Back to Journals » Drug Design, Development and Therapy » Volume 15
Tetramethylpyrazine Improves Cognitive Function of Alzheimer’s Disease Mice by Regulating SSTR4 Ubiquitination
Authors Weng G, Zhou B, Liu T, Huang Z, Huang S
Received 2 November 2020
Accepted for publication 12 April 2021
Published 1 June 2021 Volume 2021:15 Pages 2385—2399
DOI https://doi.org/10.2147/DDDT.S290030
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Anastasios Lymperopoulos
Guohu Weng,1 Bo Zhou,2 Tao Liu,3 Zhengxin Huang,2 Shixiong Huang3
1Department of Encephalopathy, Hainan Provincial Hospital of Traditional Chinese Medicine, Haikou, 570203, Hainan, People’s Republic of China; 2Department of Cardiology, Hainan Provincial Hospital of Traditional Chinese Medicine, Haikou, 570203, Hainan, People’s Republic of China; 3Department of Neurology, Hainan General Hospital, Hainan Affiliated Hospital of Hainan Medical University, Haikou, 570311, Hainan, People’s Republic of China
Correspondence: Shixiong Huang
Department of Neurology, Hainan General Hospital, Hainan Affiliated Hospital of Hainan Medical University, No. 19 Xiuhua Road, Xiuying District, Haikou, 570311, Hainan, People’s Republic of China
Tel/Fax +86-0898-68642129
Email [email protected]
Purpose: Many researches have investigated the functions of tetramethylpyrazine (TMP) in Alzheimer’s disease (AD). This study aimed to discuss the underlying mechanism of TMP in AD mice.
Methods: TMP (200 mg/kg) was administered to 6-month-old APP/PS1 transgenic mice, and behavioral changes and hippocampal nerve injury in AD mice were detected. Apoptosis and autophagy-related protein levels were detected. Changes in gene expression before and after TMP treatment were compared using transcriptome sequencing. The effects of Cullin 4B (CUL4B) overexpression and somatostatin receptor 4 (SSTR4) silencing on AD symptoms and SSTR4 ubiquitination in APP/PS1 mice were observed. SH-SY5Y and PC12 cells were treated with 25 μmol/L Aβ25– 35 and TMP to observe cell viability, apoptosis, and autophagy. Cell viability and apoptosis were measured again after treatment with proteasome inhibitor MG132 or lysosomal inhibitor 3-mA.
Results: TMP treatment improved the behavioral cognition of APP/PS1 mice and improved the neuronal apoptosis and damage in brain tissue. CUL4B was significantly upregulated in APP/PS1 mouse brain tissue, and SSRT4 protein was downregulated, and the levels of CUL4B and SSRT4 were negatively correlated. TMP treatment downregulated CUL4B, inhibited SSRT4 ubiquitination and upregulated SSRT4 protein level in APP/PS1 mouse brain tissue, while CUL4B overexpression or SSRT4 silencing reversed the effect of TMP. TMP and MG132 improved the decreased activity, increased apoptosis and increased SSRT4 protein in SH-SY5Y and PC12 cells treated with Aβ25– 35, but not 3-mA. CUL4B overexpression promoted the ubiquitination of SSTR4 in cells, which partially reversed the effect of TMP.
Conclusion: TMP could improve the cognitive ability of AD mice by inhibiting CUL4B expression and the ubiquitination degradation of SSTR, and alleviating neuronal apoptosis and injury. This study may offer a new therapeutic option for AD treatment.
Keywords: Alzheimer disease, tetramethylpyrazine, somatostatin receptor 4, ubiquitination, Cullin 4B
Introduction
As is estimated, over 45 million people over the world are afflicted by dementia and the number of cases is predicted to triple by 2050, in which, Alzheimer’s disease (AD), a progressive neurodegenerative disease, accounts for 60–80% of cases.1 Patients suffering from AD may experience a true loss of semantic knowledge, prominent deficits in episodic and semantic memory, and deficits in executive functions, attention, visuospatial and cognition.2 The pathological features for AD diagnosis were neuronal and synapse loss, granulovacuolar degeneration, glial responses, amyloid plaques and intracellular neurofibrillary tangles.3 Additionally, vascular disease, smoking, hypertension, diabetes, hyperinsulinemia and neuroinflammation are confirmed to increase the risk of developing clinical AD.4 Mediterranean diet, regular aerobic exercise and recreational physical activities are proven to reduce the risk of developing AD and possibly to prevent overall cognitive decline.5 Over the past 20 years, advances have been made in therapies by directing at symptoms rather than at underlying pathology, which leaves the underlying mechanism of AD still remains unclear.6 Therein, intensive researches should be performed to make a thorough inquiry into the underlying mechanism of AD, thereby providing diagnostic biomarkers and better therapies for AD.
Tetramethylpyrazine (TMP) is a natural compound extracted from the Chinese herb medicine Ligusticum wallichii, which is first considered as a calcium antagonist7 and widely used in the treatment of cerebrovascular, cardiovascular diseases, and central nervous system degenerative diseases, such as AD, Parkinson’s disease and multiple sclerosis.8–10 The possible mechanism of TMP treatment may include inhibiting the metastasis of vascular smooth muscle cells, preventing platelet aggregation, eliminating reactive oxygen species, and modulating inflammation and apoptosis, by which TMP is beneficial for cardiac inotropic and vascular functions.9 TMP derivative T-006 can significantly reduce the loss of SNPC tyrosine hydroxylase (TH-) positive neurons and striatum dopaminergic nerve fibers in AD, and increase the concentration of dopamine and its metabolites in striatum.11 In addition, TMP could enhance mitochondrial biosynthesis, protect excitotoxicity, block calcium channels and prevent neuronal death induced by oxidative stress, which are all associated with AD pathogenesis.8
The neurotoxicity caused by amyloid-β (Aβ) aggregation is an important pathogenesis of AD. The evidence shows that somatostatin (SRIF) can regulate the activity of Aβ-degrading enzyme in the brain, thereby regulating the level of Aβ in the brain.12 Besides, somatostatin receptors (SSTRs) play roles in physiological processes, reproductive function or emotion, and are also associated with the pathogenesis of glioma, neurodegenerative diseases and changes after hemorrhagic stroke.13 Among 5 SSTRs, silencing of SSTR4 is reported to contribute to depression behaviors and increased susceptibility to stress in mice, indicating that SSTR4 inactivation is associated with chronic stress-induced behavioral and neuroendocrine changes.14 SSTR4 is mainly concentrated in the cortex and hippocampus, which are vital to learning and memory, and affected by Aβ accumulation in AD patients.15 Moreover, it has been reported that a traditional Chinese medicine can interact with SSTR4 to exert analgesic effect.16 Additionally, ubiquitination is crucial for cell survival and differentiation, as well as innate and adaptive immunity in cancers and neurodegenerative diseases, including AD.17 Yu et al reported that PSD-93, a postsynaptic scaffold protein, may reduce its ubiquitination and increase the expression of SSTR4 by binding with SSTR4, thereby reducing the content of Aβ and improving the cognitive impairment of AD model mice.18 It is suggested that the ubiquitination of SSTR4 is involved in the pathogenesis of AD. Whether TMP regulates the function of SSTR4 through ubiquitination in AD has not been reported. Thereby, we aimed to investigate the underlying mechanism of TMP and SSTR4 ubiquitination in AD, with the purpose to provide some novel therapies against AD.
Materials and Methods
Ethics Statement
This study was performed with the approval of the Clinical Ethical Committee of Hainan Provincial Hospital of Traditional Chinese Medicine. All animal experiments were conducted with the approval of the Institutional Animal Care and Use Committee. Great efforts were made to minimize the animals and their pains. The animal experiments in this study follow the 3R principle, which is to replace experimental animals, reduce the number of experimental animals, and refine experimental procedures to reduce the harm of animals.
Animal Model
APP/PS1 mice (6-month-old) were treated as previously described.19 All mouse strains were backcrossed extensively to the C57BL6 mice and maintained on this background.
