (PI3K/Akt) pathways.17,21,45 In our study, we showed that doxorubicin treatment resulted in both Smad-dependent and non-canonical pathway (PI3K/Akt) alteration. Here, our results indicated that TMEPAI positive cells had lower activity in Smad3 activation. In contrast, TMEPAI positive cells had a higher expression of PI3K and AKT phosphorylation. Doxorubicin alone was known to reduce Smad3 phosphorylation. Filyak et al reported that doxorubicin inhibits TGF-β signal transduction by downregulating SMAD2/3/4 phosphorylation in lung carcinoma.46 In our study, we observe that the presence of TMEPAI mediates a shift in doxorubicin action from Smad3 phosphorylation inhibition to inactivation of PI3K/AKT pathway. This result suggests that doxorubicin behaves differently in TNBC cells and acts stronger on the individual cells’ predominant expressions. Our findings agree with Lou et al, who reported that the lack of TMEPAI led to autophagy inhibition as shown by reduced expression of beclin-1 and generated cancer cells’ resistance to doxorubicin. In breast cancer, autophagy helps cancer cells to eliminate doxorubicin-induced damage. Down-regulation of beclin-1, an inducer of autophagy in cancer cells, decreased autophagy initiation via PI3K/AKT pathways.47
Upregulation of Smad phosphorylation in canonical TGF-β pathways results in the activation of transcription factors of EMT, including snail, zeb1, twist, and slug. Further, the transcription factors induce vimentin and inhibit E-cadherin.48,49 In their recent study, Singha et al have suggested that in a Smad3 knock-down system in TNBC, there were declining EMT transcription factors, E-cadherin. Increased vimentin expressions followed the results.50 Additionally, EMT can also be generated by non-Smads: PI3K/AKT, Traf4/Traf6, RhoA, MAPK, and Ras/Raf.51 In our study, we demonstrate that doxorubicin strongly increased all EMT transcription factors studied in TMEPAI positive cells but not in TMEPAI knock-out cells. Increased expressions of snail, zeb1, and twist were followed by the increased vimentin expression and reduced expressions of E-cadherin. Several studies have shown that EMT was induced after only a short-term treatment of doxorubicin, resulting in a more resistant feature to drug treatment.52,53 A study by Zhang et al in colorectal cancer had also described that TMEPAI induces EMT by activating BMP signaling.54
Studies have described that doxorubicin resistance is related to the overexpression of drug efflux transporters, P-glycoprotein, BCRP, and MRP-1. Increased expressions of drug efflux transporters reduced intracellular drug concentrations, which resulted in a decreased drug efficacy.12,14,55 Takano et al have reported that EMT caused an increased expression of drug efflux transporters and further induced drug resistance.56 In their studies, Saxena et al and Jiang et al described that EMT transcription factors (snail, zeb, twist, and slug) regulate drug efflux transporters via the TGF-β signaling pathway.57,58 The present study demonstrates that TMEPAI positive cells have higher mRNA expressions of P-gp, but not BCRP and MRP-1 compared to knock-out cells. In doxorubicin-treated cells, TMEPAI promotes the elevation of P-gp and BCRP, while we observed minimum effect with MRP1. P-gp was known to be the predominant transporter that causes TNBC resistance to anthracycline vs BCRP and MRP1. Tsou et al suggested that P-gp, as a resistance marker in breast cancer cells, is elevated due to the up/down-regulation of genes involved in apoptosis, EMT, and ABC transporters.59
As suggested by the findings of several researchers that showed that overexpression of TMEPAI is more likely to be found in TNBC, our results may add some concerns regarding the choice of cytotoxic treatments, doxorubicin in particular. In our present study, our experiment used TGF-β stimulation. Yet, our results have to be carefully interpreted, considering that only about half of TNBC patients express high levels of TGF-β.34
Conclusion
Taken together, we suggest that TMEPAI plays an essential role in the decreased sensitivity of TNBC to doxorubicin, as shown by the elevation cell viability and Ki-67 expressions, downregulation of apoptosis expression markers, increased EMT and drug efflux transporters. TMEPAI mediates all the effects partly by causing a shift in doxorubicin action from Smad3 to PI3K/Akt inhibition.
