Back to Journals » Journal of Inflammation Research » Volume 19
Temporal Evolution of Brain Inflammation in the Bilateral Common Carotid Artery Stenosis Model of Chronic Cerebral Hypoperfusion
Authors Cox MF ![]() , Roberts JM, Shahidehpour RK, Dornbos III DL, Bachstetter AD
, Roberts JM, Shahidehpour RK, Dornbos III DL, Bachstetter AD ![]()
Received 14 October 2025
Accepted for publication 20 January 2026
Published 19 February 2026 Volume 2026:19 574221
DOI https://doi.org/10.2147/JIR.S574221
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
MaKayla F Cox,1,2 Jill M Roberts,2,3 Ryan K Shahidehpour,4 David L Dornbos III,3 Adam D Bachstetter1,2,4
1Spinal Cord and Brain Injury Research Center, University of Kentucky, Lexington, KY, USA; 2Department of Neuroscience, University of Kentucky, Lexington, KY, USA; 3Department of Neurosurgery, University of Kentucky, Lexington, KY, USA; 4Sanders-Brown Center on Aging, University of Kentucky, Lexington, KY, USA
Correspondence: Adam D Bachstetter, Spinal Cord and Brain Injury Research Center and Department of Neuroscience, University of Kentucky, 741 S. Limestone Street, BBSRB Room B459, Lexington, KY, 40536-0509, USA, Tel +1 859 218 4315, Email [email protected]
Abstract: Chronic cerebral hypoperfusion is a major contributor to vascular cognitive impairment and dementia, primarily through its disruption of white matter integrity and promotion of neuroinflammatory cascades. The bilateral common carotid artery stenosis (BCAS) model has emerged as the predominant experimental approach for studying the effects of chronic cerebral hypoperfusion, particularly in aging-relevant contexts. This review synthesizes current knowledge of inflammatory responses in the BCAS model, with an emphasis on their temporal progression and functional consequences. We begin by outlining the clinical burden of hypoperfusion, the rationale for BCAS, and the hemodynamic phases it produces. We then examine early neurovascular unit dysfunction, including coordinated responses among endothelial cells, pericytes, and astrocytes. These changes converge on blood-brain barrier compromise, followed by loss of tight junctions, activation of matrix metalloproteinases, and routes of leukocyte entry via venules and the choroid plexus. Inflammatory mediators such as IL-1β, TNF-α, and IL-6 amplify injury, while microglia transition from acute responders to chronic effectors of white matter damage. Although many reports have catalogued inflammatory changes, causal tests remain sparse. Few studies manipulate specific cytokines, border-associated macrophages, or temporally restricted cellular pathways, leaving unvalidated the checkpoints that govern the shift from adaptive responses (angiogenesis, gliosis, debris clearance) to maladaptive, self-sustaining inflammation. Clarifying the temporal dynamics of neuroinflammation in BCAS is critical for defining therapeutic windows and developing strategies that preserve compensatory mechanisms while preventing chronic pathology. Future research should prioritize cell-specific and time-resolved approaches to identify mechanistic checkpoints that can be targeted therapeutically to mitigate hypoperfusion-driven cognitive decline.
Keywords: microglia, leukocytes, white matter lesions, neurovascular unit, matrix metalloproteinases, cytokines, VCID, vascular dementia, subcortical ischemic vascular dementia, cerebrovascular disease
Introduction
Chronic cerebral hypoperfusion (CCH) arises when cerebral blood flow (CBF) persistently falls below metabolic demand.1–3 The most common causes are atherosclerotic cardiovascular disease and arteriolosclerosis, which progressively narrow cerebral arteries and impair perfusion in both cortical and deep brain regions.1,4 Other contributors include moyamoya disease, cardiac dysfunction, and chronic hypertension.5,6 These conditions are highly prevalent in older adults, where age-related vascular stiffening exacerbates hemodynamic decline, placing white matter at particular risk.1,6 White matter lesions, basal ganglia injury, and hippocampal vulnerability are frequent consequences, often coexisting with hippocampal sclerosis of aging and other neurodegenerative changes.7
Together, large- and small-vessel pathologies make chronic cerebral hypoperfusion the rule rather than the exception in aging brains, and a major driver of vascular contributions to cognitive impairment and dementia (VCID). In the United States, atherosclerotic cardiovascular disease alone affects over 24 million adults, often with comorbid hypertension and diabetes, amplifying hypoperfusion risk.8,9 Clinical imaging consistently demonstrates white matter lesions associated with reduced CBF, and histopathological studies confirm progressive arteriolosclerosis with concentric vessel wall thickening, lumen narrowing, and parenchymal rarefaction (Figure 1).
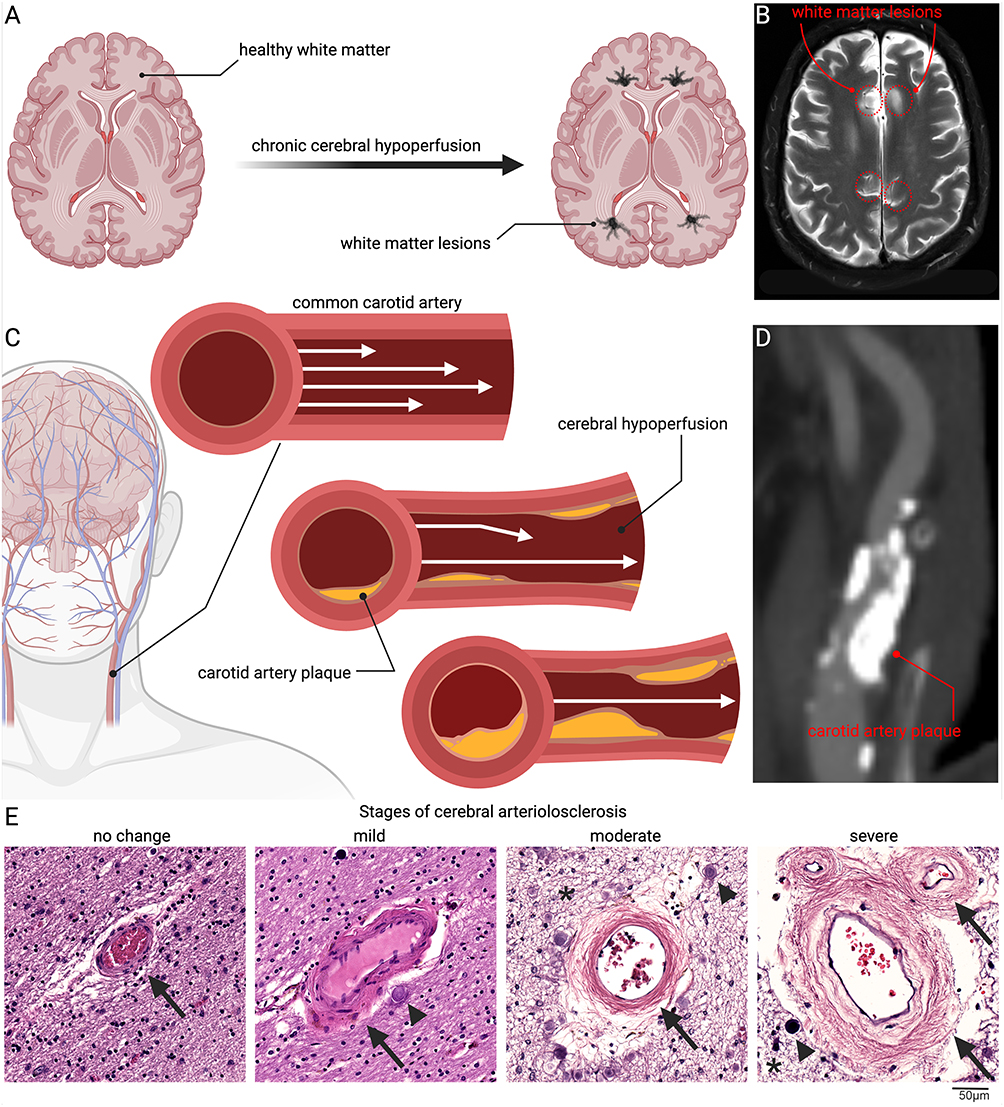 |
Figure 1 Clinical and pathological features of chronic cerebral hypoperfusion. Chronic reductions in cerebral blood flow drive progressive white matter injury, beginning with the transition from healthy tissue to the development of white matter lesions (A), a process readily observed in human MRI scans where hyperintense lesions mark areas of damage (B) (red dashed line indicates areas of white matter lesions). A major contributor is atherosclerotic narrowing of the carotid arteries, where plaque accumulation progressively restricts laminar flow and reduces cerebral perfusion (C), as confirmed in vascular imaging of carotid plaques (D). In parallel, small penetrating vessels in the brain undergo arteriolosclerosis, characterized by concentric wall thickening, hyaline change, and eventual near-occlusion. Histological sections from human amygdala periventricular white matter illustrate this continuum (E). In the normal state, arterioles have thin vessel walls, preserved surrounding parenchyma, and an intact Virchow–Robin space. With mild disease, the vessel wall begins to thicken and develop hyaline change, accompanied by early perivascular alterations and the appearance of corpora amylacea. As pathology progresses, concentric wall thickening and narrowing of the lumen become apparent, with increased corpora amylacea, mild parenchymal rarefaction, and evidence of neuronal and glial loss. In severe arteriolosclerosis, the arteriole is nearly occluded, characterized by abundant corpora amylacea and marked parenchymal rarefaction (Black arrows denote vessel walls and wall changes, arrowheads indicate corpora amylacea, and asterisks mark parenchymal rarefaction). Together, these vascular and parenchymal alterations establish chronic cerebral hypoperfusion as a central mechanism linking systemic vascular disease to white matter vulnerability and cognitive decline. |
By the time symptoms manifest, irreversible structural and functional brain changes are often established. This highlights the importance of mechanistic studies in identifying early biomarkers and therapeutic windows. Experimental models, particularly the bilateral carotid artery stenosis (BCAS) model in mice, have become indispensable for linking chronic cerebral hypoperfusion to cellular and molecular mechanisms of neuroinflammation. While BCAS remains widely used to study CCH, it is important to acknowledge its inherent limitations when considering translational capacity. The BCAS model is limited in variability for stenosis severity, incomplete recapitulation of gradual vascular decline seen in humans, and strain dependent differences in white-matter vulnerability as highlighted by Ishikawa et al.10 Recognizing these model-specific constraints provides important context for evaluating cascades triggered by CCH. Various experimental approaches exist to model ischemic injury; the present review focuses specifically on the Bilateral Common Carotid Artery Stenosis (BCAS) model to delineate the temporal evolution of inflammatory processes associated with chronic cerebral hypoperfusion.
Clinical Relevance and Pathophysiological Overview of Chronic Cerebral Hypoperfusion
Clinical imaging consistently links white matter hyperintensities, lacunes, and hippocampal atrophy to reduced cerebral blood flow, and neuropathological studies demonstrate vessel wall thickening, lumen narrowing, and parenchymal rarefaction as characteristic substrates (Figure 1).9,11 Chronic cerebral hypoperfusion represents the final common pathway for multiple systemic and cerebrovascular conditions, occurring when cerebral blood flow remains persistently below metabolic requirements. Large-vessel disease, particularly atherosclerotic cardiovascular disease, contributes through progressive plaque accumulation and arterial stiffening, which together reduce global cerebral perfusion.1–3,7 In particular, narrowing of the carotid arteries can significantly limit blood flow to cortical and subcortical regions.3,12 Age-related vascular stiffening exacerbates the hemodynamic consequences of arterial narrowing, increasing the risk of widespread cortical hypoperfusion.13 Small-vessel disease, most commonly arteriolosclerosis, impairs perfusion to deep brain regions such as the white matter, basal ganglia, and hippocampus, where it produces regional hypoxia and increased vulnerability to neurodegeneration.1–3,7 These vascular pathologies are highly prevalent in older adults and are further accelerated by systemic conditions such as hypertension, diabetes, heart failure, and metabolic dysfunction, making hypoperfusion the rule rather than the exception in the aging brain.14,15 Additional, less common causes, including moyamoya disease and congenital or acquired cardiac dysfunction, can also compromise cerebral blood flow.5,6 Multi-lumen vascular profiles are common in brain aging, although their effect on chronic cerebral hypoperfusion remains undefined.16 Collectively, these overlapping processes establish chronic cerebral hypoperfusion as both an initiator and perpetuator of cerebrovascular pathology, linking systemic vascular disease to white matter damage, hippocampal vulnerability, and the clinical spectrum of vascular contributions to cognitive impairment and dementia (VCID)1,4,9,17,18 (Figure 2).
