Back to Journals » OncoTargets and Therapy » Volume 11
TERT copy gain predicts the outcome of high-dose interferon α-2b therapy in acral melanoma
Authors Yu S, Xu T, Dai J, Ma M, Tang H, Chi Z, Si L, Cui C, Sheng X, Kong Y, Guo J
Received 27 November 2017
Accepted for publication 6 April 2018
Published 17 July 2018 Volume 2018:11 Pages 4097—4104
DOI https://doi.org/10.2147/OTT.S158239
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr XuYu Yang
Sifan Yu,* Tianxiao Xu,* Jie Dai, Meng Ma, Huan Tang, Zhihong Chi, Lu Si, Chuanliang Cui, Xinan Sheng, Yan Kong, Jun Guo
Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education/Beijing), Department of Renal Cancer and Melanoma, Peking University Cancer Hospital & Institute, Beijing 100142, China
*These authors contributed equally to this work
Background: Asian populations are more likely to develop acral melanoma (AM) than Caucasians. The high-dose interferon (HD-IFN) α-2b regimen is the main adjuvant treatment for AM. TERT encodes the catalytic subunit of telomerase reverse transcriptase, which plays an important role in melanoma. Frequent TERT mutation and increased TERT gene expression have been described in AM. Our study aimed to investigate the status and the clinical significance of TERT copy number in a large cohort of patients with AM and to analyze the relationship between TERT copy number gain and the efficiency of HD-IFN.
Patients and methods: A total of 573 melanoma samples were retrospectively collected and analyzed for TERT copy number via Sanger sequencing. Clinical data of patients were also collected.
Results: TERT copy gain (copy number >2) was detected in 257 of the 573 patients with AM (44.9%). Of the 573 patients, 81 (14.1%) had a high copy gain (copy number >4). Patients with ulceration showed a significantly higher copy gain rate of TERT compared to the patients without ulceration (P=0.028). Patients with a tumor thicker than 4 mm also had a higher copy number rate of TERT than those with <4 mm (P=0.048). Our results showed that the overall survival (OS) was not significantly different between patients with and without TERT copy gain (P=0.890). However, among the 278 patients who received an HD-IFN regimen, Kaplan–Meier survival analysis demonstrated a significant correlation between TERT copy gain and relapse-free survival (RFS) (P=0.008). In addition, multivariate Cox regression assays validated TERT copy gain to be an independent prognostic factor of RFS for patients with AM undergoing HD-IFN therapy (hazard ratio =1.50; P=0.019).
Conclusion: The copy number status of TERT might be a predictor for HD-IFN efficacy, but it is not a prognostic factor of OS in patients with AM.
Keywords: acral melanoma, TERT, gene copy number, interferon α-2b, relapse-free survival
Introduction
Acral melanoma (AM) is the most predominant melanoma subtype among non-Caucasians,1–3 particularly among Chinese, accounting for almost 50% of all cases of melanomas.4 Compared with common cutaneous melanoma, AM has a poorer prognosis2,4,5 and shows a markedly different genomic landscape, with a far lower mutation burden dominated by larger-scale genomic rearrangements.5–7 Vemurafenib and imatinib are more effective against advanced AM harboring BRAF and C-KIT mutations than traditional chemotherapies.8–11 However, the mutation frequencies of BRAF and C-KIT in AM are only approximately 16%12,13 and 12%,13–15 respectively; therefore, a validated targeted therapy is unavailable for the majority of patients. Meanwhile, adjuvant HD-IFN is a year-long treatment modality associated with improved relapse-free survival (RFS)16 and is currently the primary treatment regimen for stage II/III AM after surgery.17 Despite the optimization of this adjuvant treatment, most patients will still develop distant metastases and eventually die. Moreover, biomarkers for identifying patients who would derive significant prognostic benefit from HD-IFN treatment have yet to be determined. To sum up, developing more effective therapeutic strategies and validating new candidate biomarkers for predicting treatment response are urgently needed.
Human telomerase contains two essential components, a functional human telomerase RNA (hTR, also named TERC) and a human telomerase reverse transcriptase (hTERT).18 TERT is located at 5p15.33, which regulates telomeric length. TERT upregulation plays a critical role in oncogenesis.19 TERT promoter mutations have been reported in up to 50% of all cases of cutaneous melanoma,20,21 but only in 0%–7% of AM.22–24 In addition, gene amplification is the most frequent mechanism for TERT activation and significantly reduced overall survival (OS) in AM.25 TERT gene amplification has been evaluated via fluorescence in situ hybridization in a series of AM patients, and such amplification was detected in 6 of 34 cases (17.6%).26 Another previous study detected TERT gene amplification in 5 of 16 (31.2%) patients with AM.27 However, a small sample size was one of the limitations of these studies.