Funding
The study was funded by the PUTI 2020 Grant from the Universitas Indonesia Contract No. NKB-1556/UN2.RST/HKP.05.00/2020.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–249. doi:10.3322/caac.21660
2. Gupta GK, Collier AL, Lee D, et al. Perspectives on triple-negative breast cancer: current treatment strategies, unmet needs, and potential targets for future therapies. Cancers. 2020;12:2392. doi:10.3390/cancers12092392
3. Reddy SM, Barcenas CH, Sinha AK, et al. Long-term survival outcomes of triple-receptor negative breast cancer survivors who are disease-free at 5 years and relationship with low hormone receptor positivity. Br J Cancer. 2018;118:17–23. doi:10.1038/bjc.2017.379
4. Ali AM, AK Ansari J, M Abd El-Aziz N, et al. Triple-negative breast cancer: a tale of two decades. Anticancer Agents Med Chem. 2017;17:491–499. doi:10.2174/1871520616666160725112335
5. Sharma P. Update on the treatment of early-stage triple-negative breast cancer. Curr Treat Options Oncol. 2018;19:22. doi:10.1007/s11864-018-0539-8
6. Malhotra MK, Emens LA. The evolving management of metastatic triple-negative breast cancer. Semin Oncol. 2020;47:229–237. doi:10.1053/j.seminoncol.2020.05.005
7. Lyons TG. Targeted therapies for triple-negative breast cancer. Curr Treat Options Oncol. 2019;20:82. doi:10.1007/s11864-019-0682-x
8. Martin M, Ramos-Medina R, Bernat R, et al. Activity of docetaxel, carboplatin, and doxorubicin in patient-derived triple-negative breast cancer xenografts. Sci Rep. 2021;11:7064. doi:10.1038/s41598-021-85962-4
9. Hyder T, Bhattacharya S, Gade K, Nasrazadani A, Brufsky AM. Approaching neoadjuvant therapy in the management of early-stage breast cancer. Breast Cancer. 2021;13:199–211.
10. Cheng SW, Chen PC, Ger TR, Chiu HW, Lin YF. GBP5 serves as a potential marker to predict a favorable response in triple-negative breast cancer patients receiving a taxane-based chemotherapy. J Pers Med. 2021;11:197. doi:10.3390/jpm11030197
11. Blum JL, Flynn PJ, Yothers G, et al. Anthracyclines in early breast cancer: the ABC trials-USOR 06-090, NSABP B-46-I/USOR 07132, and NSABP B-49 (NRG oncology). J Clin Oncol. 2017;35:2647–2655. doi:10.1200/JCO.2016.71.4147
12. Nedeljković M, Damjanović A. Mechanisms of chemotherapy resistance in triple-negative breast cancer-how we can rise to the challenge. Cells. 2019;8:957. doi:10.3390/cells8090957
13. Guestini F, McNamara KM, Ishida T, Sasano H. Triple-negative breast cancer chemosensitivity and chemoresistance: current advances in biomarkers identification. Expert Opin Ther Targets. 2016;20:705–720. doi:10.1517/14728222.2016.1125469
14. O’Reilly EA, Gubbins L, Sharma S, et al. The fate of chemoresistance in triple-negative breast cancer (TNBC). BBA Clin. 2015;3:257–275. doi:10.1016/j.bbacli.2015.03.003