Mice were allocated into sham group, APP/PS1 group, APP/PS1 + TMP group, APP/PS1 + TMP + Scramble group, APP/PS1 + TMP + sh-SSTR4 group, APP/PS1 + TMP + Empty group, and APP/PS1 + TMP + ovexpressed (OE)-CUL4B group, with 12 mice in each group. Sham group consisted of the untreated wild-type mice. The 6-month-old mice were anesthetized using isoflurane and fixed on the stereotactic instrument. After disinfection with iodophor, the skin was cut along the midline of the head. Stereotactic drilling was performed 2 mm in the posterior, 2.6 mm in the lateral and 2.3 mm in the ventral side of the skull. Using a microinjection system (World Precision Instruments, Sarasota, FL, USA), the CUL4B overexpressing lentivirus (Lv-CUL4B, 5×108 TU/mL) and SSTR4 inhibition lentivirus (Lv-sh-SSTR4, 5×108 TU/mL), or control lentivirus (Lv-con, 5×108 TU/mL) were injected into the lateral ventricle (1 μL; 0.125 μL/min). The needle was maintained for 10 min and then withdrawn. The skin was sutured and the mice were placed next to the heater for recovery. APP/PS1 mice were intraperitoneally injected with 20 mg/kg TMP (San Lian Pharm, Harbin, China) every day for 30 days.20 The lentivirus mediating overexpression of Cullin 4B (CUL4B) or inhibition of SSTR4 was provided by Shanghai Genechem Co., Ltd. (Shanghai, China).
Behavior tests were carried out 4 weeks after TMP treatment. T-shaped maze test was conducted on the 1st–7th day after TMP administration, including 2 days of adaptation, 4 days of training and 1 day of test; Morris water maze test was conducted on the 11th–18th day after TMP administration; contextual and cued fear (FC) conditioning was conducted on the 22nd-24th day.
After the last TMP injection for 30 days, mice were euthanized by an intraperitoneal injection of pentobarbital (800 mg/kg). Six mice in each group were randomly selected and perfused with 0.1M phosphate buffer saline (PBS; pH = 7.4) and then 4% paraformaldehyde into the heart. The brain was stereotaxically located in the skull, taken out and fixed in 4% paraformaldehyde at 4°C for 2 h. Next, the sample was frozen and sectioned. Sections were used for subsequent histological examination. The brain tissues of the remaining 6 mice were ground into homogenate for subsequent experiments.
Cognition Behavior Tests
We conducted T-shaped maze and Morris water maze (MWM) tests, as well as contextual and FC conditioning test to evaluate the motor ability, cognitive ability, hippocampus-dependent/independent memory ability of mice in each group (N=12).
The T-shaped maze consists of a start arm and two goal arms (30 cm in length, 10 cm in width and 20 cm in height). The spontaneous alternation of the T-maze is on the basis that mice are more likely to explore new arms than familiar ones. The mice to be tested were first placed in the start arm, and then chose to enter the goal arm on the left or on the right. Each mouse’s choice was recorded for calculating the percentage of alternations of 8 trials. The interval between each trial was 15 min. The latency time for each mouse to reach the goal arm was also recorded.21
The MWM test was performed in a circular tank (120 cm diameter) full of opaque water at 25°C. The tank was surrounded by dark walls with geometric patterns as visual clues. The hidden platform (at 7.5 diameters) was placed 0.5 cm below the water surface. On the day before the first test day, each mouse underwent 90-s free swimming in the tank. On the 1st day of the training, each mouse was placed in two different quadrants of the tank and needed to find the hidden platform for two trials, 120 s at most for each trial. Over the next 5 days, each mouse was placed in three different quadrants to find the hidden platform and 120 s at most was allowed. During the 6-day training, for those mice that could not find the platform within 120 s, they were physically guided and underwent a 20-s stay on the platform. On the 7th day, the platform was removed and the mice were subjected to a probe trial with 45 s for each mouse. Mouse performances in all tasks (escape latency and distance to target quadrant during training phase; average crossings on platform during probe trial) were recorded by a computer-based video tracking system.21
The training for contextual and cued FC included an exploration period of 3 min, followed by conditioned stimulus (85 dB white noise; 30 s) and unconditioned stimulus (0.6 mA footshock; 2 s), with an interval of 1 min. After 24 h of training, mice were subjected to contextual memory test in the training chamber and cued memory test in a distinct chamber.22
Tissue Experiments
After the cognition behavior tests, mice (N=6 in each group) were sacrificed for Nissl staining and terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labeling (TUNEL) staining using a Nissl staining kit (C5042, Sigma-Aldrich, Merck KGaA, Darmstadt, Germany)23 and a TUNEL staining kit (11684809910, Roche Ltd., Basel, Switzerland)24 as per the instructions.
The sections were stained according to the manufacturer’s instructions and observed under the light microscope after Nissl staining. After that, the sections were stained with TUNEL reaction buffer, followed by DAPI staining. Anti-fluorescence quenching agent was added to each brain section, and then the fluorescence microscope was used to photograph within 24 h. The results were analyzed.
Transcriptome Sequencing Analysis
RNA-Seq was sequenced in mouse brain tissue using Illumina high-throughput sequencing technology. Total RNA extraction, quality inspection and library construction were completed by Beijing Biomarker Biotechnology Co., Ltd. RNA sequencing was carried out using the Illumina Novaseq 6000 platform. After filtering the low-quality sequences, the total number of high-quality sequences was obtained. Gene alignment was carried out using Hisat 2.0 software to obtain the Fragment Per Kilobase of transcript per Million mapped reads (FPKM). R software was used to screen differential genes, and the standard was fold change > 1.5 and P < 0.05.
Reverse Transcription Quantitative Polymerase Chain Reaction (RT-qPCR)
Total RNA was extracted from brain tissue (N=6) or neurons using a TRIzol kit (Invitrogen Inc., Carlsbad, CA, USA)25 and reversely transcribed into cDNA (Takara, Dalian, China). qPCR was performed with the StepOne system (Applied Biosystems, USA) and a SYBR green kit (Takara, Dalian, China) based on the instructions. The value of each sample was normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The primer sequences are exhibited in Table 1.
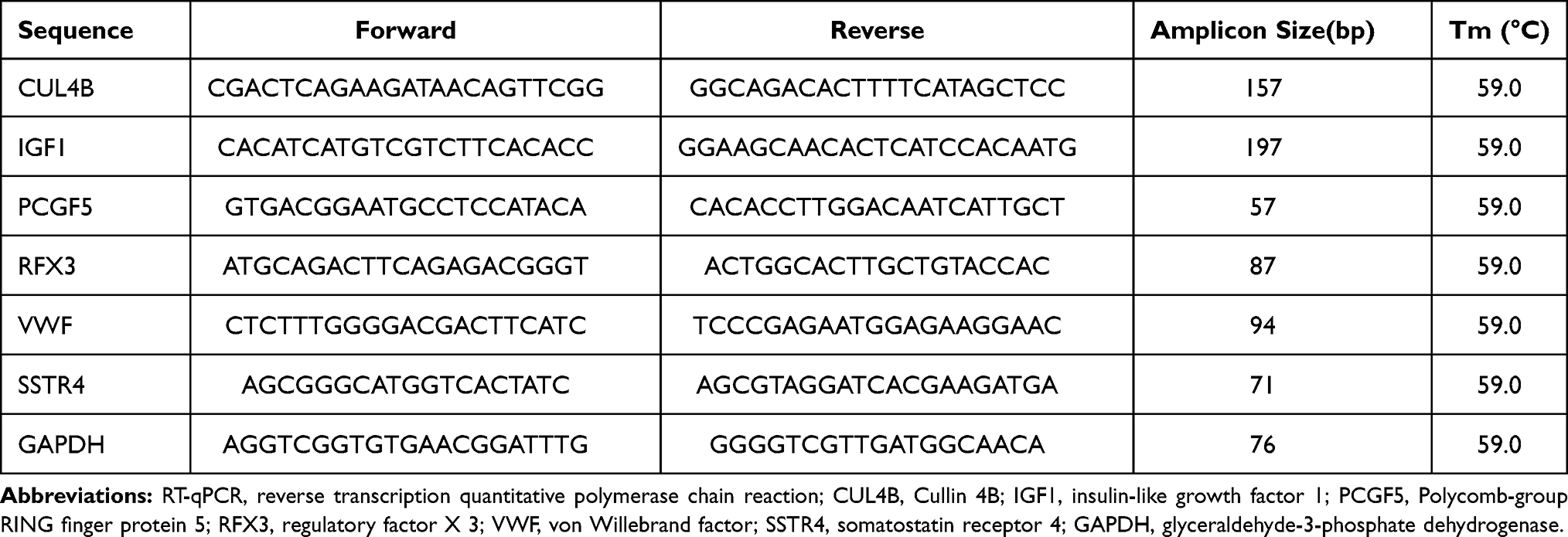 |
Table 1 Primer Sequences of RT-qPCR |
Western Blot Analysis
Western blot analysis was carried out as previously presented.26 The levels of each sample were normalized to β-actin. The antibodies used in Western blot analysis are shown in Table 2.
