")
Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
TAT-LHRH conjugated low molecular weight chitosan as a gene carrier specific for hepatocellular carcinoma cells
Authors Liu L, Dong X, Zhu D , Song L, Zhang H, Leng X
Received 27 January 2014
Accepted for publication 12 April 2014
Published 10 June 2014 Volume 2014:9(1) Pages 2879—2889
DOI https://doi.org/10.2147/IJN.S61392
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Lanxia Liu, Xia Dong, Dunwan Zhu, Liping Song, Hailing Zhang, Xigang G Leng
Laboratory of Bioengineering, Institute of Biomedical Engineering, Chinese Academy of Medical Sciences and Peking Union Medical College, Tianjin Key Laboratory of Biomedical Materials, Tianjin, People’s Republic of China
Abstract: To develop a chitosan-based nonviral gene carrier capable of delivering genes specifically into hepatoma cells, a bifunctional peptide composed of the TAT (transactivator of transcription) peptide and luteinizing hormone-releasing hormone (LHRH) was conjugated with low molecular weight chitosan, resulting in a TAT-LHRH-chitosan conjugate (TLC). TLC/DNA nanoparticles (TLCDNPs) were characterized by agarose gel retardation, atomic force microscopy, and dynamic light scattering analysis. In vitro targeting specificity and transfection efficiency were analyzed with a GE IN Cell Analyzer 2000 High-Content Cellular Analysis System. The results demonstrated that TLC had stronger DNA condensing power than unmodified chitosan, and that TLCDNPs were of roughly round shape with average diameter of 70–85 nm and zeta potential of +30 mV and were relatively stable in solution. The in vitro study demonstrated TLC was highly selective for hepatoma cells and essentially nontoxic.
Keywords: nanoparticles, cancer treatment, gene therapy, hepatoma, targeted gene delivery
Introduction
Hepatocellular carcinoma (HCC) is the major primary malignant tumor of the liver. Currently, it is the fifth most prevalent malignancy and the third leading cause of cancer-related deaths worldwide.1,2 Despite advances in therapy against HCC such as recent modifications in chemotherapy and modern surgical innovations, the overall clinical outcome has not been substantially improved.3 Long-term survival of patients with HCC is uncommon due to the frequent presence of reoccurrence, metastasis, or the development of new primaries.4 Curative treatment such as hepatic resection and liver transplantation can be utilized when HCC is diagnosed at an early stage.5,6 Unfortunately, when diagnosed the vast majority of liver cancers are inoperable,7–9 and thus the patients have to receive chemotherapy, which has limited success due to the fact that HCC is intrinsically resistant to standard chemotherapeutic agents. Therefore, it is urgently needed to develop more effective cures for HCC patients, of which gene therapy is among those with the most potential.10–12
Gene therapy involves transferring genetic materials (DNA or RNA) into a target cell so the diseased genes can be corrected. The difficulty of employing gene therapy as a cure for HCC is the ability to design an efficient vector that is able to deliver therapeutic genes specifically into the cancer cells but not the surrounding benign cells. Cancer targeting is usually achieved by adding to the gene carriers a ligand moiety specifically directed to certain types of binding sites on cancer cells. Antibodies, epidermal growth factor, aptamers, and small molecules such as galactose have been reported as potential targeting moieties for specific delivery of genes and drugs to HCC cells.11,13–15 Overexpression of the luteinizing hormone-releasing hormone (LHRH) receptor has been reported in breast, ovarian, and prostate cancer cells,16–19 whereas no detectable expression of LHRH receptors has been observed in most visceral organs. Previous reports by Dharap et al17,20 and Tang et al19 demonstrated that LHRH peptide could be used as a targeting moiety on drug-delivery systems to enhance drug uptake by breast, ovarian, and prostate cancer cells, and reduce the relative availability of the toxic drug to normal cells. These studies confirmed the high anticancer activity of LHRH-targeted carrier–drug conjugates against the aforementioned cancer cells, and that the cytotoxicity of the LHRH-targeted conjugates against the human cancer cells could be competitively inhibited by free LHRH peptide. Tang et al further demonstrated that treating mice bearing xenografts of human ovarian carcinoma with LHRH-targeted carrier–drug conjugates led to a dramatic reduction in the tumor size compared with untargeted carrier–drug conjugates. LHRH receptor was also reported to be overexpressed in hepatocellular cells21,22 and thus could be used as a target for hepatoma-specific gene or drug delivery.
Chitosan is a cationic polymer derived from chitin, a naturally occurring polysaccharide, and has been extensively studied in nonviral gene delivery owing to its excellent biocompatibility and biodegradability, and low immunogenicity.23–28 Unfortunately, gene delivery mediated by chitosan is nonspecific and the transgenic efficacy is considerably lower than what is needed for sufficient gene delivery.29,30 In order to improve the transgenic efficacy and specificity, we conjugated low molecular weight chitosan (LMWC) with the bifunctional peptide (TAT-LHRH) composed of a cell penetrating peptide TAT (transactivator of transcription) and an LHRH analog, and evaluated the transgenic efficacy and hepatoma-targeting specificity of the conjugate (TAT-LHRH-chitosan, known as TLC).
Materials and methods
Peptide synthesis
The bifunctional peptide (TAT-LHRH) used in the present study, a fusion peptide composed of a TAT peptide derived from human immunodeficiency virus (HIV)-1 linked through a cysteine residue with a part of an LHRH analog (sequence: pGlu-His-Trp-Ser-Tyr-D-Trp-Leu-Arg-Gly-Cys-Arg-Arg-Arg-Gln-Arg-Arg-Lys-Lys-Arg-NH2), was synthesized by SBS Genetech Co., Ltd (Beijing, People’s Republic of China) with a Liberty Microwave Peptide Synthesizer (CEM Corporation, Matthews, NC, USA) and purified by reverse-phase high performance liquid chromatography (HPLC) using a C18 column (Waters Corporation, Milford, MA, USA). The molecular weight of the peptide was determined by liquid chromatography–mass spectrometry (1100 Series Lc-MSD-Trap-VL machine; Agilent Technologies Inc., Santa Clara, CA, USA).
