Back to Journals » OncoTargets and Therapy » Volume 11
Targeting VEGF pathway to normalize the vasculature: an emerging insight in cancer therapy
Received 23 April 2018
Accepted for publication 17 July 2018
Published 17 October 2018 Volume 2018:11 Pages 6901—6909
DOI https://doi.org/10.2147/OTT.S172042
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjeev K. Srivastava
Jing-biao Wu, Ya-ling Tang, Xin-hua Liang
State Key Laboratory of Oral Diseases, National Clinical Research Center for Oral Diseases, West China Hospital of Stomatology (Sichuan University), Chengdu Sichuan, People’s Republic of China
Abstract: Vascular normalization is a new concept of targeting angiogenesis to restore vessel structure and function and to increase blood perfusion and delivery of drugs. It has been confirmed that vascular normalization can decrease relapse and benefit other cancer therapy, including chemotherapy, radiotherapy, and immune cell therapy. The key point of this therapy is to inhibit pro-angiogenic factors and make it be balanced with anti-angiogenic factors, resulting in a mature and normal vessel characteristic. Vascular endothelial growth factor (VEGF) is a key player in the process of tumor angiogenesis, and inhibiting VEGF is a primary approach to tumor vessel normalization. Herein, we review newly uncovered mechanisms governing angiogenesis and vascular normalization of cancer and place emphasis on targeting VEGF pathway to normalize the vasculature. Also, important methods to depress VEGF pathway and make tumor vascular are discussed.
Keywords: vascular normalization, vascular endothelial growth factor (VEGF), anti-angiogenesis, treatment resistance, cancer therapy
Introduction
Angiogenesis has been defined as the formation of new blood vessels from pre-existing blood vessels, which has a great impact on tumor process.
It is widely believed that tumor cannot grow beyond 2 mm3 without adequate supply of oxygen and nutrients via vessels. A number of blood vessels in solid tumor support tumor growth rapidly. However, these vessels are abnormal and characterized by leakiness on the vessel wall,1–3 increased interstitial fluid pressure (IFP),4 poor blood perfusion,5,6 drug delivery limit,7 hypoxia,8 treatment resistance,9 and so on. Folkman articulated the concept of tumor anti-angiogenesis therapy in 1970s. He proposed that blocking the formation of new blood vessels could deprive the supply of tumor nutrients, thus keeping tumor at a dormant state and tiny volume.10 The theory indicated that anti-angiogenesis therapy could completely block pro-angiogenic factors, which were overexpression in pathological angiogenesis, and contributed to a large number of vessel formation in tumor. It was confirmed that anti-angiogenesis therapy reduced tumor volume and has the ability of anti-tumor.11 However, the benefits remain modest. Excessive pruning of vessels after anti-angiogenesis therapy has been shown to increase intratumoral hypoxia, which in turn triggers pathological angiogenesis, inflammation, increased migration, and others. Moreover, it leads to radioresistance and chemoresistance, even metastasis and relapse.12
To overcome the disadvantages of anti-angiogenesis, Jain proposed a novel concept aimed at tumor angiogenesis, which was named “vascular normalization.”6 Unlike anti-angiogenesis obliterating vessels completely, this theory advocated to revert the grossly abnormal structure and function of the tumor vasculature toward a more normal state. This hypothesis suggested that sustaining the balance of pro-angiogenic and anti-angiogenic factors could lead to vascular normalization via appropriate dose of anti-angiogenic treatment. Compared to anti-angiogenesis therapy, vascular normalization is characterized by reducing vascular permeability and IFP and improving blood flow and tumor perfusion. These characteristics enhance the delivery of drugs, immune cells, and oxygen for radiation therapy.13,14 Dickson et al used bevacizumab, an inhibitor of angiogenesis via neutralizing human VEGF, to examine the vascular normalization in mice bearing human neuroblastoma xenograft.15 The result showed that vessels were more normal within 24 hours of therapy, including reductions in microvessel density (MVD) and vessel length, diameter, and tortuosity, a reduction in vascular permeability, a drop in tumor IFP, and an improvement in perfusion. Moreover, it improved tumor delivery of chemotherapeutics (topotecan and etoposide). These indicated that making vessel normalization was an effective approach to treat solid tumor.