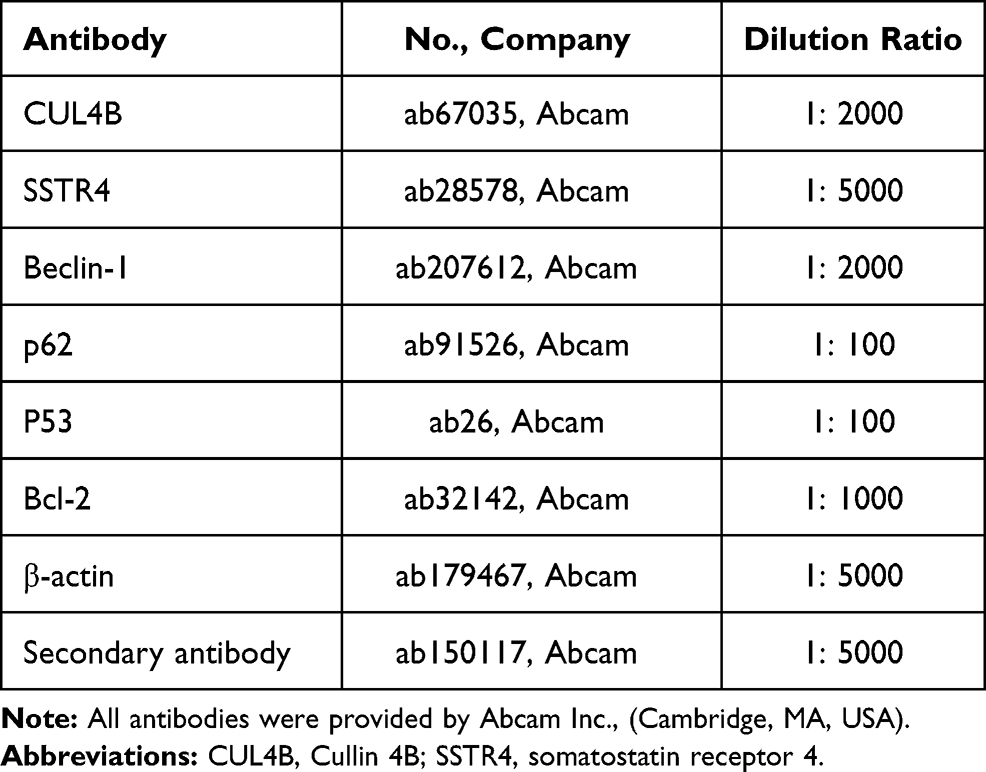 |
Table 2 Antibodies for Western Blot Analysis |
BCA protein assay kit (Thermo Scientific Pierce, Rockford, IL, USA) was used to determine the protein concentration in brain homogenate (N=6) or cells. The extracted protein was added with a loading buffer and boiled at 95°C for 10 min. Each protein sample (30 μg) was taken for sodium dodecyl sulfate-polyacrylamide gel electrophoresis (10% (w/v)) with electrophoresis voltage changed from 80 V to 120 V. Protein samples were loaded onto polyvinylidene fluoride membranes by wet transfer method at a voltage of 100 mV for 45–70 min. After that, the membranes were blocked with 5% bovine serum albumin at room temperature for 1 h, and incubated with primary antibodies at 4°C overnight. Following 3 washes with Tris-buffered saline-Tween 20 (TBST), the membranes were incubated with the secondary antibody goat anti-rabbit IgG (HRP, 1:3000, ab6721) at room temperature for 1 h. After being fully washed with TBST, the membranes were developed using enhanced chemiluminescence and visualized using Bio-Rad GelEZ imager (Bio-Rad Inc., Hercules, CA, USA). Image J software (National Institutes of health, Bethesda, Maryland, USA) was used to analyze the gray value of the target band. The primary antibodies were as follows: PUMA rabbit polyclonal antibody (2 μg/mL, ab9643, 23kDa), Bcl-2 rabbit monoclonal antibody (1:2000, ab182858, 26kDa), p62 rabbit monoclonal antibody (1:10000, ab109012, 62kDa), Beclin-1 rabbit monoclonal antibody (1:1000, ab210498, 52kDa), CUL4B rabbit polyclonal antibody (1:1000, ab227724, 104kDa), Ubiquitin rabbit monoclonal antibody (1:3000, ab134953, 8kDa), βactin rabbit monoclonal antibody (1:2000, ab8227, 42kDa), tubulin rabbit polyclonal antibody (1 µg/mL, ab18207, 55kDa), and SSTR4 rabbit polyclonal antibody (1:500, SAB4502863, 41kDa, Sigma).
Cell Culture
Human neuroblastoma cells SH-SY5Y and rat pheochromocytoma PC12 cells (the Chinese Academy Cell Resource Center, Shanghai, China) were cultured in RMPI-1640 supplemented with 10% fetal bovine serum at 37°C. When the cell confluence reached 60–70%, cells were treated with 25 μmol/L Aβ25–35 for 24 h.27 Control cells were cultured under normal conditions. Then, CUL4B overexpressing lentivirus (Lv-CUL4B, 5×108 TU/mL), SSTR4 inhibition lentivirus (Lv-sh-SSTR4, 5×108 TU/mL) or control lentivirus (Lv-con, 5×108 TU/mL) was transfected into SH-SY5Y and PC12 cells, respectively. RT-qPCR and Western blot analysis were performed for transfection validation.
3-(4, 5-Dimethylthiazol-2-Yl)-2, 5-Diphenyltetrazolium Bromide (MTT) Assay
Cell viability was measured using an MTT kit (11465007001, Roche)28 as per the manufacturer’s instructions.
The differently treated SH-SY5Y or PC12 cells were seeded into 96-well plates (5 × 103 cells/well). MTT assay was performed at 0, 24, 48, 72 and 96 h according to the instructions, and the optical density at 570 nm was measured.
Apoptosis Measurements
Apoptosis detection was determined by Hoechst 33258 staining and flow cytometry as previously described.29 The cells of different treatments were placed on 24-well plates, stained with 10 mg/mL Hoechst 33258 for 15 min, washed once with PBS, and observed and photographed under a fluorescence microscope. The cells in each group were plated in 6-well plates, washed twice with PBS, and stained with annexin V/fluorescein isothiocyanate (FITC) and propidium iodide (PI) (annexin V: FITC apoptosis detection kit, BD Pharmingen), and then analyzed by flow cytometer (FACS Calibur, Becton Dickinson).