Synthesis and characterization of TLC
TLC was synthesized by covalently coupling a TAT-LHRH peptide onto LMWC (MW: 5,000–8,000, degree of deacetylation of 90%; Golden-Shell Biochemical Co., Ltd., Yuhuan, People’s Republic of China) with N-succinimidyl-3-(2-pyridyldithio)-propionate (SPDP) (Thermo Fisher Scientific Inc., Rockford, IL, USA) as the linking agent, using the method described by Cumber et al.31 Briefly, LMWC was dissolved in distilled water and reacted with 20 mM SPDP in dimethyl sulfoxide for 1 hour, with the molar ratio of primary amines in LMWC to SPDP being 3:1. After removing the reaction byproducts and excessive nonreacted SPDP using a desalting column (MW 5,000; Merck Millipore, Billerica, MA, USA) pre-equilibrated with phosphate buffered saline – ethylenediaminetetraacetic acid (PBS–EDTA), the reaction mixture was mixed with peptide at the molar ratio of primary amines (-NH2) to peptide being 3:1, followed by incubation with stirring overnight at room temperature and purification again as described above. The resultant TLC was characterized with an infrared spectroscope (Nicolet 2000; Thermo Fisher Scientific, Waltham, MA, USA). The substitution degree of TLC was estimated by H-NMR (proton nuclear magnetic resonance) spectroscopic analysis (Avance 600; Bruker BioSpin AG, Fällanden, Switzerland), and the isoelectric point of TLC was measured by acid-base titration using a Zetasizer Nano ZS (Malvern Instruments, Malvern, UK).32
Preparation and characterizationof TLC/DNA nanoparticles
Preparation of TLC/DNA nanoparticles
pEGFP-C1 plasmid (Clontech Laboratories, Inc., Mountain View, CA, USA) was purified using EndoFree Plasmid Mega Kits (Qiagen Co. Ltd., Shanghai, People’s Republic of China) according to the manufacturer’s instructions. To prepare the TLC/DNA nanoparticles ( TLCDNPs), equal volume of plasmid DNA solution and TLC solution in distilled water were mixed together at an N/P ratio of 1:4, 1:2, 1:1, 2:1, 4:1. or 10:1, followed by preheating at 37°C for 10 minutes and subsequent vortexing for 40–60 seconds.
Agarose gel retardation assay
For this assay, 15 μL each of TLCDNPs at different N/P ratios were loaded into the sample well of agarose gel and electrophoresis was conducted in Tris-acetate/EDTA (TAE) buffer at 60 V for 40 minutes using 0.25 mg/mL DNAGREEN (Tiandz Inc., Beijing, People’s Republic of China) as fluorescent dye, with unmodified LMWC/DNA nanoparticles at the corresponding N/P ratios as the experimental control.
Atomic force microscopy and dynamiclight scattering
The size and surface morphology of TLCDNPs were analyzed with an atomic force microscope (AFM) (Nanoscope Multimode AFM; Veeco Instruments Inc, Plainview, NY, USA) in ScanAsyst mode. A Zetasizer Nano-ZS instrument (Malvern Instruments) was utilized to measure the size distribution and zeta potential of the nanoparticles, with each sample measured three times.
Stability of TLCDNPs
The tendency of TLCDNPs to aggregate in solution was evaluated by measuring the nanoparticle size distribution and zeta potential over time. Briefly, the nanoparticles were suspended in distilled water and placed at room temperature, and the size distribution and zeta potential were measured after 1, 2, 3, 7, and 14 days using the Zetasizer Nano ZS (Malvern Instruments). Each test was repeated three times and the results were presented as the mean values of the three measurements.
Cell lines and culture
Human hepatoma cell line BEL-7402 cells and human normal liver cell line L02 cells purchased from the Cell Bank of Chinese Academy of Sciences (Shanghai, People’s Republic of China) were maintained in RPMI-1640 medium (HyClone Laboratories, Inc., Logan, UT, USA) supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 100 units/mL penicillin, and 100 μg/mL streptomycin, and incubated at 37°C in a humidified incubator with 5% CO2.
Cytotoxicity assessment
The cytotoxicity of TLCDNPs was assessed using the Cell Counting Kit-8 (Dojindo Molecular Technologies, Inc., Kumamoto, Japan) according to the manufacturer’s instructions. In brief, BEL-7402 or L02 cells were seeded in a 96-well plate at a density of 8×103 cells/mL and grown overnight at 37°C in a humidified 5% CO2 incubator. Afterwards, the cells were divided into three groups, two of which were treated with TLCDNPs or lipoplexes (Lipofectamine® 2000 [Thermo Fisher Scientific]/DNA complexes) at various concentrations (DNA concentrations of 5, 10, 20, and 50 μg/mL, respectively), leaving one group of cells untreated. After 24 hours of incubation, 10 μL of WST®-8 solution (Dojindo Molecular Technologies, Inc., Kumamoto, Japan) was added to each well, and the optical absorbance at 450 nm was measured 1 hour later with a microtiter plate reader (Varioskan™ Flash; Thermo Fisher Scientific). Cell viability was presented as the ratio of OD450 of the nanoparticle-treated cells to that of the untreated cells. Six wells were measured for each test.
Labeling of DNA with fluorescein Cy5
To assess the in vitro and in vivo delivery of plasmid DNA by TLC, pGL3-control plasmid DNA (Promega Corporation, Fitchburg, WI, USA) was labeled with Cy5 at a 0.25:1 (volume:weight) ratio of Label IT® Tracker Reagent (Mirus Bio LLC, Madison, WI, USA) to DNA according to the manufacturer’s instructions. Briefly, the DNA was incubated with Label IT® Tracker Reagent at 37°C for 1 hour. The labeled DNA was precipitated with two volumes of ice cold ethanol and 0.1 volume of 5 M sodium chloride by placing at −20°C for 30 minutes. The DNA was pelleted by centrifugation at 12,000× g, 4°C, for 10 minutes, washed with 70% ethanol and centrifuged again. The dried pellet was reconstituted in sterile water at a concentration of 1 mg/mL as verified by measuring the optical absorbance at 260 nm using a NanoVue Plus Spectrophotometer (GE Healthcare Bio-Sciences Corp., Piscataway, NJ, USA). The absence of protein in the solution was confirmed by measuring optical absorbance at 280 nm (A260/A280 =1.82).
In vitro targeting specificity and transfection efficiency of TLCDNPs
BEL-7402 cells or L02 cells were seeded in a 96-well culture plate at the density of 5×104 cells/mL and incubated overnight in a humidified incubator with 5% CO2 at 37°C. To investigate the targeting specificity of TLCDNPs for hepatoma cells, the cells were co-incubated with TLCDNPs prepared with Cy5 labeled plasmid at an N/P ratio of 10:1 (DNA content of 5 μg/mL), with the lipoplexes containing the same amount of Cy5 labeled DNA as an experimental control. After 24 hours of co-incubation, the cells were washed two times with PBS and fixed in 4% paraformaldehyde for 10 minutes, followed by labeling the intracellular microfilament with the phalloidin-fluorescein isothiocyanate provided in the Actin-Tracker Green Kit (Beyotime Biotech Inc., Jiangsu, People’s Republic of China) and subsequent staining of the nucleus with 4′,6-diamidino-2-phenylindole (DAPI; Beyotime Biotech Inc.). Internalization of TLCDNPs or lipoplexes by the cells was analyzed with the GE IN Cell Analyzer 2000 High-Content Cellular Analysis System (GE Healthcare Bio-Sciences Corp.). To test the transfection efficiency of TLCDNPs, BEL-7402 or L02 cells were incubated with TLCDNPs containing the luciferase-encoding pGL3-control plasmid at an N/P ratio of 10:1 (DNA content of 5 μg/mL). After 24 hours of incubation, the cells were lysed with lysis reagent (Promega Corporation) and 20 μL of the supernatant was subsequently incubated with 100 μL of luciferase detection reagent followed by measuring with a chemiluminometer (TD-20/20n; Turner BioSystems Inc., Sunnyvale, CA, USA).