In the present study, we examined TERT copy gain in 573 melanoma samples. To the best of our knowledge, this is by far the largest study of patients with AM focusing on the copy gain of TERT, and we determined the association between TERT copy gain and the clinicopathological features of AM. Our study demonstrated prognostic value of TERT copy gain for different patient subgroups. These findings may also help to discover new potential molecular indicators for patients who may benefit from HD-IFN treatment.
Patients and methods
Study population
A total of 573 patients with AM who were treated at Peking University Cancer Hospital & Institute between 2008 and 2017 were included in this study (Table 1). These samples were evaluated to confirm the diagnosis of melanoma via H&E staining and immunohistochemistry for melanoma markers (S-100, HMB-45, or MART-1). Clinical data, including age, sex, tumor-node-metastases (TNM) stage, tumor thickness (Breslow), ulceration, and survival period (follow-up persisted until patient was lost to follow-up or death), were collected. Patients who received radiation treatment were excluded from our study. This investigation was performed after approval by the Ethics Committee of Peking University and was conducted according to the Declaration of Helsinki Principles. Written informed consent was obtained from each patient.
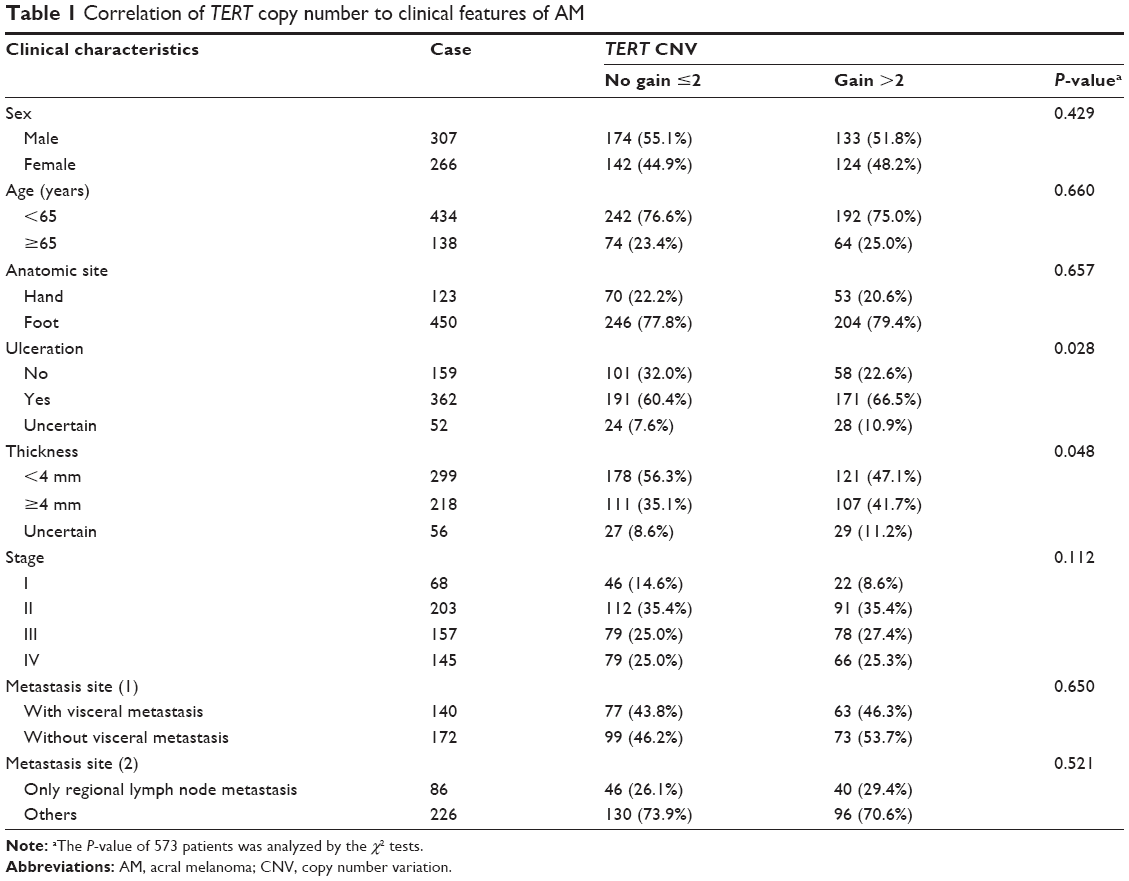 | Table 1 Correlation of TERT copy number to clinical features of AM |
Quantification of DNA using QuantiGene Plex 2.0 assay in FFPE tissues
Genomic DNA was extracted from formalin-fixed, paraffin-embedded (FFPE) specimens using the QIAamp DNA FFPE Tissue Kit (Qiagen, Hilden, Germany). The QuantiGene Plex 2.0 reagent system (Thermo Fisher Scientific, Waltham, MA, USA) was used to detect the copy number of TERT according to the manufacturer’s recommended protocols. Briefly, each patient’s DNA sample was mixed with an oligonucleotide probe. The mixture was then added to a 96-well plate, and the reagent system captured the target DNA and control DNA. Afterward, working samples were incubated overnight at 54°C. After washing unbound material with 200 μL of wash buffer, sequential hybridization of DNA amplifier molecules, preamplifier hybridization, amplifier hybridization, and label probe hybridization were then performed. The plate was then prepared for analysis by a professional analyzer. After dividing by the sample value, the gene copy numbers of the patients were calculated as follows: a copy number of 2 or less was considered no gain, while a copy number >2 was considered a copy number gain of TERT. In these amplified populations (copy number >2), we further classified the copy numbers as median gain (>2 but <4) or high copy number gain (4 or greater).