Immunoprecipitation Assay
Immunoprecipitation assay was performed as previously mentioned to characterize the ubiquitination of SSTR4.30 The brain tissues (N=12) or cells were dissolved in an immunoprecipitation buffer supplemented with a protease inhibitor, and the supernatant was incubated with anti-SSTR4 or IgG antibody, followed by 3-h incubation with Protein A/G Agarose beads (Fast Flow, Shanghai Beyotime Biotechnology Co. Ltd., Shanghai, China) at 4°C. The agarose beads were washed with cold PBS and the binding protein was eluted. The ubiquitination of SSTR4 was analyzed by Western blotting with anti-Ub antibody.
Statistical Analysis
Groups were compared with Prism V8 software (GraphPad Software, San Diego, CA, USA) using a two-tailed unpaired Student’s t-test, or one-way analysis of variance (ANOVA) and Tukey’s multiple comparison test. Pearson correlation analysis was used to observe the correlation between the two continuous variables. The p < 0.05 inferred statistically significant differences. Data are presented as mean, or mean ± standard deviation. Refer to figure legends for the specific statistical tests used in each experiment. Sample sizes were determined based on previous experiments.
Results
TMP Treatment Attenuates AD Symptoms in APP/PS1 Mice
The effects of TMP on the behavioral cognitive ability of APP/PS1 mice were analyzed. T-shaped maze test was used to assess cognitive ability, MWM test was used to evaluate spatial memory ability, and FC test was used to evaluate the hippocampal memory ability. The T-shaped maze test results showed that the TMP-treated mice had a faster moving speed (Figure 1A) and a longer moving distance (both p < 0.05) (Figure 1B). MWM test showed that TMP treatment could reduce the search time of mice (Figure 1C), and the time spent in the target quadrant (Figure 1D) and the number of times entering the target quadrant (Figure 1E) of TMP-treated mice were higher than those of untreated mice when the hidden platform was removed on the 6th day (all p < 0.05). The results of FC test showed that more mice were frightened to a stagnant state (p < 0.05) (Figure 1F) after TMP treatment. These results suggest that TMP can improve the learning and memory function of mice and alleviate the cognitive impairment caused by AD.
 |
Figure 1 TMP treatment attenuated AD symptoms in APP/PS1 mice. APP/PS1 mice were treated with 20 mg/kg TMP or saline for 30 days. (A–F), for cognition behavior determination, T-shaped maze, MWM and FC tests were performed on each group of mice (N=12). After FC tests, mice were sacrificed and TUNEL staining (G) (N=6), Nissl staining (H) (N=6) and Western blot analysis (I) (N=6) were performed to determine apoptosis, viability and autophagy index. Each symbol represents an individual mouse. Data were analyzed with one-way ANOVA, and Tukey’s multiple comparison test. *p < 0.05, **p < 0.01, ***p < 0.001. |
After TMP treatment, TUNEL staining showed that the number of apoptotic neurons was decreased (p < 0.05) (Figure 1G), Nissl staining showed that the loss of neurons in hippocampal CA3 region was decreased (p < 0.05) (Figure 1H), and Western blot showed that the autophagy-related protein levels in APP/PS1 + TMP group were lower than that in the APP/PS1 group (all p < 0.05) (Figure 1I). These results suggest that TMP treatment can reduce the apoptosis, loss and autophagy of neurons in APP/PS1 mouse brain.
TMP Treatment Reduces the Ubiquitination of SSTR4 by Inhibiting CUL4B Expression
To further study the mechanism of TMP treatment in AD, we took the brain tissues of mice for transcriptome sequencing. The results showed that compared with APP/PS1 group, APP/PS1+TMP group has 331 upregulated genes and downregulated 240 genes (Figure 2A). Subsequently, 5 genes with the most significant downregulation were selected for RT-qPCR validation in mouse brain tissue. Compared with APP/PS1 group, the expression of CUL4B in APP/PS1 + TMP group was significantly downregulated (Figure 2B). Western blot analysis found that CUL4B protein level in mouse brain tissue was significantly increased, while TMP treatment inhibited CUL4B protein level (both p < 0.05) (Figure 2C). As Del Prete et al mentioned, CUL4B is a component of Cullin-RING ubiquitin ligase (CRL) and is involved in regulating the function of amyloid precursor protein and may be involved in a variety of neurodegenerative diseases.31 Additionally, a functional mutation in CUL4B gene was found to cause an X-linked mental retardation syndrome.32 Therefore, we speculate that TMP may play a role in the treatment of AD by regulating ubiquitin protease system. It has been reported that the ubiquitination level of SSTR4 in Aβ-treated cells was significantly increased.18 We speculated that TMP may play a role in the treatment of AD by affecting the ubiquitination of SSTR4. Subsequently, we detected SSTR4 mRNA expression in the brain tissues of APP/PS1 mice. No significant change was found (p < 0.05) (Figure 2D), while the protein level of SSTR4 decreased significantly. After TMP treatment, the protein level of SSTR4 increased noticeably (p < 0.05) (Figure 2E). Additionally, we detected the ubiquitination level of SSTR4 by immunoprecipitation assay and found that TMP treatment inhibited the ubiquitination of SSTR4 (p < 0.05) (Figure 2F). We compared the protein levels of CUL4B and STR4 in the brain tissues of APP/PS1 mice and found a negative correlation (p < 0.05) (Figure 2G).
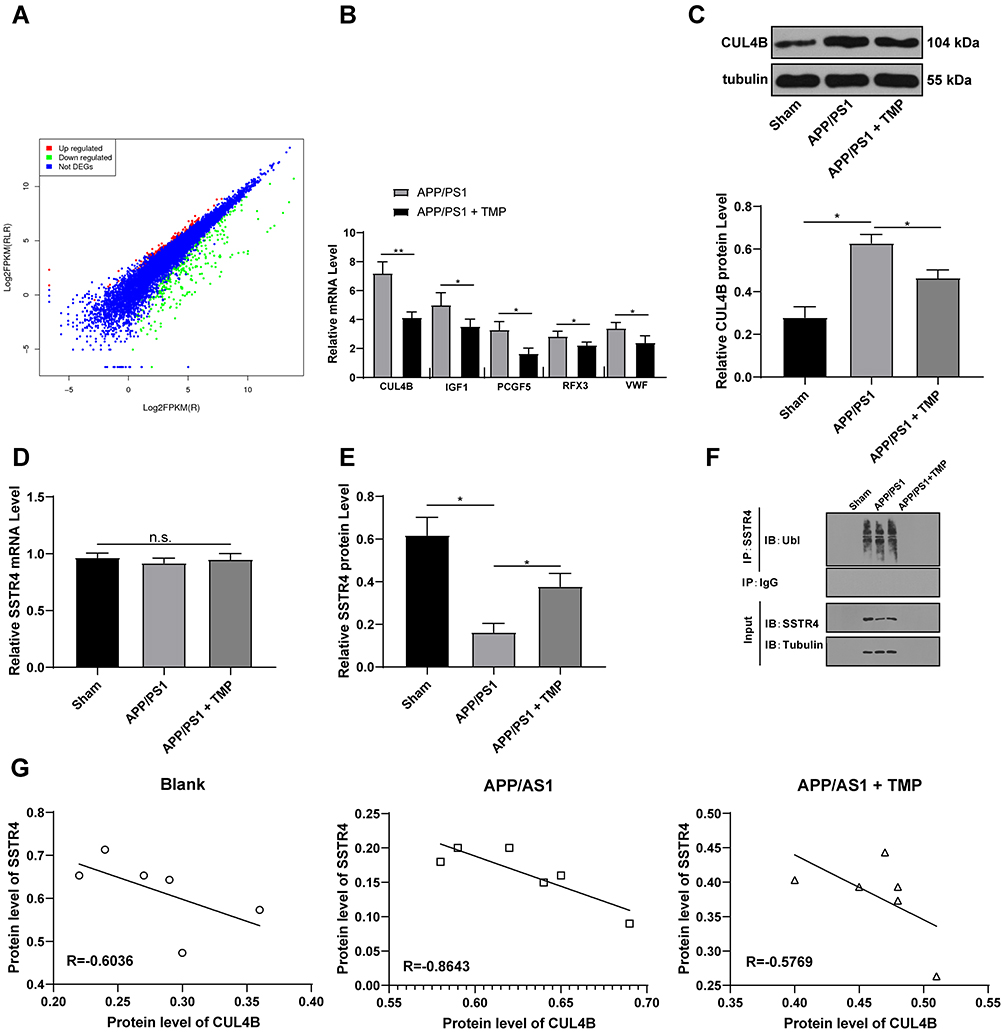 |
Figure 2 TMP treatment inhibits CUL4B expression. APP/PS1 mice were sacrificed and their brain tissues were extracted for transcriptome sequencing (A). RT-qPCR was performed to validate CUL4B, IGF1, PCGF5, RFX3, VWF and SSTR4 mRNA expression (B and D). Then, Western blot analysis was performed to determine CUL4B and SSTR4 protein levels (C and E). Representative images of immunoprecipitation assay of overexpression of CUL4B in SH-SY5Y and PC12 affected ubiquitination of SSTR4 (F). Correlation analysis of CUL4B and SSTR4 protein levels was performed using Western blot analysis (G). Each symbol represents an individual mouse. N=6. In panel B, data were analyzed with the t-test; in panels (C–E), one-way ANOVA and Tukey’s multiple comparison test were utilized for determining significance values. Data in panel (G) were analyzed with Pearson’s correlation analysis. *p < 0.05, **p < 0.01. |
Inhibition of SSTR4 or Overexpression of CUL4B Partially Reverses the Alleviation of AD Symptoms in APP/PS1 Mice After TMP Treatment
After injection of sh-SSTR4 or OE-CUL4B lentiviral vector to the lateral ventricles of mice, the levels of SSTR4 and CUL4B in the brain tissues of mice were verified by RT-qPCR and Western blot analysis, and the transfection was successful (all p < 0.05) (Figure 3A and B). Subsequently, 30 days after 20 mg/kg TMP treatment, we found that CUL4B overexpressing or SSTR4 silencing partially averted the improvement effect of TMP on the behavioral cognitive ability of APP/PS1 mice (all p < 0.05) (Figure 3C–H), and inhibited the viability of hippocampal neurons and promoted neuronal apoptosis and autophagy (all p < 0.05) (Figure 3I–K).