LHRH receptor blocking
After being seeded and incubated in a 96-well plate as described in the previous section, BEL-7402 cells were co-incubated with 1 μM, 10 μM, and 50 μM LHRH analog peptide (sequence: pGlu-His-Trp-Ser-Tyr-D-Trp-Leu-Arg-Gly-NH2), respectively. Following removing the excessive peptide at 1 hour post co-incubation, the cells were co-incubated for 24 hours with TLCDNPs or lipoplexes containing 5 μg/mL pGL3-control DNA. The internalization of TLCDNPs or lipoplexes by the cells was analyzed by the GE IN Cell Analyzer 2000 High-Content Cellular Analysis System (GE Healthcare Bio-Sciences Corp.), as aforementioned.
Statistical analysis
Data were presented as the mean of six individual observations with standard deviation. The statistical analysis was performed using the Bonferroni t-test. Statistical significance was determined at P<0.05.
Results
Identification and purity of the synthesized TAT-LHRH peptide
The synthesized TAT-Cys-LHRH peptide was analyzed with HPLC mass spectrometry to guarantee that a peak visible in the HPLC profile corresponds to the target mass. As shown in Figure 1, mass spectrometry and HPLC analysis demonstrated that the purity of the synthesized peptide was up to 99%, and the molecular mass was 2,657 daltons, which was identical to the expected molecular mass of the target peptide.
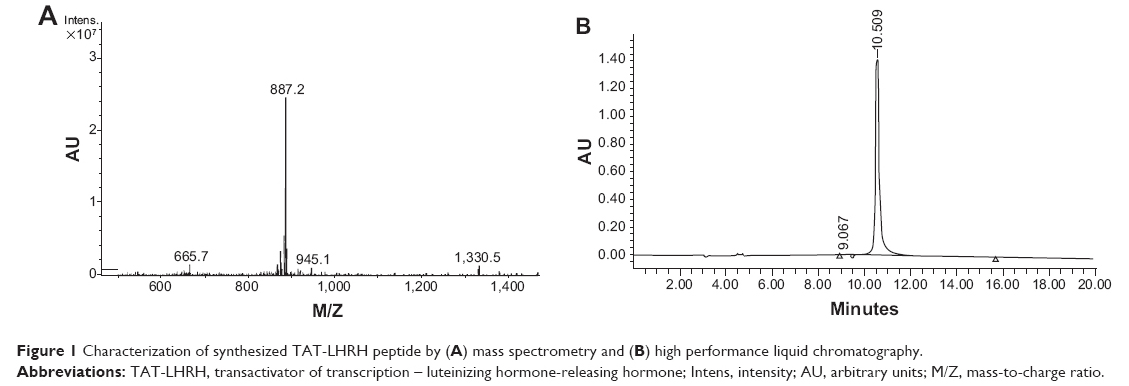 | Figure 1 Characterization of synthesized TAT-LHRH peptide by (A) mass spectrometry and (B) high performance liquid chromatography. |
Characterization of TLC
As designed in the current study, TLC should be formed through the disulfide bond linkage between the sulfydryl group from the cysteine residue in the peptide and the pyridyldithiol group generated by the reaction of SPDP with the primary amines (-NH2) in the chitosan. Therefore, the number of primary amines should be reduced and the secondary amines would increase in the resultant conjugates. Figure 2 shows the result of infrared spectroscopy analysis, which revealed that the intensity of the primary amine bond at 1,658.07 cm−1 in the resultant TLC decreased while the secondary amine bond at 1,555.56 cm−1 increased as compared with the unmodified chitosan, indicating the successful conjugation of TAT-LHRH peptides with chitosans. As shown in Figure 3, NMR spectroscopic analysis demonstrated the spectrum of H on the benzene ring of tyrosine peak at 6.8 ppm and that of the H on the primary amines of chitosan at 4.8 ppm. It was estimated that the substitution degree of TLC was about 3% according to the area ratio above the peak. Data from Figure 4 reveal that the isoelectric point of TLC was 11.3, which was significantly higher than that of the unmodified chitosan,33 suggesting that TLC would be more positively charged than the unmodified chitosan.