Statistical analysis
All statistical analyses were performed using SPSS 21.0 software (IBM Corporation, Armonk, NY, USA). OS was calculated and plotted using the Kaplan–Meier method. Differences between groups were compared via log-rank test. A 2-sided χ2 test was used to examine the correlations between gains of TERT gene and clinicopathological parameters. The Cox proportional hazard regression model was used for multivariate analysis.
Results
Copy gain of TERT in patients with acral melanoma
We used pretreatment biopsy tissue samples from 573 patients with AM. The tumor specimens were derived from primary lesions. All the tumor sections were collected at the time of diagnosis before initiating the treatments. The patients’ characteristics are shown in Table 1.
To study the copy number variation of TERT, we performed QuantiGene Plex 2.0 Assays (Thermo Fisher Scientific) for AM specimens. We found that 257 of the 573 patients (44.9%) displayed TERT copy gain (ie, a gain >2-fold change, Figure 1A). Of them, 81 patients (31.5%) had a high copy number (ie, a gain >4-fold change) (Figure 1B). Patients with ulceration showed a significantly higher copy gain rate of TERT than those without ulceration (P=0.028). Patients with tumors thicker than 4 mm also had a higher copy gain rate of TERT than those with tumors <4 mm thick (P=0.048). No relationship was noted between TERT copy gain and sex, age, clinical stage, or metastasis site (Table 1).
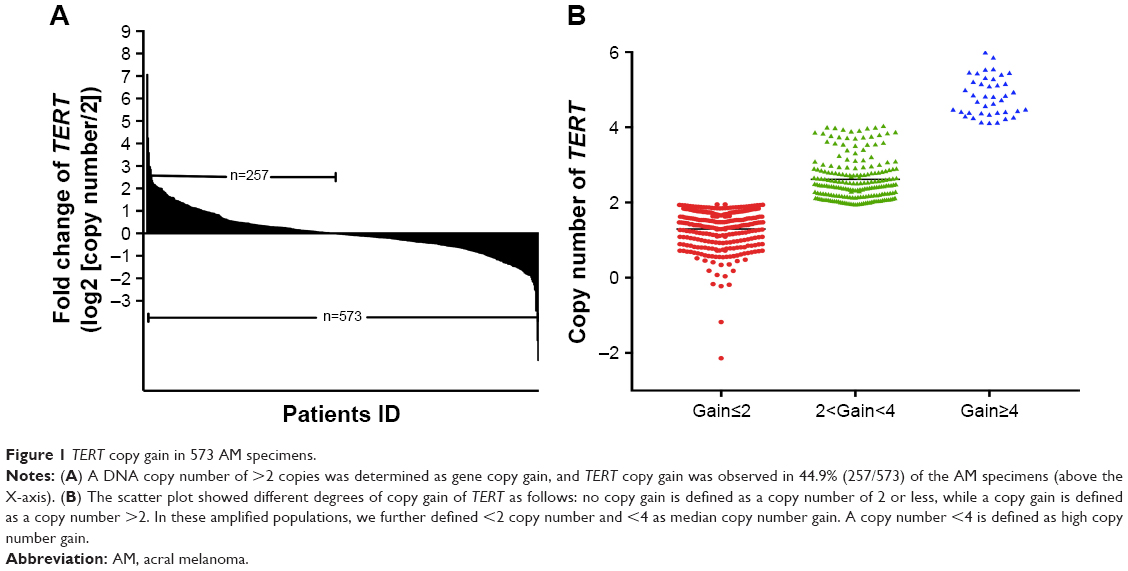 | Figure 1 TERT copy gain in 573 AM specimens. |
Relationship between TERT copy gain and representative gene of melanoma
The rates of BRAF wild-type and mutant genes among the 257 patients with TERT copy gain were 88.6% and 11.4%, respectively, while they were 84.4% and 15.6%, respectively, among patients with no TERT copy gain. No correlation between BRAF mutation and TERT copy gain was observed (P=0.143). In addition, we also analyzed the relationship between 3 other common mutations (KIT, PDGFRA, and NRAS) and TERT copy gain in 573 patients with AM. No correlation was found between TERT copy gain and common gene mutation (Table 2).