Vascular endothelial growth factor A (VEGF-A, also called VEGF, vascular permeability factor) was isolated by Ferrara’s group,16 Plouet’s group,17 and Dvorak’s group.18 VEGF-A was demonstrated to play a dominant role in angiogenesis when Carmeliet et al showed that VEGF haplo-insufficiency in mice led to the complete absence of blood vessels and embryonic lethality.19 Blocking VEGF-A reduced tumor growth and vascular density in animal models.20 Therefore, VEGF-A quickly became the preferred target for anti-angiogenesis drugs. Importantly, recent increasing preclinical studies have provided an insight that anti-VEGF therapy can initiate vascular normalization. The wide application in clinic to target VEGF is pharmacological inhibitors including anti-VEGF agents, anti-VEGFR,14 and targeting VEGF pathway such as gene model or some upstream factors. And the regulation of VEGF expression is complex and is affected by multiple factors such as hypoxia, signal transducers and activations of transcription, NO gradient, microRNA, and other factors.21,22 Hence, we review the mechanism of targeting VEGF pathway (Figure 1) and summarize some effective methods to normalize vasculature via VEGF targets, in order to provide a novel therapeutic strategy to improve treatment of anti-tumor.
 | Figure 1 Effects of HIF-1α, STAT3, NO, and miRNA in VEGF expression. |
Mechanism of inhibiting VEGF expression to normalize tumor vessels
In tumor, the overexpression of VEGF could be triggered by many factors, among which we summarize some mechanisms that being inhibited might lead to vascular normalization. When we target these key factors associated with VEGF expression, the VEGF expression would be decreased directly or indirectly to promote vascular normalization.23
Hypoxia-inducible factor 1α (HIF-1α)
As a hallmark of tumor metabolic microenviroment, hypoxia plays a critical role in abnormal angiogenesis which induces VEGF expression via HIF-1α. HIF-1α is one of subunits of HIF-1, and another is HIF-1β.24 In the presence of oxygen, beta subunit and alpha subunit were degraded, but in hypoxia, alpha subunit was accumulated and dimerized with beta subunit.25 Then HIF-1 complex can bind to VEGF gene 5′ hypoxia-responsive element (HRE) to contribute to transcription and VEGF overexpression.26 It has been proved that increase of HIF-1α promotes tumor angiogenesis while deletion of HIF-1α impairs tumor angiogenesis and leads to vessel normalization.27,28 Zhu et al discovered that silencing HIF-1α reduces hypoxia-induced angiogenesis and promotes sensitivity in lung cancer xenograft model.29 Lee et al used prostate cancer xenograft model and revealed that blocking of HIF-1α inhibited tumor growth and reduced vessel density which was identified by immunohistochemistry for CD31.30 It has been shown that HIF-1α-VEGF axis is a primary pathway of HIF-1 inducing VEGF, inhibiting of which leads to vessel normalization.
Nitric oxide (NO)
In tumor microenvironment, NO is an important gaseous mediator for vessels maturation. Perivascular NO mediates angiogenesis, PVC recruitment, and stabilization of blood vessel.22,31 The effects of NO regulation of vessel maturation depend on localization, concentration, and duration of NO exposure and cellular sensitivity to NO.22 NO induces the expression of multiple endogenous angiogenic factors, such as VEGF and basic fibroblast growth factor.32–34 NO can upregulate the activity of VEGF promoter, and the primary target of NO is the HRE, which indicated that the role of NO in vessel normalization may be relative to VEGF.35
Nitric oxide synthase (NOS) exists in three isoforms: neuronal NOS (nNOS, encoded by NOS1), inducible NOS (iNOS, encoded by NOS2), and endothelial NOS (eNOS encoded by NOS3). In normal tissues, eNOS mediates vessel maturation which is mainly expressed in vascular endothelium and its catalyzed product NO generated well gradients around vessels. But in tumor tissues, one or multiple NOSs are expressed by tumor cells. The extra NO not only induces VEGF expression and promotes angiogenesis but also interferes the perivascular NO gradients and destroys the vessels stabilization.36 Tumor cells express iNOS or nNOS or both of them but not eNOS, depending on the tumor type and stage. NO which is produced by iNOS or nNOS contribute to the perivascular NO gradients distraction and abnormalities of vessels.
Kashiwagi et al proposed a hypothesis that localizing NO selectively around blood vessels and restoring the NO gradients could normalize tumor vasculature.36 The theory indicated that if specifically silencing or blocking nNOS or iNOS in tumor cells, the morphology and function of tumor vessels would be improved. To test this hypothesis, Kashiwagi et al36 used U87MG human glioma xenograft model, in which NO is produced by nNOS in tumor cells and eNOS in vascular endothelial cells. In control U87MG tumors, NO was observed in both the vascular region and tumor parenchyma. In NOS1-silenced U87MG tumors, NO was predominantly localized around blood vessels. The results showed that in NOS1-silenced U87MG tumors, both morphology and function of vessels were improved, including reduced vessel tortuosity, improved PVCs coverage, decreased vessel diameter, lower permeability, improved tumor oxygenation, and increased benefit of radiotherapy. However, inhibition of eNOS or all NOS isoforms got one opposite result and could not make tumor vessels similar to normal tissue. These results suggested that establishment of NO gradients improved tumor vessel morphology and function, leading to a more normal and mature vasculature.