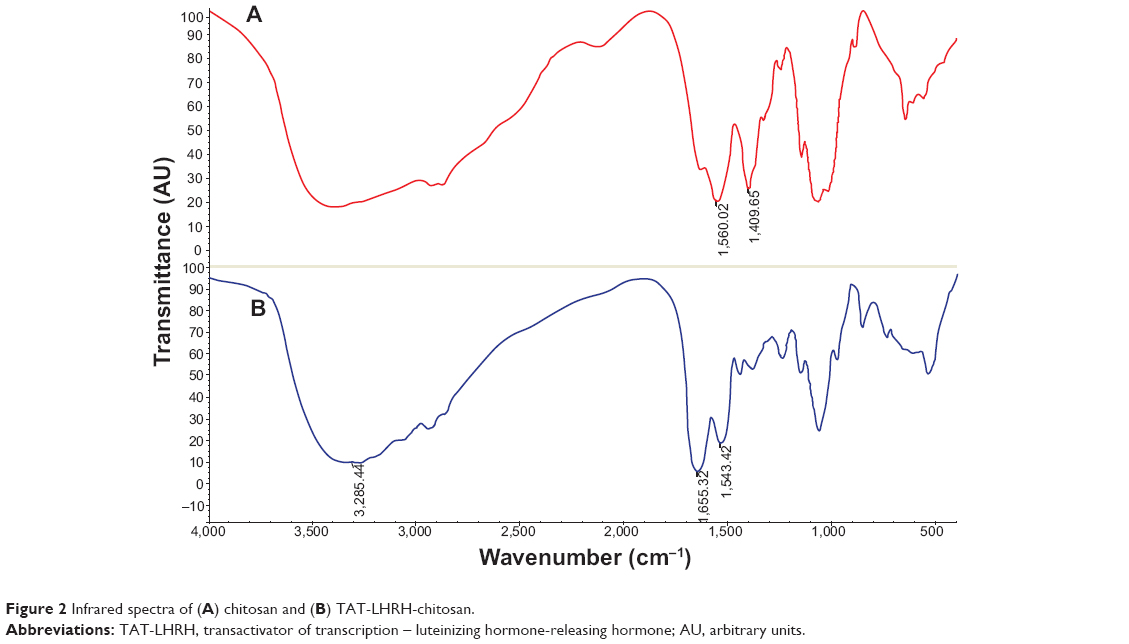 | Figure 2 Infrared spectra of (A) chitosan and (B) TAT-LHRH-chitosan. |
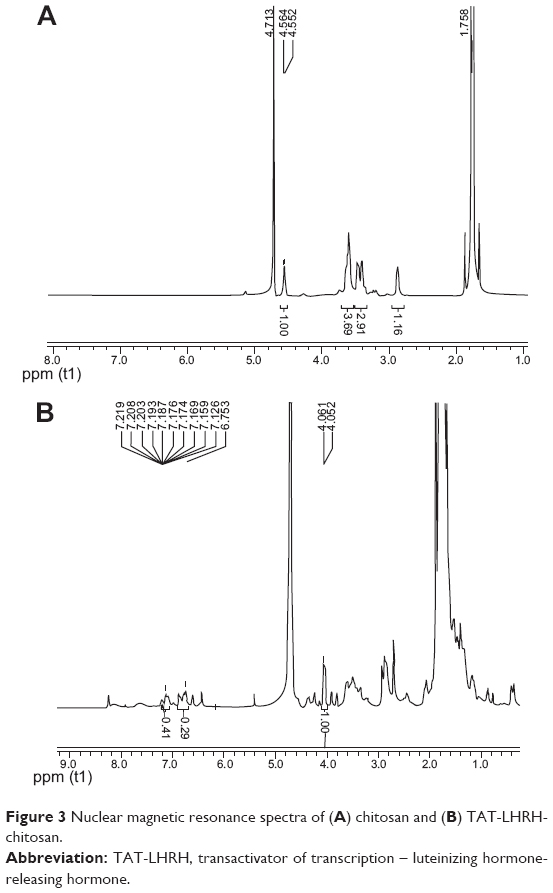 | Figure 3 Nuclear magnetic resonance spectra of (A) chitosan and (B) TAT-LHRH-chitosan. |
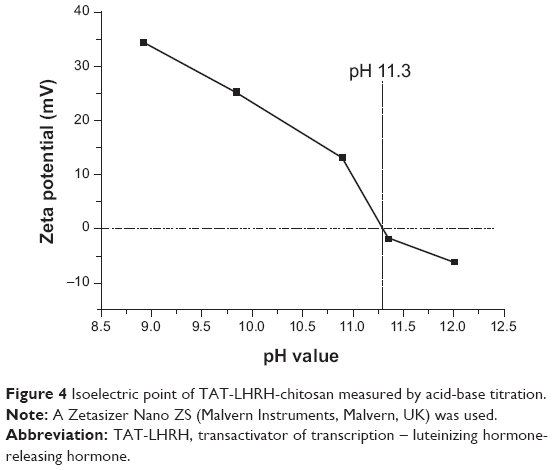 | Figure 4 Isoelectric point of TAT-LHRH-chitosan measured by acid-base titration. |
Characterization of TLCDNPs
Agarose gel retardation was performed to analyze the DNA-entrapping capability of TCL and it showed that the DNA was completely entrapped in TLC at an N/P ratio higher than 1:1 (Figure 5), while the DNA was completely entrapped by the unmodified chitosan only when the N/P ratio reached 8:1 or beyond, indicating the stronger DNA-entrapping capability of TLC in comparison with the unmodified chitosan. As shown in Figures 6 and 7, the average diameter of the TLCDNPs was 70–87 nm, the polydispersity index was about 0.12–0.4, and the zeta potential was about +30 mV as demonstrated by Zetasizer Nano ZS (Malvern Instruments), with roughly round and relatively uniformed shape as characterized with an AFM. The current study also investigated the stability of TLCDNPs in water and it demonstrated that the size and zeta potential of TLCDNPs were relatively stable during the 2 weeks of observation (Figure 8).
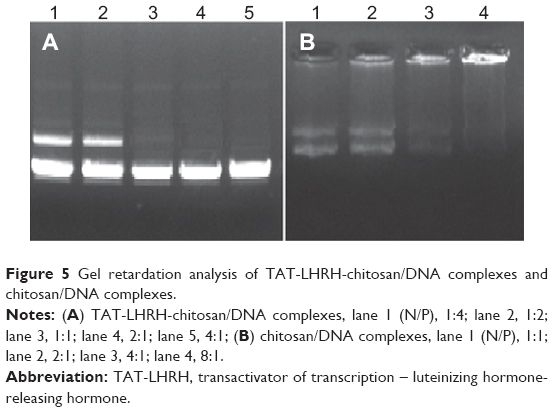 | Figure 5 Gel retardation analysis of TAT-LHRH-chitosan/DNA complexes and chitosan/DNA complexes. |
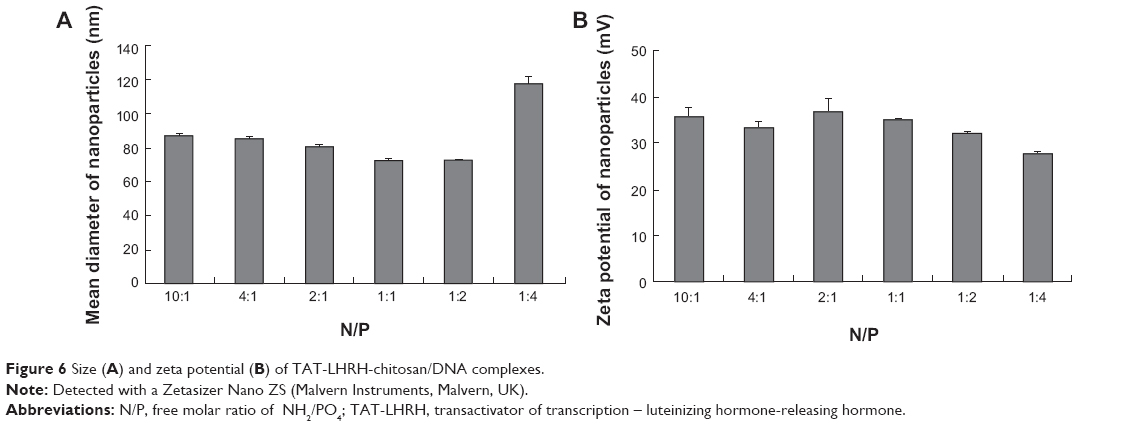 | Figure 6 Size (A) and zeta potential (B) of TAT-LHRH-chitosan/DNA complexes. |
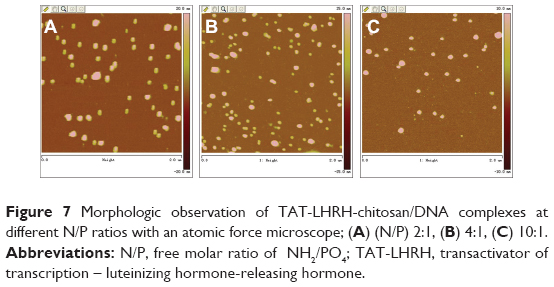 | Figure 7 Morphologic observation of TAT-LHRH-chitosan/DNA complexes at different N/P ratios with an atomic force microscope; (A) (N/P) 2:1, (B) 4:1, (C) 10:1. |
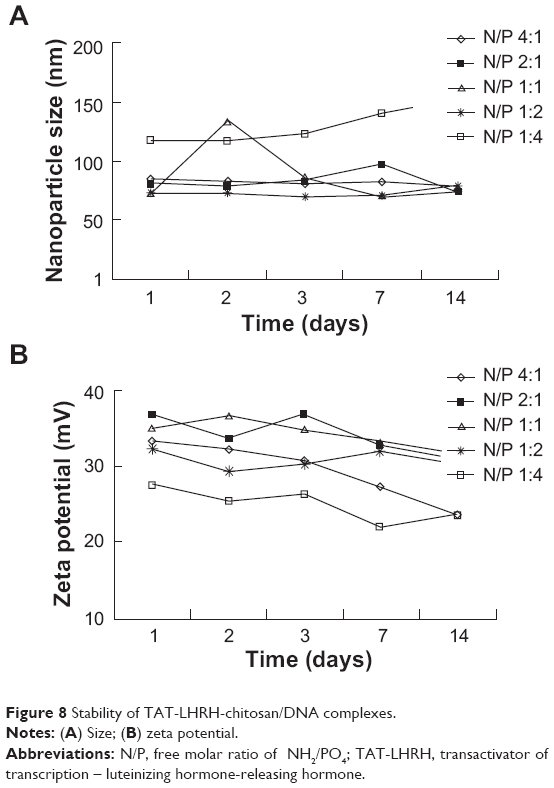 | Figure 8 Stability of TAT-LHRH-chitosan/DNA complexes. |
Cytotoxicity assessment
Figure 9 shows that TLCDNPs at the concentrations with DNA content of 20 μg/mL or less were essentially nontoxic against either normal L02 cells or BEL-7402 cells, whereas lipoplexes at the concentration containing the same amount of DNA were significantly more cytotoxic as compared with TLCDNPs. It was interesting, however, to note that TLCDNPs at the concentration containing 5 μg/mL DNA demonstrated a stimulatory effect on the viability of L02 cells. It was also worth noting that the viability of L02 cells was higher than that of BEL-7402 cells at each TLCDNP concentration, albeit the mechanism was unclear.