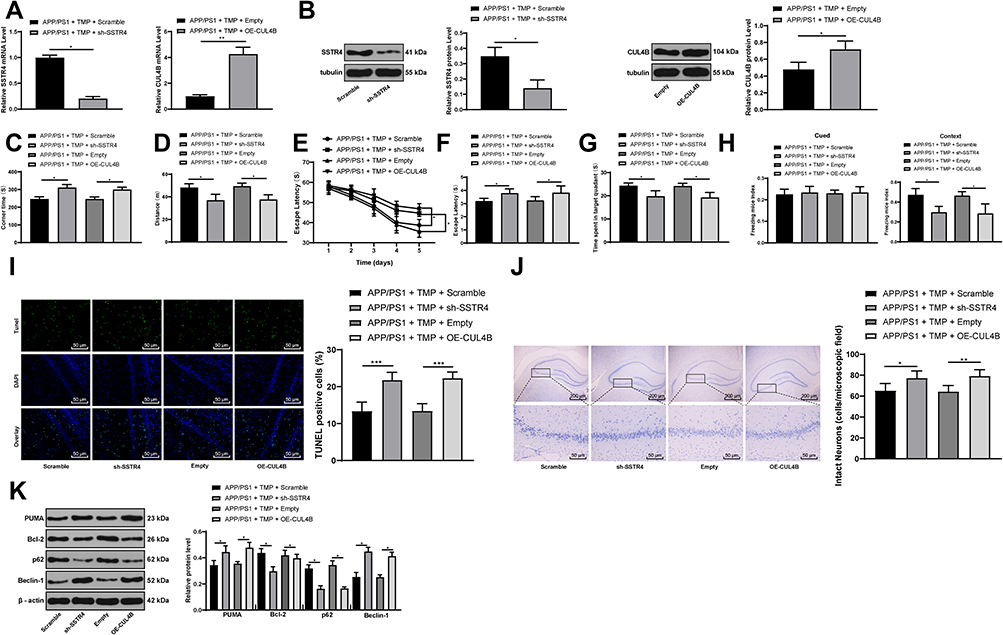 |
Figure 3 Inhibition of SSTR4 or overexpression of CUL4B attenuates TMP function in APP/PS1 mice. APP/PS1 mice were injected with sh-SSTR4 or expression vector containing CUL4B, and correspondingly injected with scramble shRNA or empty vector as control. RT-qPCR and Western blot analysis were performed to determine the mRNA expression (A) and protein levels (B) of SSTR4 and CUL4B (N=6). For cognition behavior determination, T-shaped maze, MWM and FC tests were performed on each group of mice (N=12) (C–H). After FC tests, mice were sacrificed and TUNEL staining (N=6) (I), Nissl staining (N=6) (J) and Western blot analysis (N=6) (K) of hippocampus were performed to determine apoptosis, viability and autophagy index. Each symbol represents an individual mouse. The unpaired t-test was used for data analysis. *p < 0.05, **p < 0.01, ***p < 0.001. |
TMP Treatment Could Partially Alleviate the Inhibition of SH-SY5Y and PC12 Cell Viability After Aβ25-35 Treatment
SH-SY5Y and PC12 cells were treated with 25 μmol/L Aβ25-35 for 24 h and then treated with 100 mg/L TMP. MTT kit was applied to detect cell viability and Hoechst 33258 staining and flow cytometry were applied to detect cell apoptosis. The results revealed that TMP treatment could alleviate the inhibition of cell viability and promotion of cell apoptosis induced by Aβ25-35 (all p < 0.05) (Figure 4A–C). Western blot analysis detected autophagy and apoptosis-related proteins, and found that TMP could inhibit cell apoptosis and autophagy (all p < 0.05) (Figure 4D).