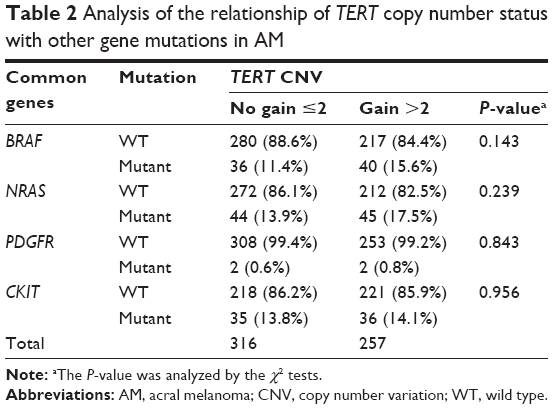 | Table 2 Analysis of the relationship of TERT copy number status with other gene mutations in AM |
Influence of TERT copy gain on OS of patients
Among the 573 patients with AM, 278 (70.4%) received HD-IFN regimen. Meanwhile, 117 patients underwent different treatment modalities, namely, dacarbazine (PUDEPHARM, Shanxi Datong, China) (n=62 patients), temozolomide (Temodal, Orion Corporation Orion Pharma, Espoo, Finland) (n=14 patients), and other chemotherapeutic drugs or targeted therapy (n=41 patients). Out of the 41 patients, 11 and 1 were treated with imatinib (Glivec, Novartis, Beijing, China) and vemurafenib (Zelbraf, Roche, Basel, Switzerland), respectively.
To evaluate the prognostic value of TERT for predicting the OS of patients, Kaplan–Meier survival analysis was performed for all the 573 patients. The median OS of patients with and without TERT gain was 73.7 months (95% CI: 69.9–78.5 months) and 62.7 months (95% CI: 59.5–67.9 months), respectively. The no TERT gain group did not show improved OS relative to the whole population (P=0.890, Figure 2A). Among the 278 patients treated with IFN α-2b, those with no TERT gain did not demonstrate better OS than those with TERT copy gain (P=0.389). Among the patients treated with IFN, we analyzed whether TERT copy gain affected survival in those with stage II and stage III AM. Our results showed that OS was not significantly different between patients with and without TERT gain (n=221, P=0.228, Figure 2B).
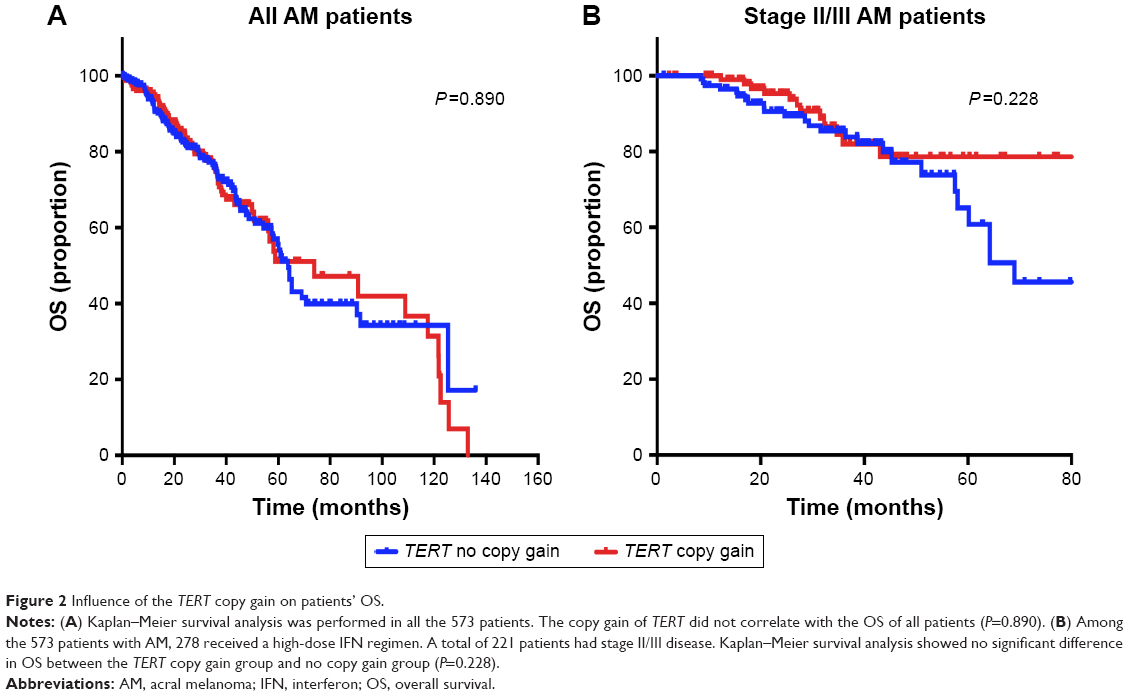 | Figure 2 Influence of the TERT copy gain on patients’ OS. |