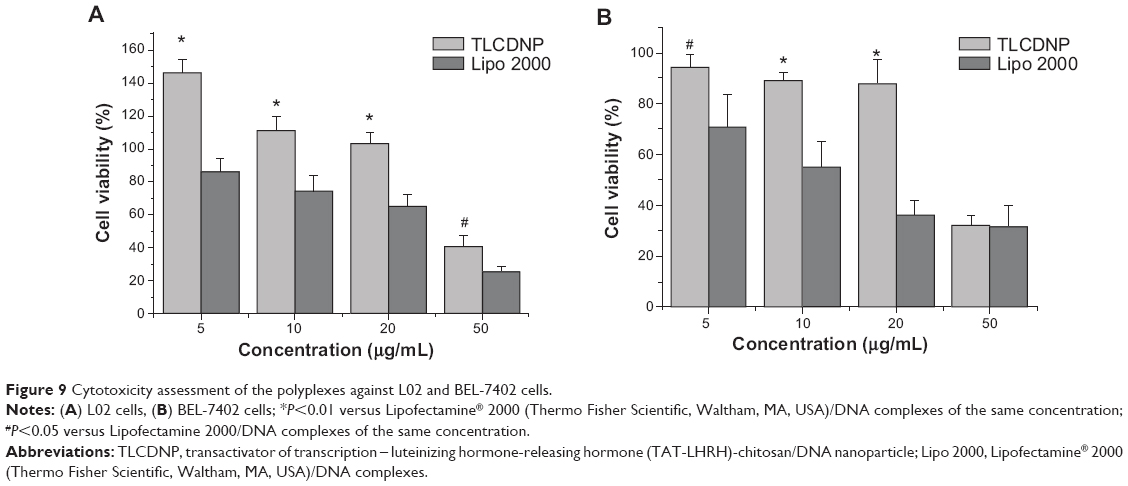 | Figure 9 Cytotoxicity assessment of the polyplexes against L02 and BEL-7402 cells. |
In vitro targeting specificityof TLCDNPs for hepatoma cells
To investigate the targeting specificity of TLC for hepatoma cells, both hepatoma cells (BEL-7402) and normal liver cells (L02) were transfected with TLCDNPs or lipoplexes prepared with fluorescein-labeled DNA. As shown in Figure 10, analysis with the GE IN Cell Analyzer 2000 High-Content Cellular Image System (GE Healthcare Bio-Sciences Corp.) demonstrated that the amount of DNA delivered into BEL-7402 cells by TLC was 14 times of that into L02 cells. It also revealed that the transfection efficiency of TCL to hepatoma cells was about 20-fold higher than that of the commercial transfection reagent Lipofectamine 2000, which demonstrated no significant specificity for hepatoma cells.
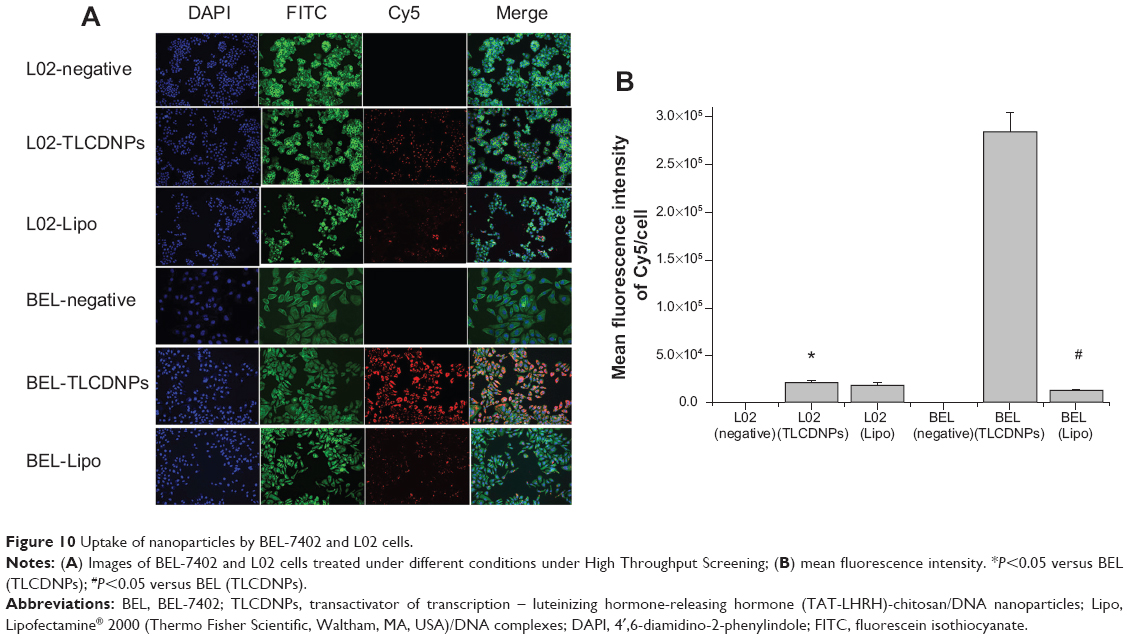 | Figure 10 Uptake of nanoparticles by BEL-7402 and L02 cells. |
An LHRH receptor-blocking experiment was performed to investigate the role that LHRH plays in the internalization of TLCDNPs by hepatoma cells. The data from Figure 11 illustrate that the uptake of TCLDNPs by BEL-7402 cells was inhibited by pre-incubating the cells with an LHRH analog in a dose-dependent manner. Blocking the LHRH receptor on BEL-7402 cells with 1 μM LHRH significantly reduced the cellular uptake of TLCDNPs by more than 50%, and pre-incubation with 10 μM or 50 μM LHRH almost completely blocked the cellular uptake of TLCDNPs, indicating that the internalization of the TLCDNPs by BEL-7402 cells was mainly mediated by LHRH.