15. Xu X, Zhang L, He X, et al. TGF-β plays a vital role in triple-negative breast cancer (TNBC) drug-resistance through regulating stemness, EMT, and apoptosis. Biochem Biophys Res Commun. 2018;502:160–165. doi:10.1016/j.bbrc.2018.05.139
16. Wardhani BW, Puteri MU, Watanabe Y, Louisa M, Setiabudy R, Kato M. TGF-β-induced TMEPAI attenuates the response of triple-negative breast cancer cells to doxorubicin and paclitaxel. J Exp Pharmacol. 2020;12:17–26. doi:10.2147/JEP.S235233
17. Singha PK, Yeh IT, Venkatachalam MA, Saikumar P. Transforming growth factor-beta (TGF-beta)-inducible gene TMEPAI converts TGF-beta from a tumor suppressor to a tumor promoter in breast cancer. Cancer Res. 2010;70:6377–6383. doi:10.1158/0008-5472.CAN-10-1180
18. Nie Z, Wang C, Zhou Z, Chen C, Liu R, Wang D. Transforming growth factor-beta increases breast cancer stem cell population partially through upregulating PMEPA1 expression. Acta Biochim Biophys Sin. 2016;48:194–201. doi:10.1093/abbs/gmv130
19. Watanabe Y, Itoh S, Goto T, et al. TMEPAI, a transmembrane TGF-beta-inducible protein, sequesters Smad proteins from active participation in TGF-beta signaling. Mol Cell. 2010;37:123–134. doi:10.1016/j.molcel.2009.10.028
20. Itoh S, Itoh F. TMEPAI family: involvement in regulation of multiple signaling pathways. J Biochem. 2018;164:195–204. doi:10.1093/jb/mvy059
21. Singha PK, Pandeswara S, Geng H, Lan R, Venkatachalam MA, Saikumar P. TGF-β induced TMEPAI/PMEPA1 inhibits canonical Smad signaling through R-Smad sequestration and promotes non-canonical PI3K/Akt signaling by reducing PTEN in triple-negative breast cancer. Genes Cancer. 2014;5:320–336. doi:10.18632/genesandcancer.30
22. Paramita P, Wardhani BWK, Wanandi SI, Louisa M. Curcumin for the prevention of epithelial-mesenchymal transition in endoxifen-treated MCF-7 breast cancer cell. Asian Pac J Cancer Prev. 2018;19:1243–1249.
23. Wardhani BW, Puteri MU, Watanabe Y, Louisa M, Setiabudy R, Kato M. TMEPAI genome editing in triple-negative breast cancer cells. Med J Indones. 2017;26:14–18. doi:10.13181/mji.v26i1.1871
24. Wardhani BW, Puteri MU, Watanabe Y, Louisa M, Setiabudy R, Kato M. Knock-out transmembrane prostate androgen-induced protein gene suppressed triple-negative breast cancer cell proliferation. Med J Indones. 2017;26:178–182. doi:10.13181/mji.v26i3.1823
25. Dai L, Wang G, Pan W. Andrographolide inhibits proliferation and metastasis of SGC7901 gastric cancer cells. BioMed Res Int. 2017;2017:6242103. doi:10.1155/2017/6242103
26. von Maltzan K, Li Y, Rundhaug JE, Hudson LG, Fischer SM, Kusewitt DF. Role of the slug transcription factor in chemically-induced skin cancer. J Clin Med. 2016;5:21. doi:10.3390/jcm5020021
27. Dai X, Ahn KS, Wang LZ, et al. Ascochlorin enhances the sensitivity of doxorubicin leading to the reversal of epithelial-to-mesenchymal transition in hepatocellular carcinoma. Mol Cancer Ther. 2016;15:2966–2976. doi:10.1158/1535-7163.MCT-16-0391