Influence of TERT copy gain on patient response to high-dose interferon treatment and RFS
IFN α-2b is the most commonly utilized adjuvant regimen for patients with AM in China. Thus, we analyzed whether TERT copy gain can affect the efficacy of IFN therapy. The median RFS of the patients with and without TERT copy gain was 18.2 months (95% CI: 10.6–25.3 months) and 29.1 months (95% CI: 21.7–36.2 months), respectively. We used Kaplan–Meier survival analysis to evaluate the predictive value of TERT and RFS for postoperative adjuvant therapy, and the results demonstrated significant correlation between TERT copy gain and RFS in cases treated with IFN α-2b. TERT copy gain significantly predicted worse RFS (P=0.008) (Figure 3A). Then, we further analyzed the efficacy of IFN therapy in patients with stage II and stage III disease. Among these patients, the median RFS was significantly different between patients with and without TERT gain (n=221, P=0.034, Figure 3B). Collectively, these results indicate that TERT copy gain in patients with AM can predict worse RFS among patients treated with HD-IFN, particularly for those with stage II and III disease.
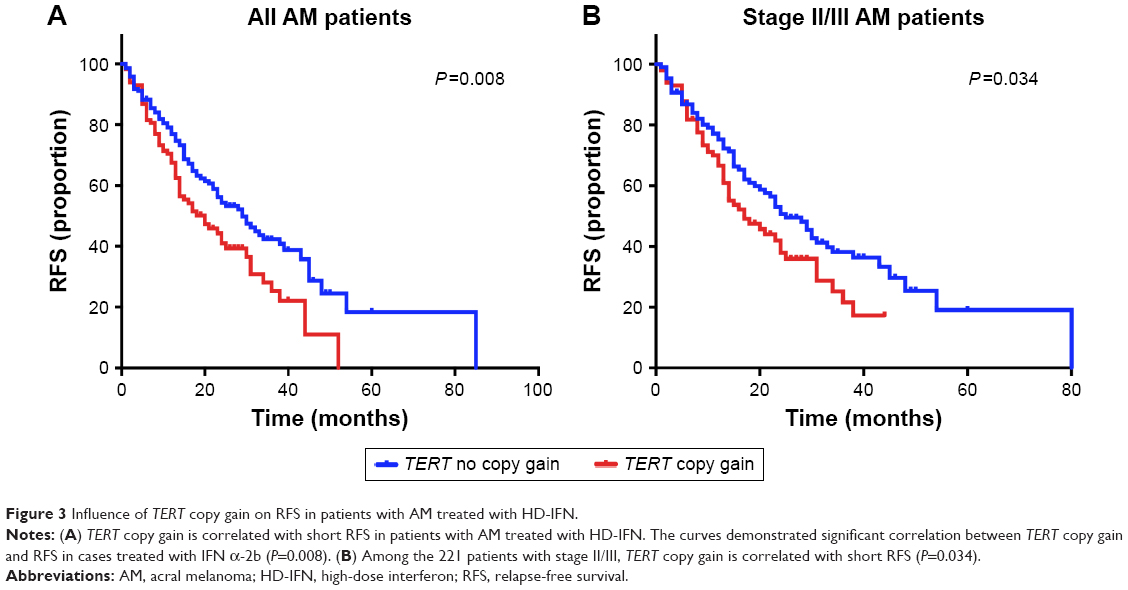 | Figure 3 Influence of TERT copy gain on RFS in patients with AM treated with HD-IFN. |
The results of multivariate Cox regression assays showed that TERT copy gain (hazard ratio =1.50; P=0.019) and TNM stage (hazard ratio =2.92; P<0.001) were independent prognostic factors of RFS for patients with AM undergoing HD-IFN therapy (Table 3).