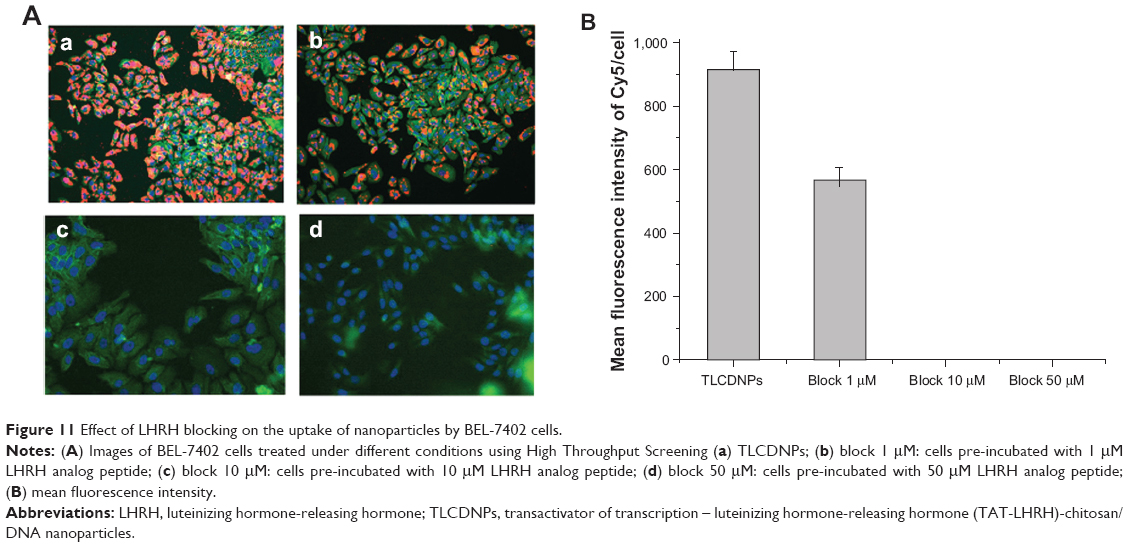 | Figure 11 Effect of LHRH blocking on the uptake of nanoparticles by BEL-7402 cells. |
In vitro transfection efficiencyof TLCDNPs for hepatoma cells
To learn whether the gene delivered by TLC could be expressed in the target cells, both BEL-7402 and L02 cells were transfected with TLCDNPs carrying the pGL3-control plasmid DNA, which encodes luciferase. Figure 12 shows that the luciferase activity in BEL-7402 cells was circa 110 times higher than that in L02 cells. Along with the aforementioned data, it indicated the superior selectivity of TLCDNPs for hepatoma cells over normal liver cells.
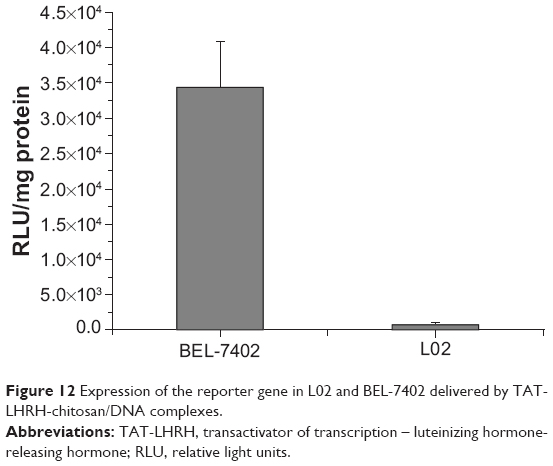 | Figure 12 Expression of the reporter gene in L02 and BEL-7402 delivered by TAT-LHRH-chitosan/DNA complexes. |
Discussion
Successful gene therapies for HCC entail developing gene carriers with low cytotoxicity, high transgenic efficacy, and strong targeting specificity for hepatoma cells. The current investigation was designed to develop a chitosan based nonviral gene carrier that is capable of delivering genes specifically into hepatoma cells. For this purpose, a bifunctional peptide composed of a cell-penetrating peptide TAT and a hepatoma-cell-targeting peptide LHRH was synthesized and conjugated with LMWC. LMWC is more water soluble than high molecular weight chitosan and thus makes the process of preparing carrier/gene polyplexes easier to handle. Unfortunately, the DNA-condensing power of LMWC is much weaker compared with high molecular weight chitosan as demonstrated in the present study by the gel retardation analysis. It was also observed in our preliminary study that the polyplexes formed between the unmodified LMWC and plasmid DNA were unstable, with non-uniform shape and much larger size (data not shown). Those reasons together hinder the application of LMWC as a highly efficient gene-delivery material. However, incorporation of TAT-LHRH into LMWC resulted in a much more positively charged TLC that showed a stronger DNA-condensing power, as demonstrated in the present study.
The size of chitosan/DNA polyplexes is one of the critical factors determining their transgenic efficacy both in vitro and in vivo. As compared with larger-sized particles, particles of smaller size are more likely to penetrate through the blood vessel to approach the target tissues and to be internalized by the cells.34 The present study showed that the average diameter of TLCDNPs was 70–85 nm, which was significantly smaller than DNA nanoparticles prepared with unmodified chitosan as demonstrated in the previous reports including ours.35,36 This is especially beneficial for a gene carrier targeting liver cancer, since it is revealed that particles with a diameter below 100 nm are required to cross liver fenestra and reach the hepatocytes.37 As expected, the polyplexes formed with unmodified LMWC and DNA were unstable with non-uniform size (data not shown); this made them unsuitable for further experiment such as gene transfection. This phenomenon is not unusual since the DNA-condensing capacity of LMWC is too weak to form stable complexes with plasmid DNA of much higher molecular mass. However, incorporation of TAT peptide provides TLC with more positive charge and thus stronger DNA-condensing power than the unmodified LMWC.
The stability of chitosan/DNA nanoparticles at physiological pH is another important factor affecting the transgenic efficiency of the nanoparticles. Due to the weak acidic property of chitosan, the optimal pH value for the formation of chitosan/DNA nanoparticles is around 5.5, and the maximum transfection mediated by chitosan/DNA nanoparticles is usually achieved at pH value lower than 7.23–25 The weak acidic pH value of the chitosan/DNA nanoparticles hinders their in vivo application for gene delivery since the physiological pH value of human body is around 7.4, which might unstabilize the nanoparticles. Therefore, it is crucial for the nanoparticles to be stable at physiological pH. The stability of TLCDNPs in water was tested by observing the changes in their size and zeta potential, and, as expected, the size and zeta potential of the nanoparticles was relatively stable for 2 weeks at room temperature, indicating considerable stability of the nanoparticles in water. The increased stability of TLCDNPs is probably the result of the elevated isoelectric point of TLC (11.3) as compared with that of unmodified chitosan (6.3),38 which confers the TLC higher positive-charge density and favors formation of more stable polyplexes with DNA at physiological pH. It is worth mentioning that TAT peptide plays an important role in the higher positive charge density of TLC, since it contains eight positively charged amino acid residues. This might be a significant advantage of TLC in comparison with conjugation of chitosan only with LHRH or other targeting moieties with lower positive charge density.