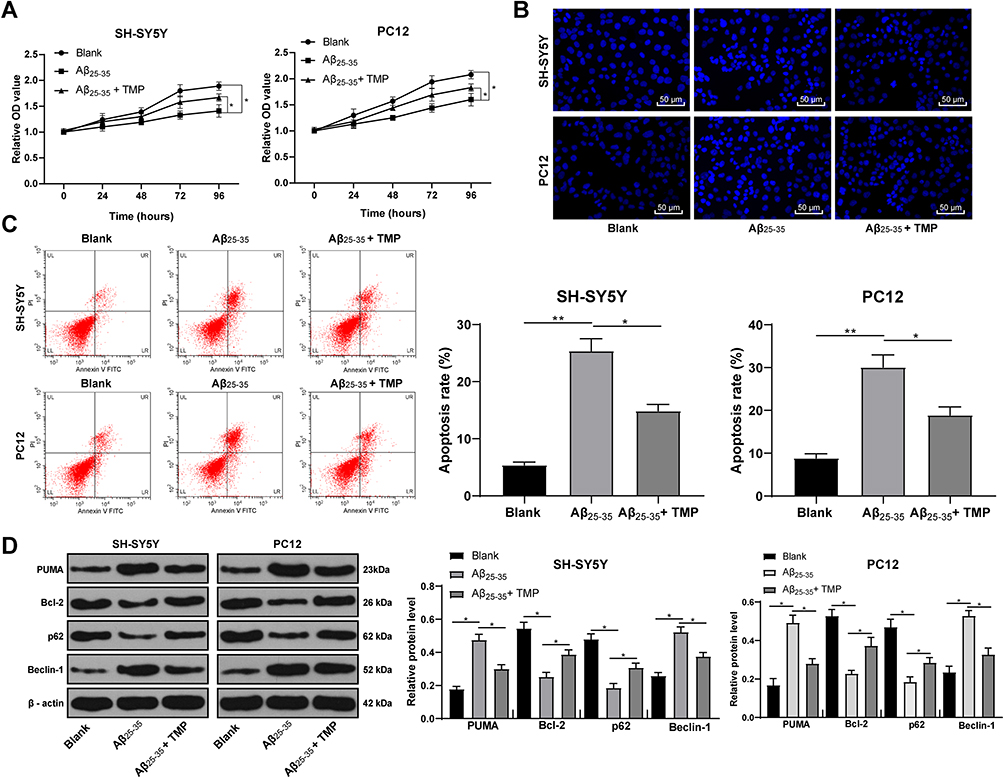 |
Figure 4 TMP treatment promoted Aβ25-35-treated SH-SY5Y and PC12 cell viability. SH-SY5Y and PC12 cells were treated with 25 μmol/L Aβ25-35 for 24 h and then 100 mg/L TMP. Cell viability (A) and apoptosis rate (B–C) were determined by MTT assay, Hoechst 33258 staining and flow cytometry, respectively. Western blot analysis was performed to determine apoptosis and autophagy-associated protein levels (D). Each symbol represents an individual experiment. Data were analyzed with one-way ANOVA and Tukey’s multiple comparison test. *p < 0.05, **p < 0.01. |
TMP Treatment Could Partially Inhibit the Ubiquitination of SSTR4 in SH-SY5Y and PC12 Cells Treated with Aβ25-35
Afterwards, we detected the levels of SSTR4 and CUL4B in cells by RT-qPCR and Western blot analysis. It was found that Aβ25-35 treatment resulted in the increase of CUL4B levels. The mRNA expression of SSTR4 did not change significantly, but SSTR4 protein level decreased remarkably (p < 0.05) (Figure 5A and B). Moreover, TMP treatment partially reversed these changes. Then, immunoprecipitation assay detected SSTR4 ubiquitination in cells and showed that the ubiquitination level of SSTR4 increased after Aβ25-35 treatment, but decreased after TMP treatment (p < 0.05) (Figure 5C).
 |
Figure 5 TMP treatment ameliorated SSTR4 ubiquitination by CUL4B via Aβ25-35 induction. RT-qPCR and Western blot analysis were performed to determine SSTR4 and CUL4B mRNA (A) and protein levels (B) in SH-SY5Y and PC12 cells. The ubiquitination of SSTR4 in SH-SY5Y and PC12 cells was detected by immunoprecipitation assay (C). Each symbol represents an individual experiment. In panel (A and B), one-way ANOVA and Tukey’s multiple comparison test was utilized for determining significance values. *p < 0.05. |
Overexpression of CUL4B Partially Reverses the Reduction of SSTR4 Ubiquitination After TMP Treatment
To further confirm the role of CUL4B in cell model, we constructed CUL4B overexpression vector and transfected it into SH-SY5Y and PC12 cells, and RT-qPCR and Western blot analysis verified the success of transfection (all p < 0.05) (Figure 6A and B). Subsequently, Western blot analysis found that the protein level of SSTR4 was further decreased after overexpression of CUL4B (p < 0.05) (Figure 6C), and the ubiquitination level was significantly increased (p < 0.05) (Figure 6D). MTT kit was utilized to detect cell viability and Hoechst 33258 staining and flow cytometry to detect cell apoptosis. Western blot analysis detected apoptosis and autophagy-related proteins. These results revealed that overexpression of CUL4B could partly reverse the effect of TMP treatment in vitro (all p < 0.05) (Figure 6E–H).
 |
Figure 6 CUL4B overexpression promoted cell apoptosis via SSTR4 ubiquitination. SH-SY5Y and PC12 cells were transfected with expression vector containing CUL4B and empty vector served as control. Then, cells were treated with 25 μmol/L Aβ25-35 for 24 h and then 100 mg/L TMP. RT-qPCR and Western blot analysis were performed to determine CUL4B and SSTR4 mRNA (A) and protein levels (B–C). (D) The ubiquitination of SSTR4 in SH-SY5Y and PC12 cells was detected by immunoprecipitation assay. Cell viability (E) and apoptosis (F–G) were determined by MTT assay, Hoechst 33258 staining and flow cytometry, respectively. Western blot analysis was performed to determine apoptosis or autophagy-associated protein levels (H). Each symbol represents an individual experiment. Data were analyzed with the t-test. *p < 0.05. |
MG132 Treatment Alleviates AD Symptoms in Cells
After adding the proteasome inhibitor MG132 or the lysosome inhibitor 3-MA, we found that the protein level of SSTR4 increased dramatically after MG132 treatment, but did not change significantly after 3-MA treatment (p > 0.05) (Figure 7A). These results suggested that SSTR4 degradation was ubiquitination-mediated. After adding MG132, AD symptoms in cells were partly relieved (all p < 0.05) (Figure 7B–E).
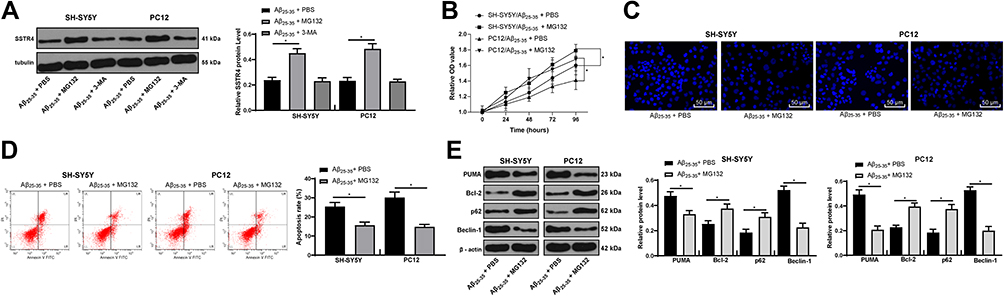 |
Figure 7 MG132 attenuates Aβ25-35-treated SH-SY5Y and PC12 cell cytotoxicity. SH-SY5Y and PC12 cells were treated with ubiquitination inhibitor MG132 or lysosome inhibitor 3-MA after 25 μmol/L Aβ25-35 treatment. SSTR4 protein level was determined by Western blot analysis (A). Cell viability (B) and apoptosis (C–D) were determined by MTT assay, Hoechst 33258 staining and flow cytometry, respectively. Western blot analysis was performed to determine apoptosis or autophagy-associated protein levels (E). Each symbol represents an individual experiment. In panel A were analyzed with one-way ANOVA and Tukey’s multiple comparison test; and the data in the rest panels were with the t-test. *p < 0.05. |
Discussion
After thousands of years of medical practice, traditional Chinese medicine has accumulated a wealth of theories and valuable experience in AD prevention and treatment33 with the advantages of relatively low toxicity, easy to cross the blood-brain barrier and a variety of synergistic effects.34 In this study, we mainly evaluated TMP, a Chinese medicine, in AD mice with the involvement of ubiquitination level of SSTR4. Collectively, we found that TMP could inhibit the ubiquitination of SSTR4 and then improve the cognitive ability of AD mice.