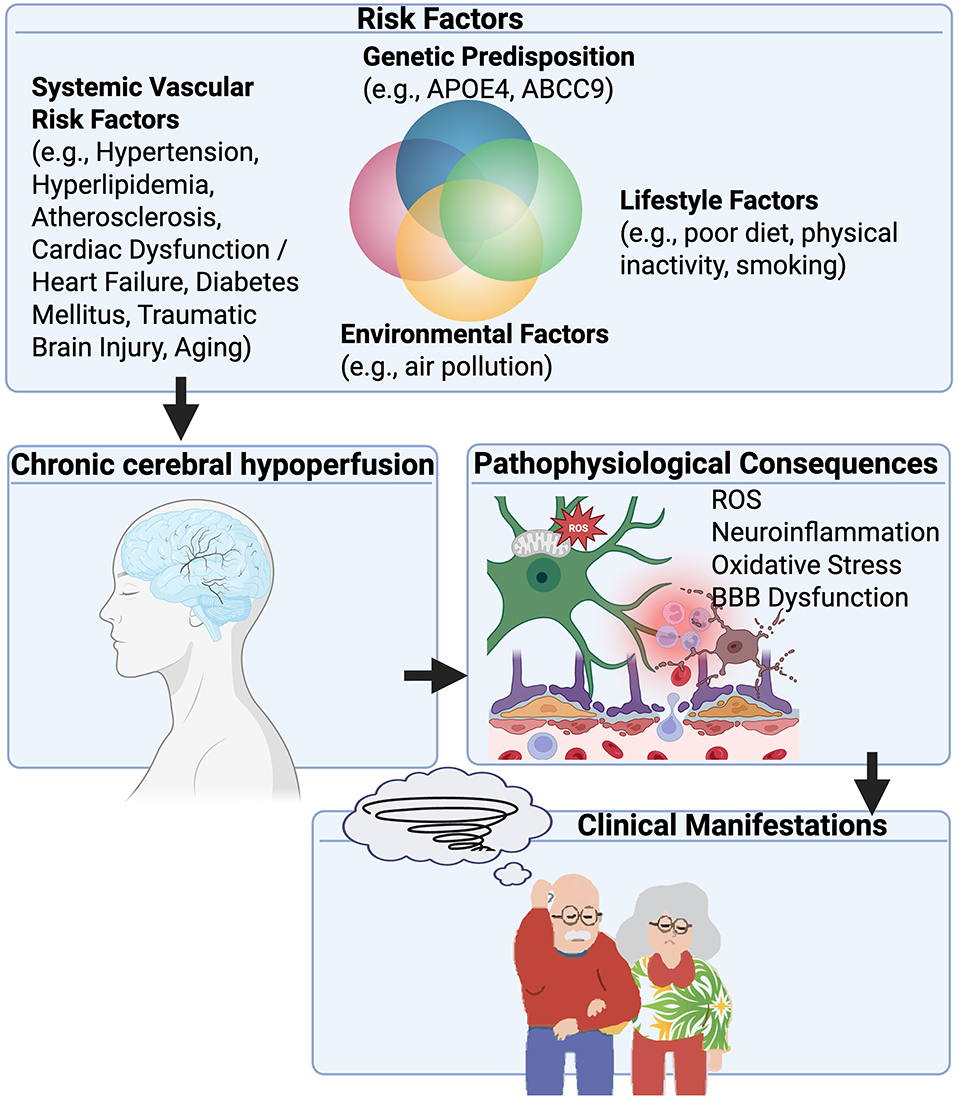 |
Figure 2 Pathophysiological cascade of chronic cerebral hypoperfusion leading to cognitive impairment. This figure illustrates the multi-factorial origins and progressive consequences of chronic cerebral hypoperfusion. Various interconnected risk factors converge to initiate and exacerbate chronic cerebral hypoperfusion. These include genetic predisposition, such as polymorphisms in APOE4 and ABCC9 genes which influence neuronal vulnerability and cerebrovascular function; lifestyle factors, including poor diet, physical inactivity, and smoking; environmental factors, such as air pollution; and systemic vascular risk factors, encompassing hypertension, atherosclerosis, hyperlipidemia, cardiac dysfunction/heart failure, diabetes mellitus, prior traumatic brain injury (TBI), and aging. These factors collectively contribute to a chronic cerebral hypoperfusion state, characterized by a sustained reduction in cerebral blood flow. This initiates a cascade of detrimental pathophysiological consequences within the brain’s microenvironment. Key interlinked pathways include oxidative stress, driven by increased reactive oxygen species (ROS) production, leading to cellular damage; neuroinflammation, characterized by activated glial cells (microglia and astrocytes) and the release of inflammatory mediators; and BBB dysfunction, resulting in increased permeability and compromised brain homeostasis. These pathological processes are highly interconnected, with oxidative stress contributing to neuroinflammation and BBB dysfunction, and inflammation exacerbating both oxidative stress and BBB compromise. Ultimately, these consequences lead to irreversible structural and functional changes in the brain, such as white matter damage (including white matter hyperintensities on MRI), neuronal injury and loss (including hippocampal sclerosis of aging), and synaptic dysfunction. These brain changes manifest clinically as a spectrum of cognitive impairments, ranging from mild cognitive impairment to vascular cognitive impairment and culminating in severe vascular dementia, often interacting with or exacerbating Alzheimer’s disease pathology. |
There are currently 8 categories of approved FDA treatments for arteriosclerosis and atherosclerotic cardiovascular disease with almost all the treatments focusing on the metabolic implications of the disease such as plaque stabilization or regression. Despite the numerous available treatments for these conditions, there are zero FDA-approved treatments for chronic cerebral hypoperfusion. Given that age is a primary risk factor for arteriosclerosis and atherosclerotic cardiovascular disease, stroke, and dementia, the increasing age of the global population supports a predicted increase in both the prevalence and incidence of chronic cerebral hypoperfusion. Therefore, discovering the physiologic role of chronic cerebral hypoperfusion is critical for developing novel therapeutics and improving cerebrovascular health.
The Bilateral Carotid Artery Stenosis (BCAS) Model of Chronic Cerebral Hypoperfusion
By the time clinical symptoms of chronic cerebral hypoperfusion become apparent, irreversible structural and functional changes in the brain have often already occurred. This underscores the critical need to understand the underlying pathophysiological mechanisms to identify early biomarkers, particularly blood-based markers, that may signal impending brain injury in individuals at risk due to a combination of genetic, lifestyle, and environmental factors. Early detection could enable proactive interventions to preserve brain health and reduce the burden of cognitive impairment and dementia.19 Achieving this goal will require experimental model systems to define the specific changes in brain structure, function, and peripheral biomarkers that can be directly linked to chronic cerebral hypoperfusion. In this review, we specifically focus on the BCAS model in mice as a key experimental system used to mimic chronic cerebral hypoperfusion. We will review the BCAS model and the physiology of normal CBF, followed by identifying potential mechanisms behind chronic cerebral hypoperfusion-induced neuroinflammation. In particular, we examine the bidirectional relationship between inflammation and hypoperfusion, BBB dysfunction, propagation of inflammatory signals across the neurovascular unit, role of pro- and anti-inflammatory mediators, and the temporal dimension of the inflammatory response, emphasizing how its effects can be either protective or detrimental depending on timing and context.20–22 Finally, this review will highlight the dynamic role of CBF in disease states and describe the implications of these mechanisms in the context of developing therapies to modify perfusion and decrease neuroinflammation.7,9,17,23,24
Previous studies have indicated that chronic cerebral hypoperfusion is associated with neuroinflammation and could contribute to inflammatory responses in both mice and humans.25,26 The mechanisms underlying chronic cerebral hypoperfusion’s contribution to neuroinflammation have yet to be understood. However, neuroinflammation is thought to be a prominent contributor to the resulting neurological dysfunction caused by chronic cerebral hypoperfusion. Proposed mechanisms for chronic cerebral hypoperfusion-induced neuroinflammation include activated microglia, inflammasome signaling, changes in blood-brain barrier/blood-CSF barrier permeability, neurovascular uncoupling, and recruitment of peripheral immune cells leading to cerebral immune dyshomeostasis.21,25,27–29 Over the past two decades, BCAS has demonstrated reproducible pathology and reliable face validity for the study of subcortical ischemic vascular dementia, vascular dementia, and other small-vessel diseases.9,23,30 The clinical and translational relevance of BCAS highlights its utility as a model system for studying the inflammatory consequences of chronic cerebral hypoperfusion.
The BCAS model was originally developed to study cerebrovascular white matter lesions but has since been repurposed to study subcortical ischemic vascular dementia and other small vessel diseases.10,30 The BCAS model was first developed and reported in 2004 by Shibata et al in Japan to address critical limitations of existing chronic cerebral hypoperfusion models.30 Previous rat models using bilateral common carotid artery occlusion (BCCAO) suffered from several significant drawbacks. First, the permanent ligation procedure damaged the ophthalmic arteries, causing visual pathway impairment that compromised behavioral testing relying on visual cues.31 Second, the limited availability of genetically modified rats in the early 2000s restricted molecular and genetic investigations. Third, the abrupt and severe CBF reduction in BCCAO models (often to 30–40% of baseline) frequently caused high mortality rates and widespread ischemic lesions extending beyond white matter into gray matter structures like the hippocampus and cortex, making it difficult to isolate white matter pathology characteristic of subcortical ischemic vascular dementia.9,10,31 The BCAS model was specifically designed to overcome these obstacles while providing a reproducible platform for studying the mechanisms of chronic cerebral hypoperfusion.
BCAS is a surgical procedure in which microcoils with a defined diameter are bilaterally wrapped around mouse common carotid arteries (Figure 3). The diameter of the coils can be modified to induce variable reductions in cerebral blood flow without causing an acute infarct. Microcoils with inner diameters ranging from 0.18–0.20 mm are surgically wrapped around both carotid arteries, with coil diameter selection determining the severity of flow restriction. The original and most commonly used microcoils have an inner diameter of 0.18 mm.30,32 This mechanical stenosis reduces cerebral blood flow to approximately 60% of baseline values within hours post-surgery, followed by partial recovery to around 70% of baseline usually by day 7 but can remain below 70% up to one month due to heterogeneity. This creates a sustained hypoperfusion state without causing acute infarction.30,32 The graduated nature of this CBF reduction progresses from severe acute hypoperfusion to moderate chronic hypoperfusion, creating distinct temporal phases that allow investigation of both immediate hypoxic responses and long-term adaptive mechanisms. This controlled, reproducible methodology enables precise manipulation of hypoperfusion severity through coil diameter modification while maintaining low surgical mortality rates and compatibility with genetically modified mouse strains.31
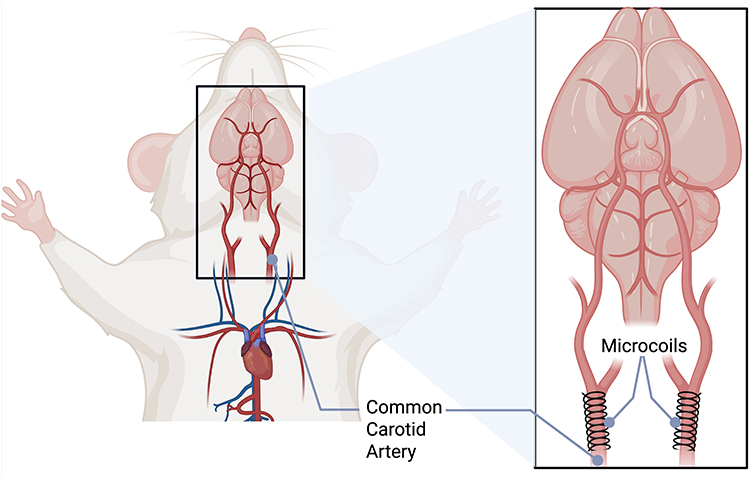 |
Figure 3 Bilateral Common Carotid Artery Stenosis (BCAS) Model Surgical Procedure. Schematic illustration of the BCAS model showing the bilateral placement of microcoils around the common carotid arteries in mice. The left panel shows the overall anatomical view of the mouse with the surgical site highlighted by the black box. The right panel provides a detailed view of the neck region showing microcoils (typically 0.18–0.20 mm inner diameter) surgically wrapped around both common carotid arteries to induce controlled cerebral hypoperfusion. The microcoils create mechanical stenosis that reduces cerebral blood flow while preserving some residual circulation, distinguishing this model from complete occlusion approaches. This surgical approach enables reproducible chronic cerebral hypoperfusion with low mortality rates and compatibility with genetically modified mouse strains. |
The BCAS model consistently produces several key pathophysiological changes that develop progressively following hypoperfusion induction. Neuroinflammation emerges rapidly, with microglial and astrocytic activation detectable as early as three days post-BCAS and persisting for at least 30 days, accompanied by elevated pro-inflammatory cytokine production.10,30,33 BBB disruption occurs in parallel, evidenced by IgG leakage in the corpus callosum within 3–7 days, driven by oxidative stress-mediated tight-junction loss and increased matrix metalloproteinase activity.21,33,34 The model demonstrates selective vulnerability of white matter structures, with the corpus callosum consistently identified as the most affected region, developing diffuse demyelination, axonal loss, and gliosis by 14 days post-BCAS.31,32,35 Other vulnerable white matter regions include the caudoputamen, internal capsule, and anterior commissure, while gray matter structures remain largely preserved when standard 0.18 mm microcoils are used.31,36 These structural changes translate into specific cognitive deficits, particularly spatial working memory impairments evident at 30 days and persisting chronically, attributed to damage in frontal-subcortical circuits. With prolonged hypoperfusion, additional deficits in spatial reference memory and hippocampal atrophy can develop.37 The specific mechanisms underlying each of these pathophysiological changes, including their inflammatory mediators, temporal dynamics, cellular responses, and functional consequences, will be examined in detail in the following sections.
The BCAS model offers several key advantages that have established it as the predominant experimental approach for studying chronic cerebral hypoperfusion. The model demonstrates exceptional reproducibility and reliability, with over 100 studies since 2004 validating its efficacy.10 Key strengths include low surgical mortality rates, preservation of visual pathways that enable accurate behavioral testing, and compatibility with genetically modified mouse strains, which are essential for mechanistic studies. The ability to precisely control hypoperfusion severity through modification of microcoil diameter provides researchers with a standardized yet flexible experimental platform. Additionally, the model’s versatility is demonstrated by successful modifications across different mouse strains, ages at surgery, and coil diameters.10,30 While the majority of BCAS studies observe mice for 8 weeks to 3 months post-surgery, studies have examined mice at as early as 30 days up to 5-, 8-, and 11-months of age to evaluate the role of chronic cerebral hypoperfusion in Alzheimer’s pathology, demonstrating the flexibility and long-term potential of the BCAS model.10,36,38
Despite these advantages, the BCAS model has notable limitations for translational studies. The relatively rapid CBF reduction differs from the gradual onset typically observed in human chronic cerebral hypoperfusion conditions, and the model does not reproduce the motor dysfunction commonly seen in clinical populations.10,31,39 Alternative models, including gradual common carotid artery stenosis (GCAS) and asymmetric common carotid artery surgery (ACAS), were developed to more closely mimic the gradual reduction in blood flow observed in humans.10,31,40,41 In GCAS, ameroid constrictors narrow the carotid arteries over several days, in contrast to the immediate reduction produced by BCAS microcoils. ACAS creates an asymmetric flow profile that similarly slows the onset of hypoperfusion. Although these models address the limitation of BCAS’s abrupt CBF reduction, the ameroid constrictors are large and obtrusive, are associated with higher mortality, and are considerably more costly. Their greatest limitation at present is the relatively small number of publications and incomplete characterization of pathophysiology compared with BCAS; however, this gap is likely to diminish as additional validation studies are completed.10,31 The variability in the BCAS phenotype is heavily influenced by the genetic background and age of the animal. Older animals typically exhibit accelerated and more severe white matter pathology compared to young adults, reflecting the synergistic impact of aging and hypoperfusion on neurovascular unit integrity.10,42 However, more investigation is required to characterize the BCAS phenotypes in different strains of mice. An important gap in current BCAS research is the limited investigation of sex differences in response to chronic cerebral hypoperfusion. Recent studies indicate that female mice experience earlier CBF recovery post-BCAS, this observation correlates with clinical data demonstrating that human females maintain higher cerebral blood flow and lower cardiac output compared to males across the adult lifespan. However, neuroinflammatory sex differences secondary to chronic cerebral hypoperfusion remain largely unknown.29,43 Addressing this limitation through sex-specific studies will be crucial for enhancing the translational validity of future BCAS research.