28. Hertanto R, Bastian W, Paramita LM. The modulation of drug efflux transporter by curcumin in MCF 7 breast cancer cells after repeated exposure of endoxifen and estradiol. Int J Appl Pharmaceut. 2018;10:102–105. doi:10.22159/ijap.2018.v10s1.21
29. Puteri MU, Watanabe Y, Wardhani BW, Amalia R, Abdelaziz M, Kato M. PMEPA1/TMEPAI isoforms function via its PY and Smad‐interaction motifs for tumorigenic activities of breast cancer cells. Genes Cells. 2020;25:375–390. doi:10.1111/gtc.12766
30. Cichon MA, Radisky DC. Cutting the brakes and flooring the gas: how TMEPAI turns TGF-β into a tumor promoter. Genes Cancer. 2014;5:303. doi:10.18632/genesandcancer.34
31. Brunen D, Willems SM, Kellner U, Midgley R, Simon I, Bernards R. TGF-β: an emerging player in drug resistance. Cell Cycle. 2013;12:2960–2968. doi:10.4161/cc.26034
32. Oshimori N, Oristian D, Fuchs E. TGF-β promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell. 2015;160:963–976. doi:10.1016/j.cell.2015.01.043
33. Katayama H, Tsou P, Kobayashi M, et al. A plasma protein-derived TGFβ signature is a prognostic indicator in triple-negative breast cancer. NPJ Precis Oncol. 2019;3:1–8. doi:10.1038/s41698-019-0082-5
34. Ding MJ, Su K, Cui GZ, et al. Association between transforming growth factor-β1 expression and the clinical features of triple-negative breast cancer. Oncol Lett. 2016;11:4040–4044. doi:10.3892/ol.2016.4497
35. Wardhani BWK, Puteri MU, Watanabe Y, Louisa M, Setiabudy R, Kato M. Decreased sensitivity of several anticancer drugs in TMEPAI knock-out triple-negative breast cancer cells. Med J Indones. 2019;28:110–115. doi:10.13181/mji.v28i2.2687
36. Amalia R, Abdelaziz M, Puteri MU, et al. TMEPAI/PMEPA1 inhibits Wnt signaling by regulating β-catenin stability and nuclear accumulation in triple negative breast cancer cells. Cell Signal. 2019;59:24–33. doi:10.1016/j.cellsig.2019.03.016
37. Imamura Y, Mukohara T, Shimono Y, et al. Comparison of 2D-and 3D-culture models as drug-testing platforms in breast cancer. Oncol Rep. 2015;33:1837–1843. doi:10.3892/or.2015.3767
38. Yong KMA, Ulintz PJ, Caceres S, et al. Heterogeneity at the invasion front of triple-negative breast cancer cells. Sci Rep. 2020;10:1–9.
39. Zhu X, Chen L, Huang B, et al. The prognostic and predictive potential of Ki-67 in triple-negative breast cancer. Sci Rep. 2020;10:1–10.
40. Ziaei E, Saghaeidehkordi A, Dill C, Maslennikov I, Chen S, Kaur K. Targeting triple-negative breast cancer cells with novel cytotoxic peptide–doxorubicin conjugates. Bioconjugate Chem. 2019;30:3098–3106. doi:10.1021/acs.bioconjchem.9b00755
41. Hsieh T, Wu J. Novel insights on the use of doxorubicin to treat chemoresistant TNBC by Immunotherapy. Int J Immunother Cancer Res. 2020;6:016–018.