Cytotoxicity is one of the major concerns in the development of gene-delivery vectors. Chitosan is generally accepted as a nontoxic and safe biomaterial and a chitosan-based wound-dressing material has been approved by the US Food and Drug Administration.39 This is one of the most significant advantages over other cationic polymers. However, concern still remains, as some investigations demonstrated increased cytotoxicity of polyplexes with increasing charge density.40–42 Therefore, we compared the cytotoxicity of TLCDNPs and lipoplexes at different concentrations with DNA content ranging from 5 μg/mL to 50 μg/mL. TLCDNPs at lower concentrations (containing 20 μg/mL or less DNA) were found to be essentially nontoxic against either normal liver cells or hepatoma cells, and were significantly less cytotoxic as compared with the lipoplexes at each concentration. It was interesting to note that TLCDNPs at the concentration containing 5 μg/mL DNA demonstrated a stimulatory effect on the viability of normal liver cells; however, the underlying mechanism remains unclear. It was also worth noting that TLCDNPs demonstrated less cytotoxicity against normal liver cells than against hepatoma cells, which would be beneficial for their application as a hepatoma-targeting gene-delivery vector.
Targeting specificity for hepatoma cells was crucial for the clinical application of gene carriers for treating liver cancer. The current study investigated both the in vitro and in vivo targeting specificity of TLCDNPs for hepatoma cells. In vitro study demonstrated that TLCDNPs were more selective for hepatoma cells than normal liver cells, whereas lipoplexes showed no obvious specificity for hepatoma cells over normal cells. It also revealed that hepatoma cells took up much more TLCDNPs than lipoplexes. All those observations indicated that TLC was superior over Lipofectamine 2000 in terms of both transfection activity and selectivity for hepatoma cells. To learn if the gene delivered by TLC could be normally expressed within the cells, both hepatoma and normal liver cells were transfected with TLCDNPs carrying the pGL3-control plasmid DNA encoding for luciferase, and it was found that the luciferase activity in hepatoma cells was circa 110 times higher than that in normal liver cells, further supporting the superior selectivity of TLCDNPs for hepatoma cells over normal liver cells.
Adherence of serum proteins onto the surface of nanoparticles might interfere with in vitro gene transfection as well as in vivo gene delivery.43 Therefore, it is crucial to ascertain that the gene-delivery systems should be able to effectively deliver genes into the cells in the presence of serum proteins. High transfection efficiency of chitosan in the presence of serum has been proved by several reports, which is a key advantage over other nonviral vectors such as polyethylenimine29 and liposomes.44 In the present study, gene transfection was conducted in the presence of 10% fetal bovine serum and, in line with the previous observations by others, it demonstrated that TLCDNPs were taken up by most of the hepatoma cells whereas the lipoplexes were internalized by only a scarce part of the hepatoma cells, indicating that TLCDNPs were able to efficiently deliver genes into the hepatoma cells even in the presence of serum proteins.
An LHRH-blocking experiment was performed to investigate the role that LHRH plays in the internalization of the TLCDNPs by hepatoma cells. It demonstrated that pre-incubation of hepatoma cells with LHRH inhibited the endocytosis of TLCDNPs in a dose-dependent manner. In addition, almost complete blockage of nanoparticle internalization was achieved by LHRH at a concentration of 50 μM, indicating that TLCDNP internalization by hepatoma cells was mainly mediated by LHRH. This would explain why TLCDNPs remained efficient as a gene-delivery vehicle even in the presence of serum protein, whereas lipoplexes failed to mediate a satisfying gene transfection.
In vivo DNA delivery by TLCDNPs was preliminarily assessed in the nude mice subcutaneously implanted with BEL-7402 cells stably expressing luciferase. Obvious accumulation of fluorescent intensity was observed in the tumor areas of mice administrated with fluorescence labelled-TLCDNPs at 3 and 5 days after nanoparticle administration and no detectable fluorescent intensity was observed in other parts of the body (data not shown), providing further evidence that TLC might find great potential as a hepatoma-targeting gene carrier.
Conclusion
By conjugating LMWC with TAT-LHRH peptide, we have developed an efficient gene carrier (TLC) that is highly specific for hepatoma cells. The significant advantages of the resultant TLC include (1) stronger DNA-condensing power, which enables the formation of stable and smaller-sized polymer/DNA nanoparticles, (2) promising transgenic efficacy and high selectivity for hepatoma cells both in vitro and in vivo, and (3) low cytotoxicity. However, several hurdles remain to be overcome before the successful application of the polymer in the gene therapy of HCC. Systemic investigation is needed on the in vivo targeting specificity and tissue/organ distribution as well as its immunogenicity.
Acknowledgments
This research was jointly supported by the Natural Science Foundation of Tianjin (Grant No: 11JCZDJC20300), the National Natural Science Foundation of China (Grant No: 81271693, 31200674 and 81100100), the Peking Union Medical College Youth Fund, and Fundamental Research Funds for the Central Universities (Grant No: 3332013060).
Disclosure
The authors have no conflicts of interest in this work.