Taken together, considering its strengths and limitations, BCAS remains the leading model for investigating chronic cerebral hypoperfusion. The next section outlines how cerebral blood flow is normally regulated and how these mechanisms fail under sustained hypoperfusion in BCAS, initiating the neurovascular responses that drive downstream inflammatory cascades.
Cerebral Blood Flow Changes and Initial Neurovascular Responses
The transition from physiological CBF regulation to chronic cerebral hypoperfusion represents a critical threshold where neuroinflammatory cascades are initiated. Despite comprising only 2% of total body weight, the brain receives approximately 15–20% of cardiac output, with this disproportionate energy demand necessitating precise vascular regulation through cerebral autoregulation, neurovascular coupling, and metabolic regulation.43,44 The neurovascular unit, consisting of endothelial cells, pericytes, astrocytes, and neurons, orchestrates these regulatory processes to maintain adequate perfusion across diverse metabolic demands. Regional CBF heterogeneity reflects metabolic differences, with gray matter receiving approximately 80 mL/100g/min compared to 20 mL/100g/min in white matter under normal conditions. Age-related CBF decline occurs at 3–5% per decade in healthy individuals, but accelerates dramatically in pathological conditions, where neuroinflammatory signaling further drives vascular deterioration.22,45
In BCAS models, CBF reduction severity depends directly on microcoil diameter selection. Standard 0.18 mm microcoils decrease CBF to approximately 60–70% of baseline within hours, while narrower 0.16 mm coils produce more severe reductions to 51% of baseline, and larger 0.20 mm coils cause milder reductions to around 77% of baseline.10,30,31,36 Following acute phase reduction, CBF undergoes gradual partial recovery through compensatory mechanisms, including collateral circulation development and increased Circle of Willis tortuosity.46 By day 7, CBF typically recovers to approximately 70% of baseline, though recovery remains incomplete and regionally heterogeneous, with white matter regions experiencing more severe and persistent hypoperfusion.36,47
Characterizing these hemodynamic requires the use of complementary measurement techniques. The accurate measurement of CBF requires techniques that provide different temporal and spatial resolution capabilities. Laser Doppler flowmetry offers real-time, continuous monitoring with high temporal resolution, ideal for studying acute hemodynamic changes, though limited to superficial cortical regions. Arterial spin labeling MRI provides non-invasive, quantitative CBF mapping with excellent spatial resolution across the entire brain, enabling longitudinal studies but with lower temporal resolution. Fluorescent microsphere techniques provide gold standard absolute CBF quantification with high spatial specificity, though this terminal method precludes longitudinal monitoring.10
Endothelial Cell Responses to Hypoperfusion
The acute reduction in CBF following BCAS initiates a rapid and coordinated disruption of the neurovascular unit, with cerebral endothelial cells among the earliest responders to hypoperfusion. Within 30 minutes, endothelial cells in the corpus callosum exhibit nuclear translocation of phosphorylated p38 MAPK, a response dependent on the oxidative stress sensor ASK1-38. This ASK1–p38 signaling cascade, triggered by hypoxia-induced oxidative stress, promotes endothelial inflammation and contributes to early BBB breakdown.48 By 48 hours, transcriptomic analyses reveal increased endothelial expression of miR-501-3p in the white matter, a change linked to barrier dysfunction. In vivo inhibition of miR-501-3p preserves BBB integrity, highlighting a mechanistic pathway connecting oxidative stress to early vascular compromise.49 These molecular alterations coincide with functional vascular deficits, including reduced myogenic tone and impaired endothelium-dependent vasodilation, attributed in part to decreased expression of TRPV4 channels in parenchymal arterioles. Impaired arteriolar flow velocity and pulsatility further compromise perivascular fluid transport, contributing to early glymphatic clearance deficits during the first days of hypoperfusion.50 Collectively, these rapid endothelial changes establish a cascade of barrier instability and impaired vascular responsiveness, setting the stage for chronic white matter injury and progressive neurovascular dysfunction in the BCAS model (Figure 4).
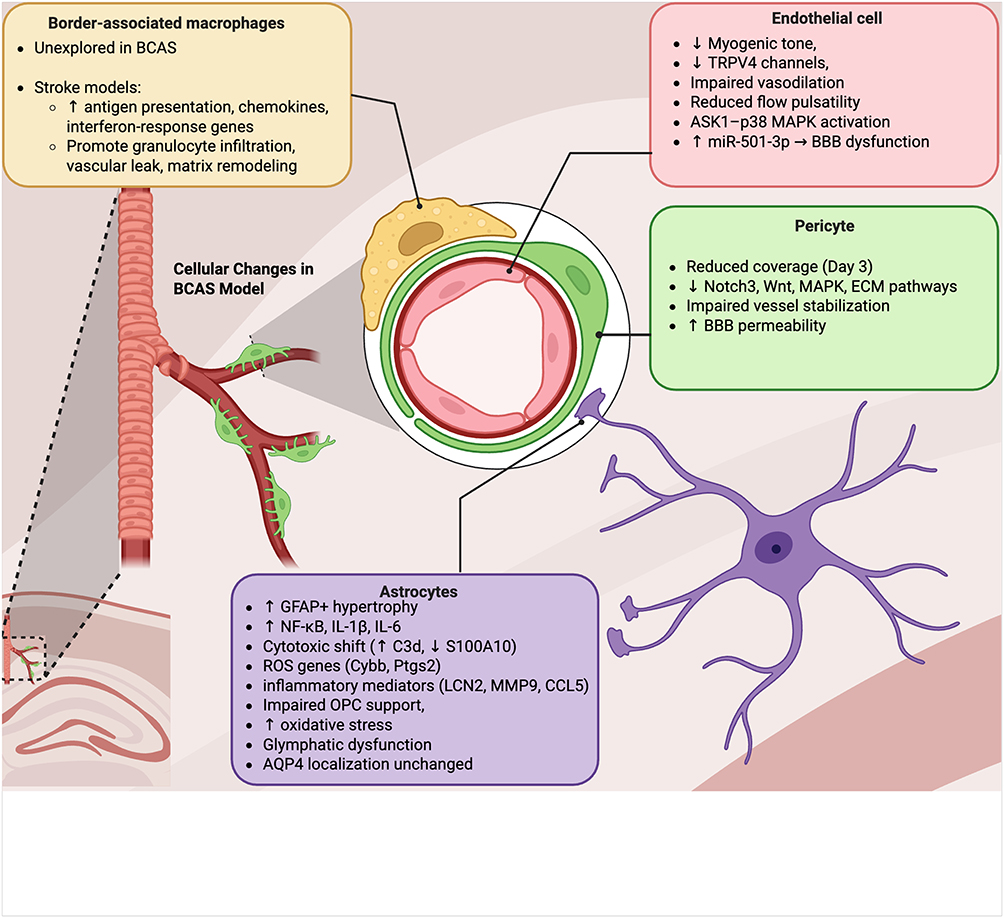 |
Figure 4 Cellular responses to chronic cerebral hypoperfusion in the BCAS model. The transition from physiological to chronically reduced cerebral blood flow triggers coordinated but cell type–specific changes within the neurovascular unit. Endothelial cells rapidly activate stress pathways, including ASK1–p38 MAPK signaling and induction of miR-501-3p, leading to impaired myogenic tone, reduced TRPV4 expression, diminished vasodilation, and BBB dysfunction. Pericytes lose vascular coverage within 3 days, accompanied by transcriptional repression of Notch3, Wnt, MAPK, and extracellular matrix pathways, weakening vessel stabilization and increasing BBB permeability. Astrocytes become reactive by day 7, with GFAP+ hypertrophy, NF-κB activation, and upregulation of pro-inflammatory mediators (IL-1β, IL-6, LCN2, MMP9, CCL5), while shifting toward a cytotoxic phenotype (increased C3d, decreased S100A10). These changes impair oligodendrocyte precursor cell support, promote oxidative stress, and disrupt glymphatic clearance, though AQP4 localization remains preserved. Border-associated macrophages (BAMs), largely unexplored in BCAS, are implicated by stroke studies as early responders at CNS interfaces, promoting granulocyte infiltration, vascular leakage, and matrix remodeling. Collectively, these cellular alterations destabilize the neurovascular unit and establish the foundation for progressive white matter injury under sustained hypoperfusion. |
Despite these advances, the molecular landscape of endothelial dysfunction in BCAS remains incompletely defined. Most studies have concentrated on candidate pathways, leaving broader transcriptional and proteomic features largely unexplored. Unbiased approaches such as single-cell and single-nuclei RNA sequencing, together with endothelial-specific proteomics, could uncover previously unrecognized regulators of stress responses, angiogenic signaling, and immune crosstalk. Time-resolved methods, including translatomics and phosphoproteomics, may further capture state transitions across the acute, subacute, and chronic phases of hypoperfusion, revealing mechanisms of adaptation, failure, or senescence. In addition, while isolated reports suggest changes in xenobiotic and solute transporters, systematic characterization is lacking. Clarifying how hypoperfusion alters endothelial transport functions could expose new metabolic vulnerabilities and potential therapeutic targets.
Mural Cell Vulnerability in BCAS
Pericytes are essential mural cells of the NVU, playing a pivotal role in stabilizing cerebral capillaries and maintaining BBB integrity by modulating endothelial permeability and preventing vascular leakage.51–53 Within 3 days of BCAS, pericyte coverage of cortical microvessels is significantly reduced, particularly in white matter regions such as the corpus callosum, as shown by decreased PDGFR-β⁺ and CD13⁺ labeling on CD31⁺ capillaries.54 This early loss of pericyte coverage coincides with the critical period of neurovascular destabilization, contributing to barrier dysfunction, impaired vascular resistance, and transition toward chronic inflammation. By 28 days, partial restoration of coverage occurs, suggesting that early pericyte dysfunction may act as a transient but pivotal amplifier of downstream injury54 (Figure 4).
At the molecular level, single-cell RNA sequencing reveals widespread transcriptional repression in mural cells, particularly pericytes, with downregulated genes enriched for Notch signaling, extracellular matrix organization, cell adhesion, and intracellular signaling.55 Disruption of Notch3 signaling in mural cells, alongside suppression of Notch1 in endothelial cells, likely disrupts the bidirectional communication required for vessel stabilization under hypoperfusion. Additional repression of MAPK, Wnt, and vascular remodeling pathways suggests that pericytes lose both structural capacity and signaling functions needed for adaptive vascular responses.53,55
The functional consequences of pericyte loss include increased BBB permeability, contributing to white matter lesions and cognitive impairment associated with chronic cerebral hypoperfusion.54 Pharmacological intervention with MLN4924, a neddylation inhibitor, has been shown to restore pericyte coverage and preserve cerebrovascular integrity in BCAS models through the ERK5-KLF2 signaling axis.54 These findings demonstrate that pericytes undergo dynamic transcriptional reprogramming in response to chronic cerebral hypoperfusion, contributing to vascular instability and BBB breakdown through coordinated downregulation of Notch3, Wnt, MAPK/ERK, and extracellular matrix-associated signaling pathways that impair pericyte adhesion, support, and interaction with endothelial cells.
However, key questions remain regarding what upstream cues, such as hypoxia, inflammatory signaling, or mechanical stress, which drive these transcriptional changes, whether pericyte subtypes exhibit distinct vulnerabilities, and to what extent these alterations are reversible or represent stable epigenetic shifts, making the identification of specific transcriptional regulators responsible for these pathway disruptions and defining whether reactivation of pathways like Notch3 or ERK5 can restore pericyte function critical goals for developing targeted interventions that preserve or restore pericyte integrity and neurovascular unit stability in chronic hypoperfusion.
Astrocytic Reactivity and Glymphatic Dysfunction in BCAS
Astrocytes undergo rapid and progressive changes in response to chronic cerebral hypoperfusion induced by BCAS, reflecting early disruptions in their homeostatic and neuroprotective functions. By 7 days post-BCAS, GFAP-positive astrocytes increase in the corpus callosum and white matter tracts, displaying hypertrophy and more complex processes.35,56,57 These reactive astrocytes display activation of pro-inflammatory pathways, including NF-κB signaling and upregulation of cytokines such as IL-1β and IL-6.35,58 By 14 days post-BCAS, astrocytes shift toward cytotoxic phenotypes, marked by elevated complement component C3d and reduced expression of neuroprotective markers such as S100A10.57,59 Transcriptomic profiling further reveals upregulation of ROS-related genes (Cybb, Ptgs2) and inflammatory mediators (LCN2, IL-1R1, TNFsf8, MMP9, CXCR2, CCL5) in astrocytes isolated from hypoperfused brains.60 Functional consequences of this reactivity include impaired trophic support for oligodendrocyte precursor cells57 and increased production of oxidative stress mediators like LCN2 and phosphorylated p47phox60 (Figure 4).