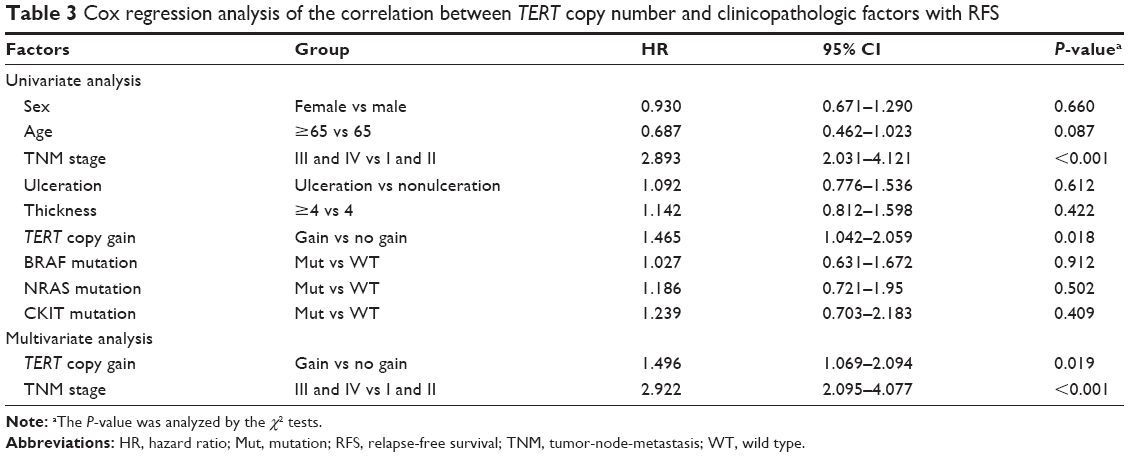 | Table 3 Cox regression analysis of the correlation between TERT copy number and clinicopathologic factors with RFS |
Discussion
AM is distinguishable from other melanoma subtypes by its unique clinical, epidemiological, and genetic features.1,2,7,28 AM is the predominant melanoma subtype in non-Caucasians and has a notably worse prognosis than common cutaneous melanoma.4,29 In recent years, AM was found to have a markedly different genomic landscape from other subtypes of melanoma, with a far lower mutation burden dominated by large-scale structural variants7 and a higher frequency of focal amplifications.13 In this study, we focused on the gene copy gain of TERT on a large scale, with an aim to determine its prognostic significance in patients with AM and to analyze the relationship between TERT and the efficiency of the HD-IFN.
Somatic mutations and amplification are significant sources of genetic diversity. In our study, the copy gain rate of TERT in AM was 44.9% (257 of the 573) (gain >2-fold change). This result was significantly higher than those of previous studies (17.6%–31.2%).26,30 This is probably due to the small sample size of these studies.
The tumor thickness, ulceration, and stage are known prognostic factors for predicting the outcomes of melanoma.31 Our results showed that ulceration (P=0.028) and thickness (P=0.048) were associated with TERT copy gain. However, TERT copy gain is not correlated with the stage of primary melanomas. In addition, unlike a previous study with a cohort of 43 AM patients,25 we found no association between TERT copy gain and OS in our study. We obtained the same result after stratified analysis according to TNM stage. Our data suggest that the TERT copy gain may not play a significant role in the OS of patients with AM.
IFN-α has been the only drug observed to improve RFS in high-risk postoperative patients with melanoma, and the US Food and Drug Administration approved HD-IFN for melanoma in 1996.32 A randomized phase II trial conducted in Chinese high-risk patients with AM showed that 1-year adjuvant HD-IFN (15×106 U/m2 for days 1–5/week for 4 weeks +9×106 U twice a week for 48 weeks) may yield clinical benefits in patients with stage IIIb–IIIc AM or those with ≥3 nodal metastases.16 The median RFS of patients with AM treated with 1-year adjuvant HD-IFN was 17.9 months; however, no specific factor can predict the treatment efficacy.