References
El-Serag HB. Hepatocellular carcinoma and hepatitis C in the United States. Hepatology. 2002;36(5 Suppl 1):S74–S83. | ||
Bosch FX, de Sanjose S, Castellsague X. The prospects of HPV vaccination in cervical cancer prevention: results of a new independent trial. Cancer Discov. 2011;1(5):377–380. | ||
Llovet JM, Bruix J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology. 2008;48(4):1312–1327. | ||
Li X, Pan Y, Fan R, et al. Adenovirus-delivered CIAPIN1 small interfering RNA inhibits HCC growth in vitro and in vivo. Carcinogenesis. 2008;29(8):1587–1593. | ||
Schwarz RE, Smith DD. Trends in local therapy for hepatocellular carcinoma and survival outcomes in the US population. Am J Surg. 2008;195(6):829–836. | ||
Deshpande R, O’Reilly D, Sherlock D. Improving outcomes with surgical resection and other ablative therapies in HCC. Int J Hepatol. 2011;2011:686074. | ||
Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362(9399):1907–1917. | ||
Levin B. Cancer prevention: new challenges and opportunities. Curr Opin Oncol. 1995;7(5):397. | ||
Okuda K. Hepatocellular carcinoma. J Hepatol. 2000;32(1 Suppl):225–237. | ||
Llovet JM, Bruix J. Novel advancements in the management of hepatocellular carcinoma in 2008. J Hepatol. 2008;48 Suppl 1:S20–S37. | ||
Arbuthnot P, Thompson LJ. Harnessing the RNA interference pathway to advance treatment and prevention of hepatocellular carcinoma. World J Gastroenterol. 2008;14(11):1670–1681. | ||
Panda JJ, Varshney A, Chauhan VS. Self-assembled nanoparticles based on modified cationic dipeptides and DNA: novel systems for gene delivery. J Nanobiotechnology. 2013;11:18. | ||
Klutz K, Schaffert D, Willhauck MJ, et al. Epidermal growth factor receptor-targeted (131)I-therapy of liver cancer following systemic delivery of the sodium iodide symporter gene. Mol Ther. 2011;19(4):676–685. | ||
Cheng MR, Li Q, Wan T, et al. Galactosylated chitosan/5-fluorouracil nanoparticles inhibit mouse hepatic cancer growth and its side effects. World J Gastroenterol. 2012;18(42):6076–6087. | ||
Meng L, Yang L, Zhao X, et al. Targeted delivery of chemotherapy agents using a liver cancer-specific aptamer. PLoS One. 2012;7(4):e33434. | ||
Minko T, Dharap SS, Fabbricatore AT. Enhancing the efficacy of chemotherapeutic drugs by the suppression of antiapoptotic cellular defense. Cancer Detect Prev. 2003;27(3):193–202. | ||
Dharap SS, Qiu B, Williams GC, Sinko P, Stein S, Minko T. Molecular targeting of drug delivery systems to ovarian cancer by BH3 and LHRH peptides. J Control Release. 2003;91(1–2):61–73. | ||
Minko T, Dharap SS, Pakunlu RI, Wang Y. Molecular targeting of drug delivery systems to cancer. Curr Drug Targets. 2004;5(4):389–406. | ||
Tang Q, Cao B, Wu H, Cheng G. Selective gene delivery to cancer cells using an integrated cationic amphiphilic peptide. Langmuir. 2012;28(46):16126–16132. | ||
Dharap SS, Wang Y, Chandna P, et al. Tumor-specific targeting of an anticancer drug delivery system by LHRH peptide. Proc Natl Acad Sci U S A. 2005;102(36):12962–12967. | ||
Yin H, Cheng KW, Hwa HL, Peng C, Auersperg N, Leung PC. Expression of the messenger RNA for gonadotropin-releasing hormone and its receptor in human cancer cell lines. Life Sci. 1998;62(22):2015–2023. | ||
Szepeshazi K, Schally AV, Treszl A, Seitz S, Halmos G. Therapy of experimental hepatic cancers with cytotoxic peptide analogs targeted to receptors for luteinizing hormone-releasing hormone, somatostatin or bombesin. Anticancer Drugs. 2008;19(4):349–358. | ||
MacLaughlin FC, Mumper RJ, Wang J, et al. Chitosan and depolymerized chitosan oligomers as condensing carriers for in vivo plasmid delivery. J Control Release. 1998;56(1–3):259–272. | ||
Richardson SC, Kolbe HV, Duncan R. Potential of low molecular mass chitosan as a DNA delivery system: biocompatibility, body distribution and ability to complex and protect DNA. Int J Pharm. 1999; 178(2):231–243. | ||
Mao HQ, Roy K, Troung-Le VL, et al. Chitosan-DNA nanoparticles as gene carriers: synthesis, characterization and transfection efficiency. J Control Release. 2001;70(3):399–421. | ||
Bloomfield VA. DNA condensation. Curr Opin Struct Biol. 1996;6(3): 334–341. | ||
Malhotra M, Tomaro-Duchesneau C, Saha S, Kahouli I, Prakash S. Development and characterization of chitosan-PEG-TAT nanoparticles for the intracellular delivery of siRNA. Int J Nanomedicine. 2013;8: 2041–2052. | ||
Oliveira AV, Silva AP, Bitoque DB, Silva GA, Rosa da Costa AM. Transfection efficiency of chitosan and thiolated chitosan in retinal pigment epithelium cells: A comparative study. J Pharm Bioallied Sci. 2013;5(2):111–118. | ||
Erbacher P, Zou S, Bettinger T, Steffan AM, Remy JS. Chitosan-based vector/DNA complexes for gene delivery: biophysical characteristics and transfection ability. Pharm Res. 1998;15(9):1332–1339. | ||
Corsi K, Chellat F, Yahia L, Fernandes JC. Mesenchymal stem cells, MG63 and HEK293 transfection using chitosan-DNA nanoparticles. Biomaterials. 2003;24(7):1255–1264. | ||
Cumber AJ, Forrester JA, Foxwell BM, Ross WC, Thorpe PE. Preparation of antibody-toxin conjugates. Methods Enzymol. 1985;112:207–225. | ||
Milani R, Monogioudi E, Baldrighi M, et al. Hydrophobin: fluorosurfactant-like properties without fluorine. Soft Matter. 2013;9:6505–6514. | ||
Bajaj G, Van Alstine WG, Yeo Y. Zwitterionic chitosan derivative, a new biocompatible pharmaceutical excipient, prevents endotoxin-mediated cytokine release. PLoS One. 2012;7(1):e30899. | ||
Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5(4):505–515. | ||
Xiao B, Wang X, Qiu Z, et al. A dual-functionally modified chitosan derivative for efficient liver-targeted gene delivery. J Biomed Mater Res A. 2013;101(7):1888–1897. | ||
Liu L, Bai Y, Zhu D, et al. Evaluation of the impact of arginine-chitosan/DNA nanoparticles on human naive CD4+ T cells. J Biomed Mater Res A.2011;96(1):170–176. | ||
Buschmann MD, Merzouki A, Lavertu M, Thibault M, Jean M, Darras V. Chitosans for delivery of nucleic acids. Adv Drug Deliv Rev. 2013;65(9):1234–1270. | ||
Li X-F, Feng X-Q, Yang S, Wang T-P, Su Z-X. Effects of molecular weight and concentration of chitosan on antifungal activity against Aspergillus niger. Iranian Polymer Journal. 2008;17(11):843–852. | ||
Wedmore I, McManus JG, Pusateri AE, Holcomb JB. A special report on the chitosan-based hemostatic dressing: experience in current combat operations. J Trauma. 2006;60(3):655–658. | ||
Garcia-Fuentes M, Alonso MJ. Chitosan-based drug nanocarriers: where do we stand? J Control Release. 2012;161(2):496–504. | ||
Kean T, Thanou M. Biodegradation, biodistribution and toxicity of chitosan. Adv Drug Deliv Rev. 2010;62(1):3–11. | ||
Huang M, Khor E, Lim LY. Uptake and cytotoxicity of chitosan molecules and nanoparticles: effects of molecular weight and degree of deacetylation. Pharm Res. 2004;21(2):344–353. | ||
Düzgünes N, De Ilarduya CT, Simões S, et al. Cationic liposomes for gene delivery: novel cationic lipids and enhancement by proteins and peptides. Curr Med Chem. 2003;10(14):1213–1220. | ||
Sato T, Ishii T, Okahata Y. In vitro gene delivery mediated by chitosan, effect of pH, serum, and molecular mass of chitosan on the transfection efficiency. Biomaterials. 2001;22(15):2075–2080. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.