Consistent with these early astrocytic changes, glymphatic influx and efflux are severely impaired by 3 days post-BCAS, remain suppressed at 10 days, and recover to baseline by 30 days, mirroring the transient reduction in CBF.61 A significant linear correlation between glymphatic influx and cerebral perfusion suggests that early astrocyte dysfunction, particularly in the regulation of perivascular fluid dynamics, contributes to glymphatic transport failure.61 Although no overt changes in aquaporin-4 (AQP4) localization are seen at 1-month post-BCAS,62 these findings imply that early glymphatic dysfunction arises more from astrocyte reactivity and altered cerebrovascular dynamics than from gross mislocalization of AQP4.
Despite these insights, major gaps remain. Cerebral perfusion pressure and edema have not been measured in BCAS models, leaving key aspects of the fluid clearance landscape unresolved. Determining how astrocyte reactivity, vascular dynamics, and glymphatic dysfunction interact across time remains an important future direction.
Border-Associated Macrophages in Chronic Hypoperfusion
Border-associated macrophages (BAMs) reside in the meninges, perivascular spaces, and choroid plexus, positioning them at key CNS interfaces where vascular injury signals are first encountered. While unexplored in BCAS, single-cell transcriptomic studies from ischemic brain injury reveal that BAMs exhibit distinct transcriptional profiles from parenchymal microglia, with upregulation of genes associated with antigen presentation, chemokine signaling, and interferon responses in the acute phase post-stroke, while these macrophages also show dynamic transcriptional reprogramming across time, suggesting that BAMs are highly plastic and sensitive to cerebrovascular stress.63 Moreover, BAMs have been shown to rapidly respond to ischemic insults by promoting granulocyte infiltration and contributing to vascular leakage.64 Their positioning at CNS interfaces, especially within perivascular Virchow-Robin spaces, places them at the frontline of sensing and amplifying vascular injury signals. Infiltration of meningeal BAMs into these perivascular spaces has been documented following stroke and is associated with matrix remodeling, fibroblast activation, and changes in vessel-associated immune signaling65 (Figure 4).
Given this evidence from ischemia models, it is plausible that BCAS-induced hypoperfusion similarly activates BAM populations, initiating or exacerbating early inflammatory and barrier-destabilizing events, yet no current studies have applied cell-type-specific sequencing, imaging, or fate-mapping techniques to BAMs in the BCAS model. Future studies addressing this gap could reveal whether BAMs serve as amplifiers of vascular inflammation or coordinators of repair during subacute and chronic hypoperfusion, as BAMs remain an underexplored but potentially critical cellular player in the neurovascular cascade initiated by chronic cerebral hypoperfusion. Integrating BAM-specific analyses into BCAS research could uncover novel immune mechanisms of vascular cognitive impairment and suggest new immunomodulatory targets to preserve neurovascular function.
Barrier Dysfunction in BCAS
Barrier systems that normally preserve brain homeostasis become progressively compromised in chronic cerebral hypoperfusion. Both the blood-brain barrier (BBB) and blood-CSF barrier (BCSFB) are disrupted through overlapping inflammatory mechanisms, converting the brain from an immune-privileged site into one permissive to peripheral immune infiltration (Figure 5).
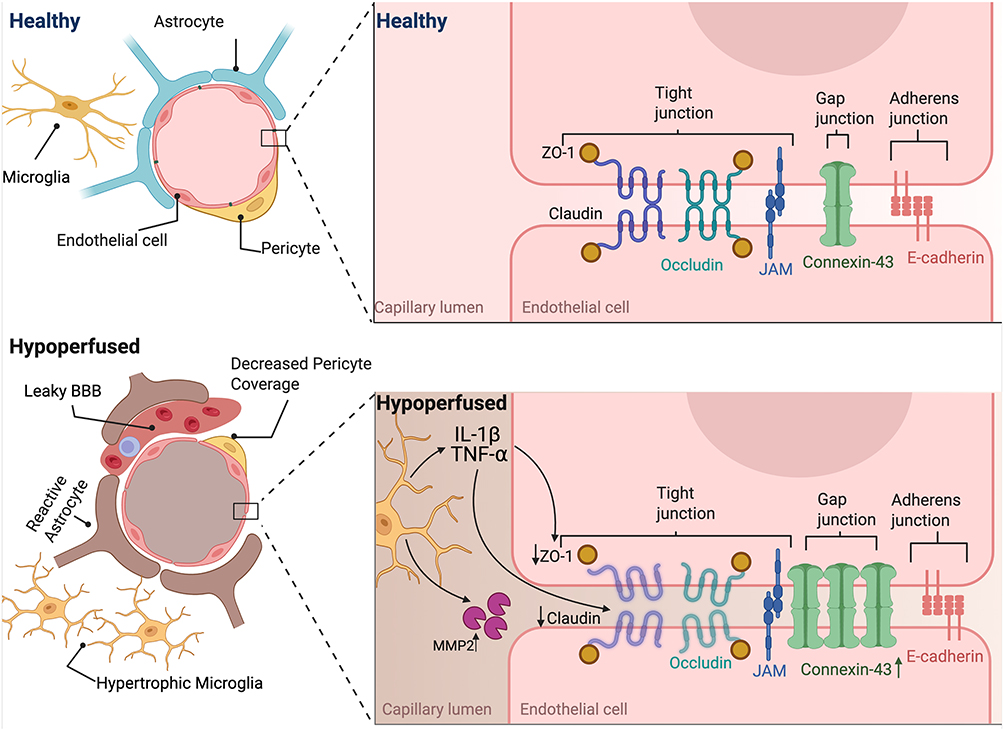 |
Figure 5 Blood-brain barrier dysfunction in chronic cerebral hypoperfusion induced by BCAS. Under healthy conditions, the blood-brain barrier (BBB) is maintained by endothelial cells joined through tight junctions (claudin-5, occludin, and ZO-1), gap junctions (connexin-43), and adherens junctions (E-cadherin, VE-cadherin), reinforced by pericytes, astrocytic end-feet, and microglia. This organization restricts paracellular permeability and preserves CNS homeostasis. Following BCAS-induced hypoperfusion, the BBB undergoes progressive disruption. Endothelial cells show reduced claudin-5, occludin, and ZO-1 expression, driven in part by TNF-α– and IL-1β–mediated inflammatory signaling and MMP-2 activity, resulting in junctional degradation and barrier instability. Concomitantly, pericyte coverage decreases, astrocytes become reactive, and microglia adopt hypertrophic morphologies, further amplifying inflammatory stress. These changes lead to increased BBB leakage, impaired vascular regulation, and reduced glymphatic clearance, establishing barrier dysfunction as an early and central feature of chronic cerebral hypoperfusion. |
The BBB, formed by specialized endothelial cells connected by tight junctions composed of transmembrane proteins including claudin-5, occludin, and zonula occludens-1 (ZO-1), along with adherens junctions containing VE-cadherin and β-catenin, creates a highly selective barrier that restricts paracellular transport.51 These endothelial cells exhibit unique characteristics including reduced pinocytotic activity, absence of fenestrations, and expression of specific efflux transporters such as P-glycoprotein.51 The BBB is further supported by pericytes embedded within the basement membrane that provide structural support and regulate capillary diameter, as well as astrocytic end-feet that form a continuous layer around the capillaries and contribute to BBB maintenance through secretion of factors like angiopoietin-1 and sonic hedgehog51,52,66 (Figure 5).
The BCSFB, composed of choroid plexus epithelial cells linked by tight junctions containing claudin-1, claudin-5, and claudin-11, forms a polarized monolayer that exhibits distinct apical and basolateral membrane domains with specialized transport systems that similarly restricts exchange between the blood and cerebrospinal fluid.67 Under normal conditions, these barriers maintain an immune-privileged environment through selective permeability mechanisms.68
In BCAS, barrier disruption develops in a time- and severity-dependent manner. By 14 days post-BCAS, Evans Blue dye extravasation reveals significant BBB disruption, which is not observed at 7 days.69 The 0.16 mm coil produces greater BBB disruption than the 0.18 mm coil, with larger cortical areas showing 2000 K-dextran leakage, coinciding with robust astrocyte and microglial reactivity.55 Mechanistically, BBB breakdown is driven by the degradation of tight junction proteins.9,21,34
Claudin-5, is downregulated both transcriptionally and post-transcriptionally in white matter by 28 days post-BCAS.48 Although some studies have reported region-specific variability or delayed claudin-5 changes,21,62,70 these differences likely reflect model-specific dynamics. Claudin-5 expression decreases are detected as early as 3 days in the corpus callosum, with reductions maintained at later time points, coinciding with increased TNF-α expression.48 Supporting evidence from in vitro studies shows that claudin-5 is also acutely reduced under oxygen-glucose deprivation in an ASK1-dependent manner48 (Figure 5).
Occludin, another integral membrane tight junction protein, is also downregulated in response to chronic hypoperfusion. In vivo, mRNA decreases are evident by 14 days post-BCAS in cortex and striatum, with protein loss confirmed by immunostaining and Western blot from Days 15 to 42.21 In vitro, TNF-α reduces occludin expression.49 From a therapeutic perspective, intermittent fasting and dl-3-n-butylphthalide (NBP) preserve occludin expression and improve BBB integrity.70,71 As with claudin-5, however, variability across regions and strains has been reported, likely reflecting model-specific dynamics72 (Figure 5).
ZO-1, a cytoplasmic scaffolding protein that links transmembrane junction proteins to the actin cytoskeleton, is especially susceptible to inflammatory regulation. ZO-1 gene expression is significantly reduced in white matter by 28 days post-BCAS,49 with protein levels falling as early as Days 1 and 3 and remaining depressed through Day 42.73 This decline is driven by TNF-α–induced upregulation of miR-501-3p, which directly binds the 3′-UTR of ZO-1 and reduces its expression, as confirmed in vitro.49 In vivo inhibition of miR-501-3p suppresses ZO-1 levels, reduces Evans Blue leakage, and improves cognitive function, highlighting ZO-1 as a key downstream effector of inflammatory microRNA signaling at the BBB49 (Figure 5).
Together, these findings support a model in which TNF-α–driven upregulation of miR-501-3p orchestrates the coordinated downregulation of claudin-5, occludin, and ZO-1, contributing to BBB compromise in the setting of chronic cerebral hypoperfusion. While TNF-α has been clearly implicated as a key upstream inflammatory signal in this pathway, it remains unknown whether this response is specific to TNF-α or if other pro-inflammatory cytokines, such as interleukin-1β (IL-1β), can elicit similar effects on tight junction protein regulation. To date, the direct impact of IL-1 on this pathway has not been tested.
Matrix metalloproteinases (MMPs) are zinc-dependent endopeptidases that degrade extracellular matrix (ECM) components and tight junction proteins, thereby promoting BBB disruption in chronic cerebral hypoperfusion.74 Among these, MMP-2 has emerged as a central mediator of vascular injury in the BCAS model. MMP-2 is upregulated in astrocytes and microglia/macrophages localized around arterioles in white matter, contributing to basal lamina degradation and ECM remodeling.75 It efficiently digests structural components such as type IV collagen, fibronectin, and gelatin, and has been shown to degrade myelin more effectively than MMP-9, implicating it directly in white matter pathology.70,76 Genetic deletion of MMP-2 or pharmacological inhibition with AG3340 significantly attenuates BBB leakage, glial activation, and white matter lesions in BCAS model75 Figure 5).
BCAS also activates membrane-type 1 MMP (MT1-MMP), an upstream activator of pro-MMP-2, further reinforcing the MT1-MMP–MMP-2 axis in barrier disruption. In the cortex, MT1-MMP is significantly elevated at 15 and 30 days post-BCAS, and pro-MMP-2 increases by day 30. In contrast, pro-MMP-9 levels decrease at both time points, suggesting isoform-specific regulation. Similar patterns are seen in the hippocampus and cerebellum, with MT1-MMP and pro-MMP-2 upregulated at day 30, while pro-MMP-9 shows significant downregulation in the cerebellum.70 These regionally distinct patterns highlight how chronic cerebral hypoperfusion differentially engages MMP regulatory programs across the brain. Intermittent fasting was shown to attenuate BCAS-induced pro-MMP-2 expression in all three brain regions but did not reverse cortical MT1-MMP elevation, suggesting only partial suppression of MMP-driven pathology through metabolic intervention.70
Notably, MMP-9 appears to play a more context-dependent role. While early transient upregulation has been observed in the cerebral white matter, originating from oligodendrocyte precursor cells (OPCs) and linked to early BBB disruption,76 other studies report negligible MMP-9 induction. For example, Nakaji et al (2006) found no significant MMP-9 immunoreactivity or enzymatic activity following BCAS, and Rajeev et al (2022) observed decreased pro-MMP-9 expression across multiple regions. These discrepancies likely reflect variations in model parameters, time points, and brain region analyzed.
Peripheral Immune Cell Infiltration Pathways in BCAS
Barrier disruption creates multiple pathways for peripheral immune cell entry into the brain during chronic cerebral hypoperfusion. BBB disruption allows circulating monocytes, T-cells, and neutrophils to cross directly from blood into brain parenchyma through compromised tight junctions and increased transcytosis. Upregulation of adhesion molecules including ICAM-1, VCAM-1, and selectins on activated endothelial cells facilitates immune cell adhesion and transmigration across the BBB.77 Additionally, connexin 43 gap junction alterations contribute to barrier dysfunction by disrupting intercellular communication between endothelial cells and supporting cells.78,79 The pattern of immune cell infiltration in BCAS models reveals important regional and temporal specificities that differ from classical neuroinflammation paradigms. In the brain, a significant increase in CD3+ T-cells and neutrophils was detected at 8 weeks (but not 4 weeks) post-BCAS, accompanied by decreased anti-inflammatory CD11b+CD45+hi/CD206+ and CD11b+CD45+lo/CD206+ myeloid cells.29 These changes were not seen in the choroid plexus.29
The earliest detectable immune responses occur at the pial vessel level, where in vivo two-photon imaging studies revealed leukocyte rolling and adhesion within 24 hours of BCAS surgery, primarily in the pial venules.80 These interactions occurred without detectable alterations in vessel structure, astrocytes, or pericytes, suggesting that immune cell activation is an early and potentially primary mechanism of vascular engagement, rather than a downstream consequence of structural damage. In addition to rolling and adhesion, transient leukocyte plugging of capillaries was observed under chronic cerebral hypoperfusion.80 These plugs, although short-lived (typically <120 seconds), caused temporary stagnation of plasma flow, often in the absence of erythrocytes, implying the potential for exacerbating parenchymal hypoxia.80 Notably, platelet activation, which is a hallmark of focal cerebral ischemia, was not observed during the same period, further distinguishing the immune profile of chronic hypoperfusion.80 While the frequency of rolling and adhesion decreased over time, it remained significantly elevated in pial venules at two weeks post-BCAS. Together, these findings suggest that leukocyte activation is among the earliest cerebrovascular responses to chronic hypoperfusion and may initiate subsequent stress responses within the neurovascular unit.