42. Wei L, Surma M, Gough G, et al. Dissecting the mechanisms of doxorubicin and oxidative stress-induced cytotoxicity: the involvement of actin cytoskeleton and ROCK1. PLoS One. 2015;10:e0131763. doi:10.1371/journal.pone.0131763
43. Sharifi S, Barar J, Hejazi MS, Samadi N. Doxorubicin changes Bax/Bcl-xL ratio, caspase-8 and 9 in breast cancer cells. Adv Pharmaceut Bull. 2015;5:351. doi:10.15171/apb.2015.049
44. Du Y, Liu Y, Xu Y, et al. The transmembrane protein TMEPAI induces myeloma cell apoptosis by promoting degradation of the c-Maf transcription factor. J Biol Chem. 2018;293:5847–5859. doi:10.1074/jbc.RA117.000972
45. Vo NT, Watanabe Y, Shiba A, Noguchi M, Itoh S, Kato M. TMEPAI/PMEPA1 enhances tumorigenic activities in lung cancer cells. Cancer Sci. 2014;105:334. doi:10.1111/cas.12355
46. Filyak Y, Filyak O, Souchelnytskyi S, Stoika R. Doxorubicin inhibits TGF-β signaling in human lung carcinoma A549 cells. Eur J Pharmacol. 2008;590:67–73. doi:10.1016/j.ejphar.2008.05.030
47. Luo S, Yang M, Lv D, et al. TMEPAI increases lysosome stability and promotes autophagy. Int J Biochem Cell Biol. 2016;76:98–106. doi:10.1016/j.biocel.2016.05.004
48. Zhang J, Tian XJ, Zhang H, et al. TGF-β–induced epithelial-to-mesenchymal transition proceeds through stepwise activation of multiple feedback loops. Sci Signal. 2014;7:ra91. doi:10.1126/scisignal.2005304
49. Wang Y, Liu J, Ying X, Lin PC, Zhou BP. Twist-mediated epithelial-mesenchymal transition promotes breast tumor cell invasion via inhibition of hippo pathway. Sci Rep. 2016;6:1–10.
50. Singha PK, Pandeswara S, Geng H, et al. Increased Smad3, and reduced Smad2 levels mediate the functional switch of TGF-β from growth suppressor to growth and metastasis promoter through TMEPAI/PMEPA1 in triple-negative breast cancer. Genes Cancer. 2019;10:134–149. doi:10.18632/genesandcancer.194
51. Wendt MK, Allington TM, Schiemann WP. Mechanisms of the epithelial-mesenchymal transition by TGF-β. Future Oncol. 2009;5:1145–1168. doi:10.2217/fon.09.90
52. Han RF, Ji X, Dong XG, et al. An epigenetic mechanism underlying doxorubicin-induced EMT in the human BGC-823 gastric cancer cell. Asian Pac J Cancer Prev. 2014;15:4271–4274. doi:10.7314/APJCP.2014.15.10.4271
53. Du B, Shim JS. Targeting epithelial-mesenchymal transition (EMT) to overcome drug resistance in cancer. Molecules. 2016;21:965. doi:10.3390/molecules21070965
54. Zhang L, Wang X, Lai C, Zhang H, Lai M. PMEPA1 induces EMT via non‐canonical TGF‐β signaling in colorectal cancer. J Cell Mol Med. 2019;23:3603–3615. doi:10.1111/jcmm.14261
55. Zheng HC. The molecular mechanisms of chemoresistance in cancers. Oncotarget. 2017;8:59950.
56. Takano M, Yamamoto C, Yamaguchi K, Kawami M, Yumoto R. Analysis of TGF-β1- and drug-induced epithelial-mesenchymal transition in cultured alveolar epithelial cell line RLE/Abca3. Drug Metab Pharmacokinet. 2015;30:111–118. doi:10.1016/j.dmpk.2014.10.007
57. Saxena M, Stephens MA, Pathak H, Rangarajan A. Transcription factors that mediate epithelial-mesenchymal transition lead to multidrug resistance by upregulating ABC transporters. Cell Death Dis. 2011;2:e179. doi:10.1038/cddis.2011.61
58. Jiang ZS, Sun YZ, Wang SM, Ruan JS. Epithelial-mesenchymal transition: a potential regulator of ABC transporters in tumor progression. J Cancer. 2017;8:2319–2327. doi:10.7150/jca.19079
59. Tsou SH, Chen TM, Hsiao HT, Chen YH. A critical dose of doxorubicin is required to alter the gene expression profiles in MCF-7 cells acquiring multidrug resistance. PLoS One. 2015;10:e0116747. doi:10.1371/journal.pone.0116747
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.