In our study, the RFS of patients with TERT copy gain was significantly shorter than those with normal TERT copy number and stage II/III patients treated with HD-IFN. The Cox proportional hazards model also revealed that TERT copy gain was an independent adverse prognostic factor of RFS for all patients with AM treated with HD-IFN. This result can be explained as follows. The molecular mechanism of IFN α-2b includes direct and indirect activities. Direct activity occurs through the inhibition of cancer cell growth via cell cycle arrest, apoptosis, or differentiation. Indirect activity occurs through the activation of immune cells, inhibition of vascularization, and induction of cytokines.33 Many studies have reported that IFN α-2b can regulate more than 300 genes in cellular signal transduction pathways.34 IFN α-2b can activate JAK-1 and TYK-2 and induce antiproliferative and antiviral activity.35,36 IFN α-2b can also inhibit the interaction of ERK with MEK or the interaction of MEK with other kinases.37
Given the mechanism by which IFN α-2b influences the growth of various cancer cells via the JAK/STAT and MAPK signal transduction pathways, some researchers have also found a relationship between TERT and these pathways. They found that the expression levels and enzymatic activity of TERT are regulated by multiple signaling molecules and pathways, including the JAK/STAT, RAS/RAF/MEK/MAPK, PI3K/Akt/mTOR, IKK/NFκB, TGFβ/Smads, and PKC pathways.38 These studies may help to explain why the HD-IFN group with no copy gain of TERT exhibited longer RFS than the TERT copy gain group. However, the exact mechanism remains unknown; thus, further research is warranted. In addition, to achieve more satisfactory effects, we also suggest that the TERT copy number of patients be detected before administering HD-IFN treatment.
Conclusion
In this study, we have shown the status and the clinical significance of TERT copy number in a large cohort of patients with AM. We found that the TERT copy gain might not be a prognostic factor of OS in patients with AM. However, TERT copy gain may predict the outcome of HD-IFN treatment for AM.
Acknowledgments
This work was supported by grants from the Major State Basic Research Development Program of China (2013CB911004), National Natural Science Foundation of China (81672696, 81772912), Beijing Municipal Natural Science Foundation (7152033), Beijing Baiqianwan Talents Project, Beijing Municipal Administration of Hospitals Clinical medicine Development of special funding support (ZYLX201603), and Beijing Municipal Science & Technology Commission (Z151100003915074).
Author contributions
All authors contributed toward data analysis, drafting, and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Piliang MP. Acral lentiginous melanoma. Surg Pathol Clin. 2009;2(3):535–541. | ||
Bradford PT, Goldstein AM, McMaster ML, Tucker MA. Acral lentiginous melanoma: incidence and survival patterns in the United States, 1986–2005. Arch Dermatol. 2009;145(4):427–434. | ||
Wu XC, Eide MJ, King J, et al. Racial and ethnic variations in incidence and survival of cutaneous melanoma in the United States, 1999–2006. J Am Acad Dermatol. 2011;65(5 Suppl 1):S26–S37. | ||
Chi Z, Li S, Sheng X, et al. Clinical presentation, histology, and prognoses of malignant melanoma in ethnic Chinese: a study of 522 consecutive cases. BMC Cancer. 2011;11:85. | ||
Tokuzen N, Nakashiro K, Tanaka H, Iwamoto K, Hamakawa H. Therapeutic potential of targeting cell division cycle associated 5 for oral squamous cell carcinoma. Oncotarget. 2016;7(3):2343–2353. | ||
Furney SJ, Turajlic S, Stamp G, et al. The mutational burden of acral melanoma revealed by whole-genome sequencing and comparative analysis. Pigment Cell Melanoma Res. 2014;27(5):835–838. | ||
Hayward NK, Wilmott JS, Waddell N, et al. Whole-genome landscapes of major melanoma subtypes. Nature. 2017;545(7653):175–180. | ||
Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363(9):809–819. | ||
Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372(1):30–39. | ||
Larkin J, Ascierto PA, Dréno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371(20):1867–1876. | ||
Carvajal RD, Antonescu CR, Wolchok JD, et al. KIT as a therapeutic target in metastatic melanoma. JAMA. 2011;305(22):2327–2334. | ||