Although barrier dysfunction facilitates peripheral immune cell entry, the extent to which this contributes to downstream injury in chronic cerebral hypoperfusion remains incompletely defined. To explore the role of Tregs in modulating injury, anti-CD25 antibody was used to deplete regulatory T cell (Treg) following BCAS.81 While this intervention produced measurable effects on inflammatory markers, its impact on structural and behavioral outcomes was limited. Anti-CD25 treatment significantly increased expression of pro-inflammatory proteins IL-1β and TNF-α, elevated iNOS, and reduced levels of the anti-inflammatory marker CD206. Microglial phenotype also shifted, with increased Iba1⁺CD16/32⁺ (pro-inflammatory) and decreased Iba1⁺CD206⁺ (anti-inflammatory) populations. However, effects on myelin integrity were modest, with no further worsening of MBP, MAG, and Olig2 levels or white matter lesion scoring via Luxol Fast Blue staining, or g-ratio measurements from electron microscopy compared to BCAS controls. Behaviorally, anti-CD25 treatment also showed no further worsening of BCAS-induced deficits on latency to platform during water maze testing and on time spent in the target quadrant in the probe trial, or novel object recognition performance.81 Together, these results suggest that while Treg depletion modestly amplifies inflammatory signaling and alters glial phenotype, it produces only marginal additional effects on white matter injury and cognitive outcomes in this model.
The BCSFB provides an alternative route for immune cell entry, as compromised choroid plexus epithelial barriers allow peripheral immune cells to enter the CSF compartment through structural changes including elevation and dislocation of tight junction proteins claudin-1 and claudin-5, regional upregulation of CSF secretory ionic regulatory proteins SPAK-NKCC1, and local activation of the NF-κB inflammatory cascade. Significant increases in CD3+ T-cells and neutrophils are observed in the choroid plexus at 4 weeks post-BCAS, demonstrating active inflammatory responses induced by chronic hypoperfusion.29 The choroid plexus emerges as a critical site of immune cell accumulation in chronic hypoperfusion. This inflammatory shift in the choroid plexus suggests that the BCSFB represents a more permeable entry route for immune cells compared to the parenchymal BBB during chronic hypoperfusion. The temporal delay in choroid plexus infiltration compared to pial vessel activation indicates that different anatomical compartments respond with distinct kinetics to chronic hypoperfusion.
Role of Cytokine-Specific Barrier Effects in BCAS
The pro-inflammatory cytokines generated during the initial neuroinflammatory cascade each contribute distinct but overlapping mechanisms to barrier dysfunction, together damaging barrier integrity.82,83 Individual cytokines demonstrate distinct receptor-mediated effects, temporal activation patterns, and regional vulnerability profiles that determine the specific characteristics of barrier dysfunction in chronic cerebral hypoperfusion.1 However, it is important to note that our understanding of cytokine changes in BCAS models remains limited, with most studies focusing on a small subset of inflammatory mediators and relatively few directly examining their causal roles in barrier dysfunction.
IL-1β demonstrates how individual cytokines create multifaceted barrier dysfunction through direct vascular targeting, via the IL-1 receptor (IL-1R1), which is highly expressed on brain endothelial cells, making cerebral vasculature particularly susceptible to IL-1β-mediated damage.84,85 In BCAS models, IL-1β levels increase significantly in the cerebellum as early as 7 days post-surgery and throughout the whole brain by 2 weeks post-surgery, with these elevations corresponding to increased inflammasome activity, including NLRP1, NLRP3, AIM2, and caspase-1 activation.82,86 IL-1β signaling through endothelial IL-1R1 directly disrupts tight junction proteins, including occludin and ZO-1, substantially increasing paracellular permeability,84 while simultaneously activating brain endothelial cells to upregulate adhesion molecules VCAM-1 and ICAM-1 through NF-κB mechanisms, facilitating leukocyte infiltration at venous sites where IL-1R1 expression peaks.84 The cytokine also activates multiple matrix metalloproteinases (MMP-1, −2, −3, −9) that degrade both tight junction components and basal lamina proteins, while stimulating VEGF release that further increases vascular permeability through endothelial protein phosphorylation,84 though these specific MMP and VEGF mechanisms have not yet been directly evaluated in BCAS models. As a potent vasodilator, chronic IL-1β elevation paradoxically contributes to sustained vasodilation that impairs normal vascular reactivity and autoregulation,87 reducing the capacity for appropriate vasoconstriction when needed and further compromising the neurovascular unit’s ability to maintain optimal perfusion pressure and respond to metabolic demands, although this vasodilatory mechanism has not been directly tested in BCAS models. Beyond direct vascular effects, IL-1β creates positive feedback loops where infiltrating leukocytes release additional IL-1β, perpetuating chronic inflammation and progressive barrier breakdown,20,85,88,89 and brain endothelial cells following injury may also produce IL-1β, suggesting autocrine regulation mechanisms. In white matter regions, IL-1β compounds vascular damage by promoting oligodendrocyte death through glutamate excitotoxicity while impeding oligodendrocyte progenitor cell recruitment, creating both immediate barrier dysfunction and long-term repair impairment that explains the progressive nature of white matter lesions observed in chronic hypoperfusion models.90,91
Working in concert with IL-1β, tumor necrosis factor-alpha (TNF-α) functions as a master regulator of endothelial dysfunction and BBB breakdown in chronic cerebral hypoperfusion (chronic cerebral hypoperfusion), acting through tightly linked oxidative stress and post-transcriptional pathways. Across multiple studies, TNF-α gene expression increases rapidly in white matter as early as 24–72 hours after BCAS, and elevated protein levels are detected in cerebral endothelial cells by 72 hours,48,49,58 with these early and sustained increases coinciding with the timing of BBB compromise, although most supporting evidence remains indirect. Direct TNF-α stimulation of cultured brain endothelial cells recapitulates key aspects of the in vivo response, including the downregulation of tight junction proteins (claudin-5, ZO-1, occludin) and activation of the ASK1-p38 MAPK cascade,48 which together promote tight junction loss and amplify inflammatory gene expression. A key post-transcriptional mediator of this response is miR-501-3p, which is upregulated in white matter endothelial cells by 48 hours post-BCAS,49 with TNF-α inducing miR-501-3p in vitro and overexpression of this microRNA reducing ZO-1 levels, while anti-miR-501-3p treatment in vivo preserves barrier integrity, suggesting that this TNFα miR-501-3p pathway may underlie persistent BBB disruption even after TNF-α levels normalize.49 Notably, this mechanism appears regionally specific, with increases in TNF-α and miR-501-3p primarily restricted to white matter, despite comparable glial activation in the hippocampus,49 and this spatial specificity may contribute to the heightened vulnerability of white matter to hypoperfusion-induced injury. Such studies would be essential to confirm the causal role of TNF-α and assess the therapeutic potential of targeting TNF-α signaling in the context of chronic cerebral hypoperfusion. Together, these findings point to TNF-α as a central upstream mediator of BBB dysfunction following BCAS, acting through both transcriptional (ASK1-p38) and post-transcriptional (miR-501-3p) mechanisms, though validation through direct intervention studies is still needed to establish TNF-α as a therapeutic target for mitigating vascular cognitive impairment.
While previously thought to act primarily in late-stage inflammation, recent studies reveal that IL-6 mRNA expression increases as early as 14 days post-BCAS in the hippocampus and white matter,81,92 with these increases being exacerbated by depletion of Tregs, indicating that loss of immunoregulatory control sensitizes the brain to early IL-6-driven inflammatory responses.81 In sham-operated mice, Treg depletion alone is sufficient to elevate IL-6, suggesting that IL-6 participates in both injury-induced and homeostatic inflammation, and together, these data revise the prior view of IL-6 as a late-acting cytokine, instead positioning it as an early amplifier of inflammation under permissive conditions. In contrast, more delayed elevations of IL-6, such as those observed at 30 days post-BCAS in A2A receptor knockout mice, likely reflect a sustained or unresolved inflammatory state,93 where in this chronic phase, IL-6 signaling via the gp130/STAT3 pathway may reinforce BBB disruption by promoting inflammatory gene expression and inhibiting tight junction repair. IL-6-mediated activation of STAT3 also intersects with oxidative and cytokine-driven pathways, potentiating NF-κB signaling and contributing to a cycle of non-resolving neuroinflammation.94 IL-6 additionally facilitates the recruitment and activation of peripheral immune cells by inducing adhesion molecules and chemokines that enhance leukocyte-endothelial interactions, sustaining inflammatory cell infiltration into perivascular and parenchymal compartments, a function that is consistent with IL-6’s role as a secondary amplifier of immune responses, where its elevation helps maintain leukocyte presence and prolongs exposure of the NVU to inflammatory mediators.81,94 Despite the growing evidence of IL-6 upregulation in both acute and chronic phases of hypoperfusion, no studies to date have directly manipulated IL-6 signaling in the BCAS model using neutralizing antibodies, receptor antagonists, or genetic approaches, leaving open critical questions about causality and the precise role of IL-6 in BBB disruption.
While multiple cytokines have been implicated in the pathogenesis of white matter injury and neurovascular dysfunction following chronic cerebral hypoperfusion induced by BCAS, direct manipulation of individual cytokines in this model has been rare. Most studies implicate TNF-α based on its early and sustained upregulation in wild-type mice from 24 hours to 4 weeks post-BCAS, with expression localized to cerebral endothelial cells and known to drive downstream activation of the ASK1–p38 cascade and miR-501-3p expression that compromise barrier integrity.48 However, selective TNF-α inhibitors such as XPro1595, which target soluble TNF-α without affecting transmembrane TNF signaling, have yet to be tested in the BCAS model.95 Similarly, IL-6 expression is elevated 14 days post-BCAS and is significantly increased in mice with regulatory T cell depletion,81 as well as in A2A receptor knockout mice at 30 days.93 However, no studies have directly targeted IL-6 or its receptor using antibodies or genetic approaches. IL-1β is also consistently upregulated during BCAS, but its role has only been inferred from indirect modulation through interventions such as colchicine, which reduces its expression in vitro and in vivo.96 The only cytokine to be directly manipulated in the BCAS model is TGF-β1, where conditional knockout in microglia exacerbated demyelination and neuroinflammation, establishing a causal link between microglial TGF-β1 and white matter protection.83 Other cytokines and chemokines, including IFN-stimulated genes, CCL5, CXCL10, and CCL21, are transcriptionally elevated during hypoperfusion, particularly in white matter and glial populations, but remain untested through selective intervention.97 Collectively, these findings highlight the need for targeted, mechanistic studies that directly manipulate cytokine signaling to validate their roles in BCAS-induced vascular and cognitive impairment.
Microglial Dynamics and Functional Contributions in Chronic Cerebral Hypoperfusion
Microglia, the brain’s resident immune cells, undergo rapid and region-specific activation following BCAS, representing one of the earliest and most sustained cellular responses to chronic hypoperfusion. Early post-BCAS time points reveal significant increases in Iba1-positive microglia exhibiting hypertrophic morphology, particularly in vulnerable white matter regions such as the corpus callosum and hippocampus, with changes evident as early as 3 days and intensifying throughout the first weeks.35,75,98 Importantly, microglial activation demonstrates spatial heterogeneity that reflects the severity of hypoperfusion, with more pronounced amoeboid, phagocytic microglia observed on the severely stenosed side and hyper-ramified morphologies on the less restricted side, indicating that microglial responses scale with local ischemic pressure.99
By 2–3 weeks post-BCAS, microglial activation intensifies with co-expression of inflammatory markers including CD68, MHCII, and CD16/32, alongside increased phagocytic phenotypes and upregulation of inflammasome components such as AIM2 and NLRP3.25,82 Single-nucleus RNA sequencing and chromatin accessibility studies confirm the emergence of neuroinflammatory and interferon-response gene signatures by 3 weeks, with upregulation of immune and phagocytic pathways, including neuroinflammation signaling and phagosome formation, predominantly in microglial populations.83,100 Notably, markers of both pro-inflammatory (CD16/32, MHCII, CD68) and anti-inflammatory or reparative phenotypes (CD206, Arg1) are elevated at 14–21 days, suggesting the co-existence of distinct functional microglial subsets rather than a uniform activation state81,82 (Figure 6).