First, we found that TMP treatment attenuated AD symptoms in APP/PS1 mice, and reduced apoptosis and autophagy in APP/PS1 mice and in Aβ25-35-treated SH-SY5Y and PC12 cells, presenting with decreased levels of PUMA and Beclin-1, and increased levels of Bcl-2 and p62. TMP increased Bcl-2 expression, but decreased Bax expression in the spinal cord, thus playing an anti-apoptotic role in spinal cord ischemia/reperfusion injury.35 TMP also suppressed neuron apoptosis in a concentration-dependent manner in ischemic hippocampal neuron injury induced by oxygen-glucose deprivation,36 which was consistent with the neuroprotective effects of TMP in the present study. Besides, TMP could effectively reverse scopolamine-induced memory and learning disorders and maintain postsynaptic protein synthesis.37 A previous study suggested that autophagy and Beclin-1 are specifically of great significance in clearing Aβ aggregation and maintaining neuronal functions in AD.38 The induction of autophagy often resulted in p62 degradation, and reduced p62 levels represented enhanced autophagy activity in AD models.39 The potential efficacy of TMP in the treatment of AD was further supported by the observations of notably improved cognitive functions and brain amyloid pathology in dementia AD transgenic mice.40
In addition, our results identified that CUL4B protein level was increased in the brain tissues of APP/PS1 mice and then decreased after TMP treatment, while SSTR4 protein level showed an opposite trend. The unneddylated CUL4B isoforms accumulated during mitosis exist in the brain and are necessary for neural progenitor cells.41 In neurons, it was reported that the accumulation of ubiquitin proteins induced by CRBN, including CUL4B, might contribute to neurodegenerative diseases.31 Additionally, a functional mutation in CUL4B gene can cause an X-linked mental retardation syndrome.32 But there are few researches on the involvement of CUL4B in AD, which on the other hand indicates the innovation of the present study. CUL4B ubiquitin ligase accelerates ubiquitination and degradation of assorted proteins.42 In this study, we found that the protein level of SSTR4 was further decreased after overexpression of CUL4B, and the ubiquitination level increased significantly. The ubiquitination level of SSTR4 in Aβ-treated cells was significantly increased.18 In addition, neprilysin activity is upregulated by SST in primary cortical neurons,12 and in the hippocampus of APP/PS1 mice, SST expression is reduced, and a linear correlation is observed between Aβ content and neuropeptide Y or SST loss in AD.43 As expected, our results revealed that overexpressing CUL4B or silencing of SSTR4 significantly inhibited the behavioral cognitive ability of APP/PS1 mice, and inhibited the activity of hippocampal neurons and promoted neuronal apoptosis and autophagy in TMP-treated mice. There is compelling evidence of a close relationship between SST depletion and Aβ accumulation in AD.44
Furthermore, Aβ25-35 treatment resulted in increased CUL4B levels, decreased SSTR4 protein level, and enhanced ubiquitination level of SSTR4. TMP treatment could partially reverse these changes. In AD mice, the reduction of SSTR4 in primary cortical neurons was proved to be the result of ubiquitination.18 Aβ can trigger a cascade leading to synaptic damage and neuron loss, and ultimately to the pathological hallmarks of AD.3 SSTR4 agonist increased the activity of neprilysin and accordingly decreased the trimer Aβ1-42 in the cortex via enhancement of enzymatic degradation, and was thought to improve learning and memory abilities in AD mice, and blocked the AD progression.15,45
Moreover, the proteasome inhibitor MG132 dramatically increased the protein level of SSTR4, and relieved AD symptoms in cells. There is evidence that protein accumulation may be the result of dysfunction of ubiquitin-proteasome system, and clear genetic and biochemical evidence indicates that ubiquitin-proteasome system is involved in AD.46 Significant positive correlations were found between ubiquitin load and histone modifications using the proteasome inhibitor MG132 in AD.47 MG132 was also reported to block rapid ischemic tolerance, suggesting that ischemic protein degradation after preconditioning resulted in neuroprotective phenotype.48
Given the rising prevalence and mortality of AD coupled with the growing total healthcare costs, there continues to be a sense of urgency in the medical community to develop effective means for the early diagnosis and effective treatment of AD and other progressive neurodegenerative diseases with the application of traditional Chinese medicines.
Funding
There is no funding to report.
Disclosure
The authors declare no conflicts of interest.
References
1. Crous-Bou M, Minguillon C, Gramunt N, Molinuevo JL. Alzheimer’s disease prevention: from risk factors to early intervention. Alzheimers Res Ther. 2017;9(1):71. doi:10.1186/s13195-017-0297-z
2. Bondi MW, Edmonds EC, Salmon DP. Alzheimer’s disease: past, present, and future. J Int Neuropsychol Soc. 2017;23(9–10):818–831. doi:10.1017/S135561771700100X
3. Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011;1(1):a006189. doi:10.1101/cshperspect.a006189
4. Briggs R, Kennelly SP, O’Neill D. Drug treatments in Alzheimer’s disease. Clin Med. 2016;16(3):247–253. doi:10.7861/clinmedicine.16-3-247
5. Weller J, Budson A. Current understanding of Alzheimer’s disease diagnosis and treatment. F1000Res. 2018;7:1161. doi:10.12688/f1000research.14506.1
6. Gao LB, Yu XF, Chen Q, Zhou D. Alzheimer’s Disease therapeutics: current and future therapies. Minerva Med. 2016;107(2):108–113.
7. Pang PK, Shan JJ, Chiu KW. Tetramethylpyrazine, a calcium antagonist. Planta Med. 1996;62(5):431–435. doi:10.1055/s-2006-957933
8. Wang M, Qin HL, Leng J, et al. Synthesis and biological evaluation of new tetramethylpyrazine-based chalcone derivatives as potential anti-Alzheimer agents. Chem Biol Drug Des. 2018;92(5):1859–1866. doi:10.1111/cbdd.13355
9. Zhao Y, Liu Y, Chen K. Mechanisms and clinical application of tetramethylpyrazine (an interesting natural compound isolated from ligusticum wallichii): current status and perspective. Oxid Med Cell Longev. 2016;2016:2124638. doi:10.1155/2016/2124638
10. Cai X, Chen Z, Pan X, et al. Inhibition of angiogenesis, fibrosis and thrombosis by tetramethylpyrazine: mechanisms contributing to the SDF-1/CXCR4 axis. PLoS One. 2014;9(2):e88176. doi:10.1371/journal.pone.0088176
11. Zhou H, Shao M, Yang X, et al. Tetramethylpyrazine analogue T-006 exerts neuroprotective effects against 6-hydroxydopamine-induced Parkinson’s disease in vitro and in vivo. Oxid Med Cell Longev. 2019;2019:8169125. doi:10.1155/2019/8169125
12. Saito T, Iwata N, Tsubuki S, et al. Somatostatin regulates brain amyloid β peptide Aβ 42 through modulation of proteolytic degradation. Nat Med. 2005;11(4):434–439. doi:10.1038/nm1206
13. Kossut M, Lukomska A, Dobrzanski G, Liguz-Lecznar M. Somatostatin receptors in the brain. Postepy Biochem. 2018;64(3):213–221. doi:10.18388/pb.2018_133
14. Scheich B, Cseko K, Borbely E, et al. Higher susceptibility of somatostatin 4 receptor gene-deleted mice to chronic stress-induced behavioral and neuroendocrine alterations. Neuroscience. 2017;346:320–336. doi:10.1016/j.neuroscience.2017.01.039