Si L, Kong Y, Xu X, et al. Prevalence of BRAF V600E mutation in Chinese melanoma patients: large scale analysis of BRAF and NRAS mutations in a 432-case cohort. Eur J Cancer. 2012;48(1):94–100. | ||
Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353(20):2135–2147. | ||
Si L, Guo J. C-kit-mutated melanomas: the Chinese experience. Curr Opin Oncol. 2013;25(2):160–165. | ||
Kong Y, Si L, Zhu Y, et al. Large-scale analysis of KIT aberrations in Chinese patients with melanoma. Clin Cancer Res. 2011;17(7):1684–1691. | ||
Mao L, Si L, Chi Z, et al. A randomised phase II trial of 1 month versus 1 year of adjuvant high-dose interferon α-2b in high-risk acral melanoma patients. Eur J Cancer. 2011;47(10):1498–1503. | ||
Coit DG, Thompson JA, Algazi A. Melanoma, version 2.2016, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2016;14(4):450–473. | ||
Liu T, Yuan X, Xu D. Cancer-specific telomerase reverse transcriptase (TERT) promoter mutations: biological and clinical implications. Genes (Basel). 2016;7(7):pii: E38. | ||
Shay JW, Wright WE. Telomerase therapeutics for cancer: challenges and new directions. Nat Rev Drug Discov. 2006;5(7):577–584. | ||
Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly recurrent TERT promoter mutations in human melanoma. Science. 2013;339(6122):957–959. | ||
Horn S, Figl A, Rachakonda PS, et al. TERT promoter mutations in familial and sporadic melanoma. Science. 2013;339(6122):959–961. | ||
Macerola E, Loggini B, Giannini R, et al. Coexistence of TERT promoter and BRAF mutations in cutaneous melanoma is associated with more clinicopathological features of aggressiveness. Virchows Arch. 2015;467(2):177–184. | ||
Liau JY, Tsai JH, Jeng YM, Chu CY, Kuo KT, Liang CW. TERT promoter mutation is uncommon in acral lentiginous melanoma. J Cutan Pathol. 2014;41(6):504–508. | ||
Heidenreich B, Nagore E, Rachakonda PS, et al. Telomerase reverse transcriptase promoter mutations in primary cutaneous melanoma. Nat Commun. 2014;5:3401. | ||
Diaz A, Puig-Butillé JA, Muñoz C, et al. TERT gene amplification is associated with poor outcome in acral lentiginous melanoma. J Am Acad Dermatol. 2014;71(4):839–841. | ||
Diaz A, Puig-Butillé JA, Valera A, et al. TERT and AURKA gene copy number gains enhance the detection of acral lentiginous melanomas by fluorescence in situ hybridization. J Mol Diagn. 2014;16(2):198–206. | ||
Puig-Butillé JA, Badenas C, Ogbah Z, et al. Genetic alterations in RAS-regulated pathway in acral lentiginous melanoma. Exp Dermatol. 2013;22(2):148–150. | ||
Phan A, Touzet S, Dalle S, Ronger-Savlé S, Balme B, Thomas L. Acral lentiginous melanoma: histopathological prognostic features of 121 cases. Br J Dermatol. 2007;157(2):311–318. | ||
Ito T, Wada M, Nagae K, et al. Acral lentiginous melanoma: who benefits from sentinel lymph node biopsy. J Am Acad Dermatol. 2015;72(1):71–77. | ||
Bennett DC. Genetics of melanoma progression: the rise and fall of cell senescence. Pigment Cell Melanoma Res. 2016;29(2):122–140. | ||
Manola J, Atkins M, Ibrahim J, Kirkwood J. Prognostic factors in metastatic melanoma: a pooled analysis of Eastern Cooperative Oncology Group trials. J Clin Oncol. 2000;18(22):3782–3793. | ||
Mocellin S, Pasquali S, Rossi CR, Nitti D. Interferon alpha adjuvant therapy in patients with high-risk melanoma: a systematic review and meta-analysis. J Natl Cancer Inst. 2010;102(7):493–501. | ||
Asmana NR. Human interferon alpha-2b: a therapeutic protein for cancer treatment. Scientifica (Cairo). 2014;2014:970315. | ||
Chawla-Sarkar M, Lindner DJ, Liu YF, et al. Apoptosis and interferons: role of interferon-stimulated genes as mediators of apoptosis. Apoptosis. 2003;8(3):237–249. | ||
Jonasch E, Haluska FG. Interferon in oncological practice: review of interferon biology, clinical applications, and toxicities. Oncologist. 2001;6(1):34–55. | ||
Samuel CE. Antiviral actions of interferon. Interferon-regulated cellular proteins and their surprisingly selective antiviral activities. Virology. 1991;183(1):1–11. | ||
Romerio F, Riva A, Zella D. Interferon-alpha2b reduces phosphorylation and activity of MEK and ERK through a Ras/Raf-independent mechanism. Br J Cancer. 2000;83(4):532–538. | ||
Yamada O, Kawauchi K. The role of the JAK-STAT pathway and related signal cascades in telomerase activation during the development of hematologic malignancies. JAKSTAT. 2013;2(4):e25256. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.