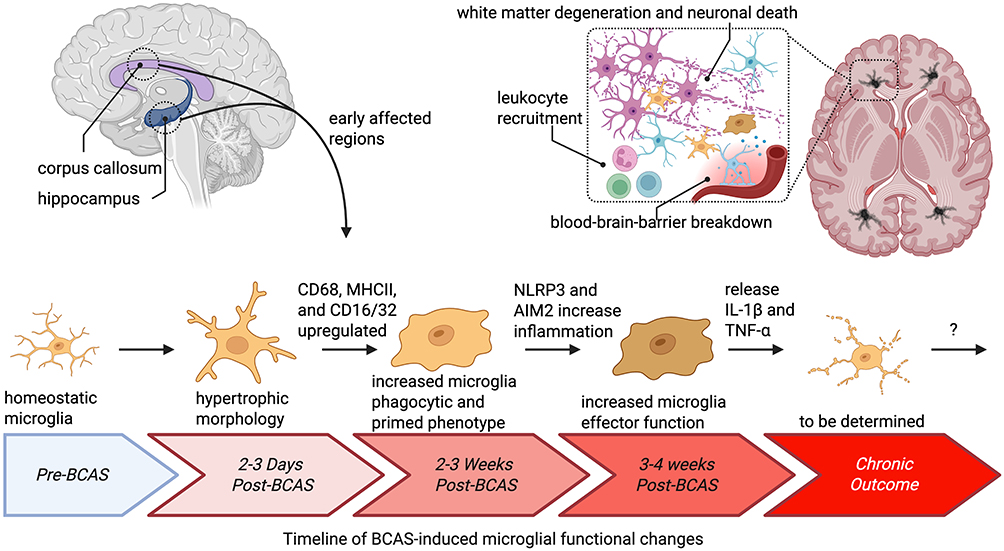 |
Figure 6 Timeline of BCAS-induced microglial functional changes. Microglia are among the earliest and most dynamic responders to chronic cerebral hypoperfusion. In pre-BCAS conditions, microglia maintain a homeostatic, ramified morphology. By 2–3 days post-BCAS, microglia in vulnerable regions such as the corpus callosum and hippocampus exhibit hypertrophic morphologies, reflecting early activation. Over the next 2–3 weeks, microglia acquire a primed phenotype, with upregulation of CD68, MHCII, and CD16/32, increased phagocytic activity, and induction of inflammasome components including NLRP3 and AIM2. By 3–4 weeks, these changes evolve into heightened effector functions with sustained release of pro-inflammatory cytokines IL-1β and TNF-α, contributing to BBB breakdown, leukocyte recruitment, white matter degeneration, and neuronal injury. Transcriptomic profiling further reveals upregulation of both pro- and anti-inflammatory markers, suggesting the coexistence of distinct microglial subsets during this period. The chronic outcomes of these responses remain incompletely defined, but accumulating evidence suggests that microglia contribute both to pathological injury and to repair processes, highlighting their dual role in the progression of hypoperfusion-induced neuroinflammation. |
Multiple studies have directly manipulated microglial activity in the BCAS model, revealing their critical role in both compensatory and pathological responses to hypoperfusion. Pharmacological interventions targeting pro-inflammatory signaling pathways have demonstrated beneficial effects and colchicine treatment suppressing TAK1/MAPK/NF-κB signaling to reduce microglial proliferation and inflammatory cytokine production.96 Genetic approaches have similarly revealed the therapeutic potential of microglial modulation, with conditional knockout of microglial angiotensin II type 1 receptors preventing Iba1+ cell accumulation and suppressing pro-inflammatory gene expression,101 while TRPM2 deficiency suppressed pro-inflammatory microglial markers and reduced cytokine release.25 Pathway-specific interventions have further highlighted the importance of transcriptional regulation, as inhibiting PU.1, a transcription factor regulating interferon response genes, reduced microglial hyperactivation and neuroinflammatory gene expression at 3 weeks post-BCAS.102
Beyond anti-inflammatory approaches, several studies have focused on enhancing beneficial microglial functions, particularly phagocytosis and growth factor expression. MFG-E8 overexpression improved microglial phagocytic clearance and increased IGF-1 levels, promoting white matter repair,103 while salidroside treatment induced a polarization shifts in microglia phenotypes, as demonstrated by decreased CD16+ and increased Arg1+ microglia.104 Even non-pharmacological interventions have shown efficacy, with enriched environmental housing reducing microglial numbers across hippocampal subfields105 and high-frequency repetitive transcranial magnetic stimulation suppressing microglial activation in the hippocampus.92
Nearly all manipulations targeting microglia in the context of chronic cerebral hypoperfusion have yielded beneficial outcomes when aimed at dampening excessive inflammatory signaling or promoting anti-inflammatory phenotypes, leading to improved neuroinflammatory profiles, preserved white matter, and enhanced cognitive function. Conversely, interventions that disrupt anti-inflammatory signaling tend to exacerbate microglial activation and worsen pathological progression. Importantly, no studies have yet employed microglial depletion strategies in the BCAS model, leaving unresolved whether microglia are ultimately harmful or if they play a necessary compensatory role across disease stages. Given that chronic cerebral hypoperfusion commonly coexists with amyloid and tau pathology in the elderly, future work should reconsider whether restoring microglial homeostatic functions or even enhancing phagocytic capacity might offer advantages over broad immunosuppression. Along these lines, strategies that promote debris clearance and immune resolution, rather than blunt activation, may more effectively support neurovascular repair. Furthermore, combining microglial modulation with senolytic therapies to eliminate senescent cells could offer a synergistic approach to rejuvenate the aged brain and preserve cognitive function.
Future Directions for Exploring the Role of Inflammation in Chronic Cerebral Hypoperfusion
While chronic cerebral hypoperfusion triggers complex inflammatory cascades that initially attempt cerebrovascular compensation through angiogenic signaling and collateral recruitment, the mechanisms governing the transition from protective to pathological inflammatory states remain poorly understood. Early responses demonstrate coordinated attempts to restore perfusion through VEGF-mediated angiogenesis and protective astrogliosis,9,106 yet the temporal triggers and molecular switches that transform these beneficial responses into sustained destructive processes represent critical knowledge gaps in BCAS research. Moreover, the field lacks a comprehensive understanding of what determines whether inflammatory responses resolve toward homeostasis or progress to chronic activation states. Future studies should seek to understand what specific temporal thresholds exist for protective microglial and astrocytic responses to become irreversibly pathological. How do regional differences in hypoperfusion severity influence the compensatory-to-maladaptive transition? What molecular checkpoints control the switch from beneficial angiogenesis to aberrant vessel formation with compromised function? Current studies describe the endpoints of chronic inflammation but provide limited insight into the decision points that determine inflammatory fate.
While self-perpetuating inflammatory cycles involving NF-κB, ASK1-p38, and oxidative stress pathways have been identified,107,108 the hierarchical relationships between these networks and their relative contributions to chronic pathology remain unclear. The field needs systematic temporal mapping studies that track the sequential activation of inflammatory pathways, identify the primary drivers versus secondary amplifiers of chronic inflammation, and determine whether specific intervention windows exist for redirecting maladaptive responses back toward resolution. Furthermore, it remains unknown whether age-related microglial priming represents a quantitative amplification of existing pathways or qualitatively different inflammatory programs that require distinct therapeutic approaches.
Several underexplored areas may hold the key to advancing this field. First, while much focus has been placed on glial and endothelial contributions to inflammation, the role of peripheral immune cells, including monocytes, T cells, and neutrophils entering through the leptomeninges or choroid plexus, remains poorly defined in BCAS models. Regulatory T cells, for example, modulate cytokine production,81 yet how peripheral immune tone influences transition toward chronicity is unknown. Second, transcriptional and epigenetic regulators such as microRNAs, and histone modifiers likely shape the long-term inflammatory phenotype but have yet to be profiled in a temporal, cell-type-specific manner. Third, neurovascular unit components may undergo asynchronous transitions, with endothelial cells or astrocytes reaching chronic dysfunction earlier or later than microglia or pericytes, and this temporal misalignment could destabilize compensatory networks.
Addressing these gaps has direct implications for the clinical translation of BCAS research and for the development of disease-modifying therapies in chronic cerebrovascular disease. By modeling graded, long-term reductions in cerebral blood flow, BCAS provides a platform to identify early, potentially reversible stages of neurovascular unit dysfunction that precede overt cognitive decline, thereby informing strategies for risk stratification and intervention in patients with vascular cognitive impairment or mixed dementia.10 Future therapeutic approaches informed by BCAS findings are likely to extend beyond symptomatic management and instead target mechanisms that preserve neurovascular resilience, including stabilization of endothelial and pericyte function, reinforcement of barrier integrity, and modulation of maladaptive inflammatory amplification while sparing protective immune responses.33,73 The model is also well suited for evaluating therapies aimed at restoring neurovascular coupling, improving microvascular perfusion, and selectively reprogramming glial or immune cell phenotypes rather than broadly suppressing inflammation.29 Crucially, integrating BCAS-derived molecular and imaging signatures with clinically accessible biomarkers, such as perfusion imaging, circulating inflammatory mediators, or markers of barrier disruption, could enable earlier diagnosis and the rational timing of interventions.41 Together, these advances position BCAS research as a critical translational bridge between mechanistic studies of chronic hypoperfusion and precision therapeutic strategies designed to delay or prevent irreversible neurovascular and cognitive decline.
Vascular pathology itself may also act as both a driver and a product of chronic inflammation, as continued reductions in perfusion, BBB leakage, and altered transporter function may reinforce immune activation. Additionally, while current work has focused on pathway-level analyses, the field lacks reliable biomarkers that signal when the tipping point from compensation to chronicity has occurred. Identifying such inflection markers through transcriptomic, proteomic, or metabolomic profiling could enable time-targeted interventions. Critically, few studies have directly tested mechanisms regulating inflammation following BCAS, with most approaches indirectly drawing assumptions regarding the role of inflammation following interventions with multiple targets. Understanding these temporal dynamics and identifying the molecular mechanisms that control inflammatory resolution versus chronicity will be essential for developing targeted therapies that preserve the brain’s compensatory capacity while preventing destructive inflammatory persistence in chronic cerebrovascular disease. Clarifying these dynamics will be key for transforming the current descriptive understanding of chronic inflammation into a temporally informed therapeutic framework that identifies both windows of opportunity and mechanisms of irreversible transition.
Declaration of Generative AI Usage in Scientific Writing
During the preparation of this work, the author(s) used ChatGPT and Claude to assist in evaluating the original text for clarity, completeness, and style. These tools provided editorial suggestions and recommendations; however, the author(s) reviewed and edited all content as necessary. The author(s) take full responsibility for the final content of the published article.
Abbreviations
ABCC9, ATP-binding cassette subfamily C member 9; ACAS, asymmetric common carotid artery surgery; AD, Alzheimer’s disease; AIM2, absent in melanoma 2; APOE4, apolipoprotein E4; AQP4, aquaporin-4; Arg1, arginase 1; ASK1, apoptosis signal-regulating kinase 1; BAMs, border-associated macrophages; BBB, blood-brain barrier; BCAS, bilateral common carotid artery stenosis; BCCAO, bilateral common carotid artery occlusion; BCSFB, blood-CSF barrier; CBF, cerebral blood flow; CCL5, C-C motif chemokine ligand 5; CCL21, C-C motif chemokine ligand 21; CD, cluster of differentiation; CNS, central nervous system; CSF, cerebrospinal fluid; CXCL10, C-X-C motif chemokine ligand 10; CXCR2, C-X-C motif chemokine receptor 2; ECM, extracellular matrix; ERK5, extracellular signal-regulated kinase 5; FDA, Food and Drug Administration; GCAS, gradual common carotid artery stenosis; GFAP, glial fibrillary acidic protein; gp130, glycoprotein 130; Iba1, ionized calcium-binding adapter molecule 1; ICAM-1, intercellular adhesion molecule-1; IGF-1, insulin-like growth factor 1; IL-1β, interleukin-1 beta; IL-1R1, interleukin-1 receptor type 1; IL-6, interleukin-6; iNOS, inducible nitric oxide synthase; JNK, c-Jun N-terminal kinase; KLF2, Kruppel-like factor 2; LCN2, lipocalin 2; LIF, leukemia inhibitory factor; MAG, myelin-associated glycoprotein; MAPK, mitogen-activated protein kinase; MBP, myelin basic protein; MFG-E8, milk fat globule-EGF factor 8; MHCII, major histocompatibility complex class II; MMP, matrix metalloproteinase; MRI, magnetic resonance imaging; MT1-MMP, membrane-type 1 matrix metalloproteinase; NBP, dl-3-n-butylphthalide; NF-κB, nuclear factor kappa B; NIH, National Institutes of Health; NLRP1, NOD-like receptor protein 1; NLRP3, NOD-like receptor protein 3; NVU, neurovascular unit; OPC, oligodendrocyte precursor cell; PDGFR-β, platelet-derived growth factor receptor beta; PPARγ, peroxisome proliferator-activated receptor gamma; ROS, reactive oxygen species; SPAK-NKCC1, STE20/SPS1-related proline/alanine-rich kinase-Na-K-Cl cotransporter 1; STAT3, signal transducer and activator of transcription 3; TAK1, transforming growth factor beta-activated kinase 1; TBI, traumatic brain injury; TGF-β1, transforming growth factor beta 1; TNF-α, tumor necrosis factor-alpha; Treg, regulatory T cell; TRPA1, transient receptor potential ankyrin 1; TRPM2, transient receptor potential melastatin 2; TRPV4, transient receptor potential vanilloid 4; VCAM-1, vascular cell adhesion molecule-1; VCID, vascular contributions to cognitive impairment and dementia; VE-cadherin, vascular endothelial cadherin; VEGF, vascular endothelial growth factor; ZO-1, zonula occludens-1.
Acknowledgments
Illustrations were created in https://BioRender.com.
Author Contributions
MaKayla F. Cox: Conceptualization, Investigation, Writing – original draft, Writing – review & editing, Visualization. Jill M. Roberts: Writing – review & editing, Validation. Ryan K. Shahidehpour: Writing – review & editing, Visualization. David L. Dornbos: Writing – review & editing, Validation. Adam D. Bachstetter: Conceptualization, Supervision, Funding acquisition, Visualization. Writing – review & editing, Project administration. All authors have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work. All authors gave final approval of the version to be published.
Funding
Salary support for this work was provided by the University of Kentucky College of Medicine MD/PhD Program (MFC). Additional salary support was provided by the National Institutes of Health under award numbers T32AG078110 (RKS), R01NS119165 (ADB), R01AG068215 (ADB), R01NS103785 (ADB), R01NS117391 (ADB), and P20GM148326 (ADB).
Disclosure
Dr Jill Roberts reports grants from National Institute of Health, outside the submitted work. Dr David Dornbos III reports consulting from Imperative Care, grants from Siemens Healthineers, consulting from Penumbra, consulting from Stryker, outside the submitted work. The author(s) report no other conflicts of interest in this work.