15. Sandoval KE, Farr SA, Banks WA, Crider AM, Morley JE, Witt KA. Somatostatin receptor subtype-4 agonist NNC 26-9100 decreases extracellular and intracellular Aβ(1)(-)(4)(2) trimers. Eur J Pharmacol. 2012;683(1–3):116–124. doi:10.1016/j.ejphar.2012.03.020
16. Rowlands DK, Cui YG, So SC, Tsang LL, Chung YW, Chan HC. Bak Foong Pills induce an analgesic effect by inhibiting nociception via the somatostatin pathway in mice. Cell Biol Int. 2012;36(1):63–69. doi:10.1042/CBI20110015
17. Popovic D, Vucic D, Dikic I. Ubiquitination in disease pathogenesis and treatment. Nat Med. 2014;20(11):1242–1253. doi:10.1038/nm.3739
18. Yu L, Liu Y, Yang H, et al. PSD-93 attenuates amyloid-β-mediated cognitive dysfunction by promoting the catabolism of amyloid-β. J Alzheimers Dis. 2017;59(3):913–927. doi:10.3233/JAD-170320
19. Gimbel DA, Nygaard HB, Coffey EE, et al. Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J Neurosci. 2010;30(18):6367–6374. doi:10.1523/JNEUROSCI.0395-10.2010
20. Yang Q, Huang DD, Li DG, et al. Tetramethylpyrazine exerts a protective effect against injury from acute myocardial ischemia by regulating the PI3K/Akt/GSK-3beta signaling pathway. Cell Mol Biol Lett. 2019;24:17. doi:10.1186/s11658-019-0141-5
21. Peng Y, Liu J, Tang Y, et al. High-Fat-diet-induced weight gain ameliorates bone loss without exacerbating AβPP processing and cognition in female APP/PS1 mice. Front Cell Neurosci. 2014;8:225. doi:10.3389/fncel.2014.00225
22. Ma T, Du X, Pick JE, Sui G, Brownlee M, Klann E. Glucagon-like peptide-1 cleavage product GLP-1 (9-36)amide rescues synaptic plasticity and memory deficits in Alzheimer’s disease model mice. J Neurosci. 2012;32(40):13701–13708. doi:10.1523/JNEUROSCI.2107-12.2012
23. Desgent S, Duss S, Sanon NT, et al. Early-life stress is associated with gender-based vulnerability to epileptogenesis in rat pups. PLoS One. 2012;7(8):e42622. doi:10.1371/journal.pone.0042622
24. Zhang QQ, Ding Y, Lei Y, et al. Andrographolide suppress tumor growth by inhibiting TLR4/NF-kappaB signaling activation in insulinoma. Int J Biol Sci. 2014;10(4):404–414. doi:10.7150/ijbs.7723
25. Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82(4):239–259. doi:10.1007/BF00308809
26. Tsokas P, Ma T, Iyengar R, Landau EM, Blitzer RD. Mitogen-activated protein kinase upregulates the dendritic translation machinery in long-term potentiation by controlling the mammalian target of rapamycin pathway. J Neurosci. 2007;27(22):5885–5894. doi:10.1523/JNEUROSCI.4548-06.2007
27. You F, Li Q, Jin G, Zheng Y, Chen J, Yang H. Genistein protects against Abeta25-35 induced apoptosis of PC12 cells through JNK signaling and modulation of Bcl-2 family messengers. BMC Neurosci. 2017;18(1):12. doi:10.1186/s12868-016-0329-9
28. Ta MH, Liuwantara D, Rangan GK. Effects of pyrrolidine dithiocarbamate on proliferation and nuclear factor-kappaB activity in autosomal dominant polycystic kidney disease cells. BMC Nephrol. 2015;16:212. doi:10.1186/s12882-015-0193-3
29. Zheng X, Xie Z, Zhu Z, et al. Methyllycaconitine alleviates amyloid-beta peptides-induced cytotoxicity in SH-SY5Y cells. PLoS One. 2014;9(10):e111536. doi:10.1371/journal.pone.0111536
30. Zhang M, Li Q, Chen L, et al. PSD-93 deletion inhibits Fyn-mediated phosphorylation of NR2B and protects against focal cerebral ischemia. Neurobiol Dis. 2014;68:104–111. doi:10.1016/j.nbd.2014.04.010
31. Del Prete D, Rice RC, Rajadhyaksha AM, D’Adamio L. Amyloid precursor protein (APP) may act as a substrate and a recognition unit for CRL4CRBN and Stub1 E3 ligases facilitating ubiquitination of proteins involved in presynaptic functions and neurodegeneration. J Biol Chem. 2016;291(33):17209–17227. doi:10.1074/jbc.M116.733626
32. Zou Y, Liu Q, Chen B, et al. Mutation in CUL4B, which encodes a member of cullin-RING ubiquitin ligase complex, causes X-linked mental retardation. Am J Hum Genet. 2007;80(3):561–566. doi:10.1086/512489
33. Wu TY, Chen CP, Jinn TR. Traditional Chinese medicines and Alzheimer’s disease. Taiwan J Obstet Gynecol. 2011;50(2):131–135. doi:10.1016/j.tjog.2011.04.004
34. Liu P, Kong M, Yuan S, Liu J, Wang P. History and experience: a survey of traditional Chinese medicine treatment for Alzheimer’s disease. Evid Based Complement Alternat Med. 2014;2014:642128. doi:10.1155/2014/642128
35. Fan LH, Wang KZ, Cheng B, Wang CS, Dang XQ. Anti-apoptotic and neuroprotective effects of tetramethylpyrazine following spinal cord ischemia in rabbits. BMC Neurosci. 2006;7:48. doi:10.1186/1471-2202-7-48
36. Gong G, Yuan L, Cai L, et al. Tetramethylpyrazine suppresses transient oxygen-glucose deprivation-induced connexin32 expression and cell apoptosis via the ERK1/2 and p38 MAPK pathway in cultured hippocampal neurons. PLoS One. 2014;9(9):e105944. doi:10.1371/journal.pone.0105944
37. Wu W, Yu X, Luo XP, Yang SH, Zheng D. Tetramethylpyrazine protects against scopolamine-induced memory impairments in rats by reversing the cAMP/PKA/CREB pathway. Behav Brain Res. 2013;253:212–216. doi:10.1016/j.bbr.2013.07.052
38. Rohn TT, Wirawan E, Brown RJ, Harris JR, Masliah E, Vandenabeele P. Depletion of Beclin-1 due to proteolytic cleavage by caspases in the Alzheimer’s disease brain. Neurobiol Dis. 2011;43(1):68–78. doi:10.1016/j.nbd.2010.11.003
39. Tian M, Lin X, Wu L, Lu J, Zhang Y, Shi J. Angiotensin II triggers autophagy and apoptosis in PC12 cell line: an in vitro Alzheimer’s disease model. Brain Res. 2019;1718:46–52. doi:10.1016/j.brainres.2019.05.002
40. Tan Z. Erratum: neural protection by naturopathic compounds-an example of tetramethylpyrazine from retina to brain. J Ocul Biol Dis Infor. 2009;2(3):137–144. doi:10.1007/s12177-009-9033-7
41. Liu HC, Enikolopov G, Chen Y. Cul4B regulates neural progenitor cell growth. BMC Neurosci. 2012;13:112. doi:10.1186/1471-2202-13-112
42. Wang HL, Chang NC, Weng YH, Yeh TH. XLID CUL4B mutants are defective in promoting TSC2 degradation and positively regulating mTOR signaling in neocortical neurons. Biochim Biophys Acta. 2013;1832(4):585–593. doi:10.1016/j.bbadis.2013.01.010
43. Ramos B, Baglietto-Vargas D, Del Rio JC, et al. Early neuropathology of somatostatin/NPY GABAergic cells in the hippocampus of a PS1xAPP transgenic model of Alzheimer’s disease. Neurobiol Aging. 2006;27(11):1658–1672. doi:10.1016/j.neurobiolaging.2005.09.022
44. Grosser C, Neumann L, Horsthemke B, Zeschnigk M, van de Nes J. Methylation analysis of SST and SSTR4 promoters in the neocortex of Alzheimer’s disease patients. Neurosci Lett. 2014;566:241–246. doi:10.1016/j.neulet.2014.02.046
45. Sandoval KE, Farr SA, Banks WA, Crider AM, Morley JE, Witt KA. Somatostatin receptor subtype-4 agonist NNC 26-9100 mitigates the effect of soluble Abeta(42) oligomers via a metalloproteinase-dependent mechanism. Brain Res. 2013;1520:145–156. doi:10.1016/j.brainres.2013.05.006
46. Oddo S. The ubiquitin-proteasome system in Alzheimer’s disease. J Cell Mol Med. 2008;12(2):363–373. doi:10.1111/j.1582-4934.2008.00276.x
47. Narayan PJ, Lill C, Faull R, Curtis MA, Dragunow M. Increased acetyl and total histone levels in post-mortem Alzheimer’s disease brain. Neurobiol Dis. 2015;74:281–294. doi:10.1016/j.nbd.2014.11.023
48. Meller R. The role of the ubiquitin proteasome system in ischemia and ischemic tolerance. Neuroscientist. 2009;15(3):243–260. doi:10.1177/1073858408327809
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.