References
1. Rajeev V, Chai YL, Poh L, et al. Chronic cerebral hypoperfusion: a critical feature in unravelling the etiology of vascular cognitive impairment. Acta Neuropathol Commun. 2023;11(1):93. doi:10.1186/s40478-023-01590-1
2. Ciacciarelli A, Sette G, Giubilei F, Orzi F. Chronic cerebral hypoperfusion: an undefined, relevant entity. J Clin Neurosci. 2020;73:8–23. doi:10.1016/j.jocn.2020.01.026
3. Khan AA, Patel J, Desikan S, et al. Asymptomatic carotid artery stenosis is associated with cerebral hypoperfusion. J Vasc Surg. 2021;73(5):1611–1621e2. doi:10.1016/j.jvs.2020.10.063
4. Park JH, Hong JH, Lee SW, et al. The effect of chronic cerebral hypoperfusion on the pathology of Alzheimer’s disease: a positron emission tomography study in rats. Sci Rep. 2019;9(1):14102. doi:10.1038/s41598-019-50681-4
5. Ma X, Ji C. Remote ischemic conditioning: a potential treatment for chronic cerebral hypoperfusion. Eur Neurol. 2022;85(4):253–259. doi:10.1159/000521803
6. Zou X, Yuan Y, Liao Y, et al. Moyamoya disease: a human model for chronic hypoperfusion and intervention in Alzheimer’s disease. Alzheimers Dement. 2022;8(1):e12285. doi:10.1002/trc2.12285
7. Blevins BL, Vinters HV, Love S, et al. Brain arteriolosclerosis. Acta Neuropathol. 2021;141(1):1–24. doi:10.1007/s00401-020-02235-6
8. Gu J, Sanchez R, Chauhan A, Fazio S, Wong N. Lipid treatment status and goal attainment among patients with atherosclerotic cardiovascular disease in the United States: a 2019 update. Am J Prev Cardiol. 2022;10:100336. doi:10.1016/j.ajpc.2022.100336
9. Duncombe J, Kitamura A, Hase Y, Ihara M, Kalaria RN, Horsburgh K. Chronic cerebral hypoperfusion: a key mechanism leading to vascular cognitive impairment and dementia. Closing the translational gap between rodent models and human vascular cognitive impairment and dementia. Clin Sci. 2017;131(19):2451–2468. doi:10.1042/cs20160727
10. Ishikawa H, Shindo A, Mizutani A, Tomimoto H, Lo EH, Arai K. A brief overview of a mouse model of cerebral hypoperfusion by bilateral carotid artery stenosis. J Cereb Blood Flow Metab. 2023;43(2_suppl):18–36. doi:10.1177/0271678x231154597
11. Wardlaw JM, Smith C, Dichgans M. Small vessel disease: mechanisms and clinical implications. Lancet Neurol. 2019;18(7):684–696. doi:10.1016/S1474-4422(19)30079-1
12. Viticchi G, Falsetti L, Potente E, Bartolini M, Silvestrini M. Impact of carotid stenosis on cerebral hemodynamic failure and cognitive impairment progression: a narrative review. Ann Transl Med. 2021;9(14):1209. doi:10.21037/atm-20-7226
13. Tarumi T, Zhang R. Cerebral blood flow in normal aging adults: cardiovascular determinants, clinical implications, and aerobic fitness. J Neurochem. 2018;144(5):595–608. doi:10.1111/jnc.14234
14. Virani SS, Alonso A, Benjamin EJ, et al. Heart disease and stroke statistics-2020 update: a report from the American Heart Association. Circulation. 2020;141(9):e139–e596. doi:10.1161/CIR.0000000000000757
15. Woodworth DC, Lou JJ, Yong WH, et al. Common neuropathologic change drivers of hippocampal sclerosis of ageing. Brain. 2025;148(7):2400–2411. doi:10.1093/brain/awaf158
16. Ighodaro ET, Shahidehpour RK, Bachstetter AD, et al. A neuropathologic feature of brain aging: multi-lumen vascular profiles. Acta Neuropathol Commun. 2023;11(1):138. doi:10.1186/s40478-023-01638-2
17. Corriveau RA, Bosetti F, Emr M, et al. The science of vascular contributions to cognitive impairment and dementia (VCID): a framework for advancing research priorities in the cerebrovascular biology of cognitive decline. Cell Mol Neurobiol. 2016;36(2):281–288. doi:10.1007/s10571-016-0334-7
18. Gorelick PB, Scuteri A, Black SE, et al. Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the American heart association/american stroke association. Stroke. 2011;42(9):2672–2713. doi:10.1161/STR.0b013e3182299496
19. van Dinther M, Hooghiemstra AM, Bron EE, et al. Lower cerebral blood flow predicts cognitive decline in patients with vascular cognitive impairment. Alzheimers Dement. 2024;20(1):136–144. doi:10.1002/alz.13408
20. Wohleb ES, Patterson JM, Sharma V, Quan N, Godbout JP, Sheridan JF. Knockdown of interleukin-1 receptor type-1 on endothelial cells attenuated stress-induced neuroinflammation and prevented anxiety-like behavior. J Neurosci. 2014;34(7):2583–2591. doi:10.1523/JNEUROSCI.3723-13.2014
21. Roberts JM, Maniskas ME, Bix GJ. Bilateral carotid artery stenosis causes unexpected early changes in brain extracellular matrix and blood-brain barrier integrity in mice. PLoS One. 2018;13(4):e0195765. doi:10.1371/journal.pone.0195765
22. Santisteban MM, Iadecola C. The pathobiology of neurovascular aging. Neuron. 2025;113(1):49–70. doi:10.1016/j.neuron.2024.12.014
23. Gooch J, Wilcock DM. Animal models of vascular cognitive impairment and dementia (VCID). Cell Mol Neurobiol. 2016;36(2):233–239. doi:10.1007/s10571-015-0286-3
24. Dinh QN, Arumugam T. The Bilateral Carotid Artery Stenosis (BCAS) model of vascular dementia. Methods Mol Biol. 2024;2746:67–72. doi:10.1007/978-1-0716-3585-8_5
25. Miyanohara J, Kakae M, Nagayasu K, et al. TRPM2 channel aggravates CNS inflammation and cognitive impairment via activation of microglia in chronic cerebral hypoperfusion. J Neurosci. 2018;38(14):3520–3533. doi:10.1523/jneurosci.2451-17.2018
26. de la Torre JC. Cardiovascular risk factors promote brain hypoperfusion leading to cognitive decline and dementia. Cardiovasc Psychiatry Neurol. 2012;2012:367516. doi:10.1155/2012/367516
27. Poh L, Fann DY, Wong P, et al. AIM2 inflammasome mediates hallmark neuropathological alterations and cognitive impairment in a mouse model of vascular dementia. Mol Psychiatry. 2021;26(8):4544–4560. doi:10.1038/s41380-020-00971-5
28. Kakae M, Tobori S, Morishima M, Nagayasu K, Shirakawa H, Kaneko S. Depletion of microglia ameliorates white matter injury and cognitive impairment in a mouse chronic cerebral hypoperfusion model. Biochem Biophys Res Commun. 2019;514(4):1040–1044. doi:10.1016/j.bbrc.2019.05.055
29. Lin L, Chen Y, He K, et al. Carotid artery vascular stenosis causes the blood-CSF barrier damage and neuroinflammation. J Neuroinflammation. 2024;21(1):220. doi:10.1186/s12974-024-03209-1
30. Shibata M, Ohtani R, Ihara M, Tomimoto H. White matter lesions and glial activation in a novel mouse model of chronic cerebral hypoperfusion. Stroke. 2004;35(11):2598–2603. doi:10.1161/01.Str.0000143725.19053.60
31. Washida K, Hattori Y, Ihara M. Animal models of chronic cerebral hypoperfusion: from mouse to primate. Int J Mol Sci. 2019;20(24):6176. doi:10.3390/ijms20246176
32. Hattori Y, Enmi J, Iguchi S, et al. Substantial reduction of parenchymal cerebral blood flow in mice with bilateral common carotid artery stenosis. Sci Rep. 2016;6:32179. doi:10.1038/srep32179
33. Liu Q, Radwanski R, Babadjouni R, et al. Experimental chronic cerebral hypoperfusion results in decreased pericyte coverage and increased blood-brain barrier permeability in the corpus callosum. J Cereb Blood Flow Metab. 2019;39(2):240–250. doi:10.1177/0271678x17743670
34. Seo JH, Miyamoto N, Hayakawa K, et al. Oligodendrocyte precursors induce early blood-brain barrier opening after white matter injury. J Clin Invest. 2013;123(2):782–786. doi:10.1172/jci65863
35. Saggu R, Schumacher T, Gerich F, et al. Astroglial NF-kB contributes to white matter damage and cognitive impairment in a mouse model of vascular dementia. Acta Neuropathol Commun. 2016;4(1):76. doi:10.1186/s40478-016-0350-3
36. Shibata M, Yamasaki N, Miyakawa T, et al. Selective impairment of working memory in a mouse model of chronic cerebral hypoperfusion. Stroke. 2007;38(10):2826–2832. doi:10.1161/strokeaha.107.490151
37. Nishio K, Ihara M, Yamasaki N, et al. A mouse model characterizing features of vascular dementia with hippocampal atrophy. Stroke. 2010;41(6):1278–1284. doi:10.1161/strokeaha.110.581686
38. Kitaguchi H, Tomimoto H, Ihara M, et al. Chronic cerebral hypoperfusion accelerates amyloid beta deposition in APPSwInd transgenic mice. Brain Res. 2009;1294:202–210. doi:10.1016/j.brainres.2009.07.078
39. Fleischman DA, Yang J, Arfanakis K, et al. Physical activity, motor function, and white matter hyperintensity burden in healthy older adults. Neurology. 2015;84(13):1294–1300. doi:10.1212/wnl.0000000000001417
40. Hattori Y, Enmi J, Iguchi S, et al. Gradual carotid artery stenosis in mice closely replicates hypoperfusive vascular dementia in humans. J Am Heart Assoc. 2016;5(2):e002757. doi:10.1161/JAHA.115.002757
41. Kakae M, Kawashita A, Onogi H, Nakagawa T, Shirakawa H. Bilateral common carotid artery stenosis in mice: a model of chronic cerebral hypoperfusion-induced vascular cognitive impairment. Biol Protoc. 2024;14(13):e5022. doi:10.21769/BioProtoc.5022
42. Wolf G, Lotan A, Lifschytz T, et al. Differentially severe cognitive effects of compromised cerebral blood flow in aged mice: association with myelin degradation and microglia activation. Front Aging Neurosci. 2017;9:191. doi:10.3389/fnagi.2017.00191
43. Xing CY, Tarumi T, Liu J, et al. Distribution of cardiac output to the brain across the adult lifespan. J Cereb Blood Flow Metab. 2017;37(8):2848–2856. doi:10.1177/0271678x16676826
44. Attwell D, Laughlin SB. An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab. 2001;21(10):1133–1145. doi:10.1097/00004647-200110000-00001
45. Graff BJ, Harrison SL, Payne SJ, El-Bouri WK. Regional cerebral blood flow changes in healthy ageing and Alzheimer’s disease: a narrative review. Cerebrovasc Dis. 2023;52(1):11–20. doi:10.1159/000524797
46. Hattori Y, Enmi J, Kitamura A, et al. A novel mouse model of subcortical infarcts with dementia. J Neurosci. 2015;35(9):3915–3928. doi:10.1523/JNEUROSCI.3970-14.2015
47. Hattori Y, Okamoto Y, Maki T, et al. Silent information regulator 2 homolog 1 counters cerebral hypoperfusion injury by deacetylating endothelial nitric oxide synthase. Stroke. 2014;45(11):3403–3411. doi:10.1161/strokeaha.114.006265
48. Toyama K, Koibuchi N, Uekawa K, et al. Apoptosis signal-regulating kinase 1 is a novel target molecule for cognitive impairment induced by chronic cerebral hypoperfusion. Arterioscler Thromb Vasc Biol. 2014;34(3):616–625. doi:10.1161/atvbaha.113.302440
49. Toyama K, Spin JM, Deng AC, et al. MicroRNA-mediated therapy modulating blood-brain barrier disruption improves vascular cognitive impairment. Arterioscler Thromb Vasc Biol. 2018;38(6):1392–1406. doi:10.1161/atvbaha.118.310822
50. Matin N, Fisher C, Jackson WF, Dorrance AM. Bilateral common carotid artery stenosis in normotensive rats impairs endothelium-dependent dilation of parenchymal arterioles. Am J Physiol Heart Circ Physiol. 2016;310(10):H1321–9. doi:10.1152/ajpheart.00890.2015
51. Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37(1):13–25. doi:10.1016/j.nbd.2009.07.030
52. Armulik A, Genove G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21(2):193–215. doi:10.1016/j.devcel.2011.07.001
53. Li G, Gao J, Ding P, Gao Y. The role of endothelial cell-pericyte interactions in vascularization and diseases. J Adv Res. 2025;67:269–288. doi:10.1016/j.jare.2024.01.016
54. Yang X, Chang L, Liu Z, et al. Neddylation in the chronically hypoperfused corpus callosum: MLN4924 reduces blood-brain barrier injury via ERK5/KLF2 signaling. Exp Neurol. 2024;371:114587. doi:10.1016/j.expneurol.2023.114587
55. Ru D, Zhang Z, Liu M, et al. Downregulation of notch signaling-stimulated genes in neurovascular unit alterations induced by chronic cerebral hypoperfusion. Immun Inflamm Dis. 2024;12(11):e70082. doi:10.1002/iid3.70082
56. Magami S, Miyamoto N, Ueno Y, et al. The effects of astrocyte and oligodendrocyte lineage cell interaction on white matter injury under chronic cerebral hypoperfusion. Neuroscience. 2019;406:167–175. doi:10.1016/j.neuroscience.2019.03.004
57. Miyamoto N, Magami S, Inaba T, et al. The effects of A1/A2 astrocytes on oligodendrocyte linage cells against white matter injury under prolonged cerebral hypoperfusion. Glia. 2020;68(9):1910–1924. doi:10.1002/glia.23814
58. Du Y, Song Y, Zhang X, et al. Leptin receptor deficiency protects mice against chronic cerebral hypoperfusion-induced neuroinflammation and white matter lesions. Mediators Inflamm. 2020;2020:7974537. doi:10.1155/2020/7974537
59. Kakae M, Nakajima H, Tobori S, et al. The astrocytic TRPA1 channel mediates an intrinsic protective response to vascular cognitive impairment via LIF production. Sci Adv. 2023;9(29):eadh0102. doi:10.1126/sciadv.adh0102
60. Liu Q, Bhuiyan MIH, Liu R, et al. Attenuating vascular stenosis-induced astrogliosis preserves white matter integrity and cognitive function. J Neuroinflammation. 2021;18(1):187. doi:10.1186/s12974-021-02234-8
61. Cao J, Yao D, Li R, et al. Digoxin ameliorates glymphatic transport and cognitive impairment in a mouse model of chronic cerebral hypoperfusion. Neurosci Bull. 2022;38(2):181–199. doi:10.1007/s12264-021-00772-y
62. Holland PR, Searcy JL, Salvadores N, et al. Gliovascular disruption and cognitive deficits in a mouse model with features of small vessel disease. J Cereb Blood Flow Metab. 2015;35(6):1005–1014. doi:10.1038/jcbfm.2015.12
63. Yu N, Zhao Y, Wang P, Zhang F, Wen C, Wang S. Changes in border-associated macrophages after stroke: single-cell sequencing analysis. Neural Regen Res. 2026;21(1):346–356. doi:10.4103/NRR.NRR-D-24-01092
64. Pedragosa J, Salas-Perdomo A, Gallizioli M, et al. CNS-border associated macrophages respond to acute ischemic stroke attracting granulocytes and promoting vascular leakage. Acta Neuropathol Commun. 2018;6(1):76. doi:10.1186/s40478-018-0581-6
65. Riew TR, Hwang JW, Jin X, Kim HL, Lee MY. Infiltration of meningeal macrophages into the Virchow-Robin space after ischemic stroke in rats: correlation with activated PDGFR-beta-positive adventitial fibroblasts. Front Mol Neurosci. 2022;15:1033271. doi:10.3389/fnmol.2022.1033271
66. Zhao Y, Xing W, Chen W, Wang Y. Integrated bioinformatics analysis and biological experiments to identify key immune genes in vascular dementia. Front Immunol. 2025;16:1560438. doi:10.3389/fimmu.2025.1560438
67. Daneman R, Prat A. The blood-brain barrier. Cold Spring Harb Perspect Biol. 2015;7(1):a020412. doi:10.1101/cshperspect.a020412
68. Galea I, Bechmann I, Perry VH. What is immune privilege (not)? Trends Immunol. 2007;28(1):12–18. doi:10.1016/j.it.2006.11.004
69. Nie Q, Zhang L, Gu Z, Li Y, Bi X. Lipocalin 2 facilitates the initial compromise of the blood-brain barrier integrity in chronic cerebral hypoperfusion. CNS Neurosci Ther. 2025;31(5):e70438. doi:10.1111/cns.70438
70. Rajeev V, Fann DY, Dinh QN, et al. Intermittent fasting attenuates hallmark vascular and neuronal pathologies in a mouse model of vascular cognitive impairment. Int J Biol Sci. 2022;18(16):6052–6067. doi:10.7150/ijbs.75188
71. Han QY, Zhang H, Zhang X, et al. dl-3-n-butylphthalide preserves white matter integrity and alleviates cognitive impairment in mice with chronic cerebral hypoperfusion. CNS Neurosci Ther. 2019;25(9):1042–1053. doi:10.1111/cns.13189
72. Guo X, Tian Y, Yang Y, Li S, Guo L, Shi J. Pituitary adenylate cyclase-activating polypeptide protects against cognitive impairment caused by chronic cerebral hypoperfusion. Mol Neurobiol. 2021;58(9):4309–4322. doi:10.1007/s12035-021-02381-2
73. Yang L, Song J, Nan D, Wan Y, Guo H. Cognitive impairments and blood-brain barrier damage in a mouse model of chronic cerebral hypoperfusion. Neurochem Res. 2022;47(12):3817–3828. doi:10.1007/s11064-022-03799-3
74. Rempe RG, Hartz AMS, Bauer B. Matrix metalloproteinases in the brain and blood-brain barrier: versatile breakers and makers. J Cereb Blood Flow Metab. 2016;36(9):1481–1507. doi:10.1177/0271678X16655551
75. Nakaji K, Ihara M, Takahashi C, et al. Matrix metalloproteinase-2 plays a critical role in the pathogenesis of white matter lesions after chronic cerebral hypoperfusion in rodents. Stroke. 2006;37(11):2816–2823. doi:10.1161/01.Str.0000244808.17972.55
76. Miyamoto N, Pham LD, Maki T, Liang AC, Arai K. A radical scavenger edaravone inhibits matrix metalloproteinase-9 upregulation and blood-brain barrier breakdown in a mouse model of prolonged cerebral hypoperfusion. Neurosci Lett. 2014;573:40–45. doi:10.1016/j.neulet.2014.05.005
77. Engelhardt B. Molecular mechanisms involved in T cell migration across the blood-brain barrier. J Neural Transm. 2006;113(4):477–485. doi:10.1007/s00702-005-0409-y
78. Yu W, Jin H, Sun W, et al. Connexin43 promotes angiogenesis through activating the HIF-1alpha/VEGF signaling pathway under chronic cerebral hypoperfusion. J Cereb Blood Flow Metab. 2021;41(10):2656–2675. doi:10.1177/0271678X211010354
79. Johnson AM, Roach JP, Hu A, et al. Connexin 43 gap junctions contribute to brain endothelial barrier hyperpermeability in familial cerebral cavernous malformations type III by modulating tight junction structure. FASEB J. 2018;32(5):2615–2629. doi:10.1096/fj.201700699R
80. Yata K, Nishimura Y, Unekawa M, et al. In vivo imaging of the mouse neurovascular unit under chronic cerebral hypoperfusion. Stroke. 2014;45(12):3698–3703. doi:10.1161/strokeaha.114.005891
81. Wang Y, Wu Q, Fang Y, et al. Depletion of regulatory T cells exacerbates inflammatory responses after chronic cerebral hypoperfusion in mice. Mol Cell Neurosci. 2022;123:103788. doi:10.1016/j.mcn.2022.103788
82. Matsuyama H, Shindo A, Shimada T, et al. Chronic cerebral hypoperfusion activates AIM2 and NLRP3 inflammasome. Brain Res. 2020;1736:146779. doi:10.1016/j.brainres.2020.146779
83. Xie Y, Wang X, Liu S, et al. Transforming growth factor β1 protects against ischemic demyelination via regulating microglial lipid metabolism pathway. Stroke. 2025;56(6):1554–1568. doi:10.1161/strokeaha.124.048206
84. Bodnar CN, Watson JB, Higgins EK, Quan N, Bachstetter AD. Inflammatory regulation of CNS barriers after traumatic brain injury: a tale directed by interleukin-1. Front Immunol. 2021;12:688254. doi:10.3389/fimmu.2021.688254
85. Liu X, Nemeth DP, McKim DB, et al. Cell-type-specific interleukin 1 receptor 1 signaling in the brain regulates distinct neuroimmune activities. Immunity. 2019;50(2):317–333e6. doi:10.1016/j.immuni.2018.12.012
86. Poh L, Razak S, Lim HM, et al. AIM2 inflammasome mediates apoptotic and pyroptotic death in the cerebellum following chronic hypoperfusion. Exp Neurol. 2021;346:113856. doi:10.1016/j.expneurol.2021.113856
87. Vila E, Salaices M. Cytokines and vascular reactivity in resistance arteries. Am J Physiol Heart Circ Physiol. 2005;288(3):H1016–21. doi:10.1152/ajpheart.00779.2004
88. Shaftel SS, Carlson TJ, Olschowka JA, Kyrkanides S, Matousek SB, O’Banion MK. Chronic interleukin-1beta expression in mouse brain leads to leukocyte infiltration and neutrophil-independent blood brain barrier permeability without overt neurodegeneration. J Neurosci. 2007;27(35):9301–9309. doi:10.1523/JNEUROSCI.1418-07.2007
89. Chung JY, Krapp N, Wu L, et al. Interleukin-1 receptor 1 deletion in focal and diffuse experimental traumatic brain injury in mice. J Neurotrauma. 2019;36(2):370–379. doi:10.1089/neu.2018.5659
90. Zhou Y, Zhang J, Wang L, et al. Interleukin-1β impedes oligodendrocyte progenitor cell recruitment and white matter repair following chronic cerebral hypoperfusion. Brain Behav Immun. 2017;60:93–105. doi:10.1016/j.bbi.2016.09.024
91. Takahashi JL, Giuliani F, Power C, Imai Y, Yong VW. Interleukin-1beta promotes oligodendrocyte death through glutamate excitotoxicity. Ann Neurol. 2003;53(5):588–595. doi:10.1002/ana.10519
92. Zou H, Bao S, Chen X, Zhou X, Zhang S. High-frequency repetitive transcranial magnetic stimulation ameliorates memory impairment by inhibiting neuroinflammation in the chronic cerebral hypoperfusion mice. Brain Behav. 2024;14(7):e3618. doi:10.1002/brb3.3618
93. Mou KJ, Shen KF, Li YL, Wu ZF, Duan W. Adenosine A(2A) receptor in bone marrow-derived cells mediated macrophages M2 polarization via PPARγ-P65 pathway in chronic hypoperfusion situation. Front Aging Neurosci. 2021;13:792733. doi:10.3389/fnagi.2021.792733
94. Zhu Y, Tong J, Jiang J, et al. Suppression of miR-218 promotes SOCS3 expression to alleviate cognitive impairment and white matter injury after chronic cerebral hypoperfusion. Metab Brain Dis. 2025;40(3):147. doi:10.1007/s11011-025-01572-3
95. MacPherson KP, Sompol P, Kannarkat GT, et al. Peripheral administration of the soluble TNF inhibitor XPro1595 modifies brain immune cell profiles, decreases beta-amyloid plaque load, and rescues impaired long-term potentiation in 5xFAD mice. Neurobiol Dis. 2017;102:81–95. doi:10.1016/j.nbd.2017.02.010
96. Liu Y, Zhai Y, Ma L, et al. Colchicine alleviates ischemic white matter lesions and cognitive deficits by inhibiting microglia inflammation via the TAK1/MAPK/NF-κB signaling pathway. Behav Brain Res. 2025;490:115619. doi:10.1016/j.bbr.2025.115619
97. Zhang Z, Ru D, Liu Z, et al. Integrative multiomics profiling of mouse hippocampus reveals transcriptional upregulation of interferon-stimulated genes through PU.1 regulator in microglial activation induced by chronic cerebral hypoperfusion. MedComm. 2025;6(5):e70157. doi:10.1002/mco2.70157
98. Chen Y, Tian H, Yao E, et al. Soluble epoxide hydrolase inhibition promotes white matter integrity and long-term functional recovery after chronic hypoperfusion in mice. Sci Rep. 2017;7(1):7758. doi:10.1038/s41598-017-08227-z
99. Zhang Z, Jin P, Guo Z, et al. Integrated analysis of chromatin and transcriptomic profiling identifies PU.1 as a core regulatory factor in microglial activation induced by chronic cerebral hypoperfusion. Mol Neurobiol. 2024;61(5):2569–2589. doi:10.1007/s12035-023-03734-9
100. Zhang Z, Guo Z, Jin P, et al. Transcriptome profiling of hippocampus after cerebral hypoperfusion in mice. J Mol Neurosci. 2023;73(6):423–436. doi:10.1007/s12031-023-02123-0
101. Li D, Zhang Q, Yang X, et al. Microglial AT1R conditional knockout ameliorates hypoperfusive cognitive impairment by reducing microglial inflammatory responses. Neuroscience. 2024;545:125–140. doi:10.1016/j.neuroscience.2024.02.002
102. Zhang H, Yang S, Lu YL, et al. Microglial Nrf2-mediated lipid and iron metabolism reprogramming promotes remyelination during white matter ischemia. Redox Biol. 2025;79:103473. doi:10.1016/j.redox.2024.103473
103. Dong X, Zhang Z, Shu X, et al. MFG-E8 alleviates cognitive impairments induced by chronic cerebral hypoperfusion by phagocytosing myelin debris and promoting remyelination. Neurosci Bull. 2024;40(4):483–499. doi:10.1007/s12264-023-01147-1
104. Ji W, Zhang Z, Jin T, et al. Salidroside attenuates cognitive deficits induced by chronic cerebral hypoperfusion via modulating microglial phenotypic transformation in mice. J Neuroimmunol. 2025;400:578544. doi:10.1016/j.jneuroim.2025.578544
105. Stevenson W, Hase Y, Wilson E, et al. Long-term effects of experimental carotid stenosis on hippocampal infarct pathology, neurons and glia and amelioration by environmental enrichment. Brain Res Bull. 2020;163:72–83. doi:10.1016/j.brainresbull.2020.07.014
106. Hu Y, Zheng Y, Wang T, Jiao L, Luo Y. VEGF, a key factor for blood brain barrier injury after cerebral ischemic stroke. Aging Dis. 2022;13(3):647–654. doi:10.14336/AD.2021.1121
107. Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-kappaB signaling pathways. Nat Immunol. 2011;12(8):695–708. doi:10.1038/ni.2065
108. Sarker KP, Biswas KK, Yamakuchi M, et al. ASK1-p38 MAPK/JNK signaling cascade mediates anandamide-induced PC12 cell death. J Neurochem. 2003;85(1):50–61. doi:10.1046/j.1471-4159.2003.01663.x
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Flavins and Flavoproteins in the Neuroimmune Landscape of Stress Sensitization and Major Depressive Disorder

Schrier MS, Smirnova MI, Nemeth DP, Deth RC, Quan N
Journal of Inflammation Research 2025, 18:681-699
Published Date: 16 January 2025