Signal transducer and activation of transcription 3 (STAT3) regulates VEGF expression
STAT3 has been referred as a critical role in oncogenesis through preventing programmed cell death and enhancing cell proliferation.37,38 STAT3 mediates tumor initiation, progression, chemoresistance, and angiogenesis and is activated in many human cancers including hepatocellular carcinoma, breast cancer, prostate cancer, multiple myeloma, head and neck squamous cell carcinoma, etc.39–41 Recently, Niu et al have shown that constitutively activated STAT3 upregulated VEGF expression in ten human cancer cell lines, which spanned three types of cancer, breast cancer, head and neck neoplasm, and melanoma.42 And previous study revealed that interrupting STAT3 signaling in B16 tumor cells inhibited VEGF expression, through B16 tumor cell transfected with an expression vector encoding a dominant negative variant of STAT3 protein, or STAT3 antisense oligonucleotide. These results suggested that STAT3 as an anti-VEGF target had ability to result in vascular normalization.
miRNAs associated with vascular normalization
miRNAs are 19–25 single-stranded non-coding RNAs that regulate mRNA stability and translation and are implicated in many tumor processes including metabolism, angiogenesis, autophagy, invasion, and metastasis.43,44 miRNAs that mediate the angiogenic switch are termed as angiomiRs, which regulate vascular integrity and modulate endothelial cells (ECs) functions tightly.45,46
It has been shown that targeting angiomiRs was an effective approach to make tumor vessels normalized.47,48 miR-126 is wildly expressed in endothelial cells from veins and arteries, which is deemed to be a master regulator of angiogenesis and vascular integrity. It promotes tumor angiogenesis through the regulation of downstream growth factors like VEGF, bFGF, and EGF.49,50 Van Solingen et al used murine ischemic hindlimb model injected with a single dose of antagomir-126 to verify whether silencing of miR-126 impairs angiogenesis.51 They analyzed CD31-stained capillaries in sections and discovered that mice treated with antagomir-126 showed a markedly lower density of capillary vessels. miR-20b was reported as a negative regulator of VEGF in nasopharyngeal carcinoma epithelioid cells and breast cancer cells under hypoxia.52,53 The mechanism was that the nuclear accumulation of HIF-1α and STAT3 were strongly inhibited by miR-20b. miR-93 promotes tumor growth and angiogenesis by targeting integrin β8.54 miR-107 is a microRNA regulated by p53. Yamakuchi et al revealed that overexpression of miR-107 decreased VEGF and HIF-1β, while knockdown of endogenous miR-107 increased VEGF levels in HCT116 cells.55 They injected nude mice with HCT116 cells that had been transduced with LV-GFP-miR-107. The results showed that the tumor volume and numbers of vessels were decreased significantly compared to control groups. miR-874 is a suppressive regulator in angiogenesis by targeting STAT3 in gastric cancer, and inhibiting miR-874 increased VEGF level and MVD.56 Therefore, targeting miRNA signaling pathways in tumor cells provides new therapeutic avenues to suppress tumor pathologic angiogenesis and make vessel normalization. We have summarized some miRNAs that might be targets of vascular normalization in Table 1.
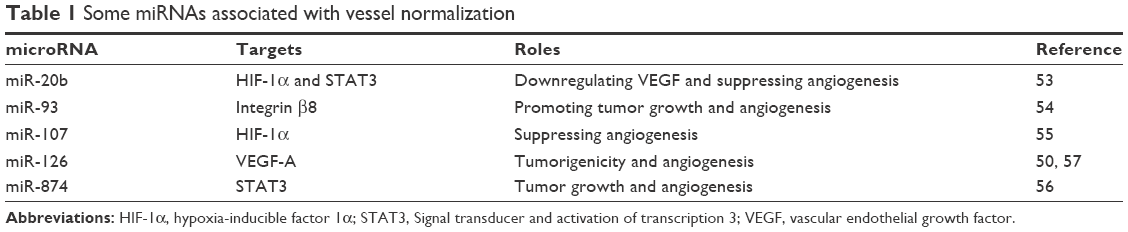 | Table 1 Some miRNAs associated with vessel normalization |
Methods of inhibiting VEGF expression to normalize tumor vessels
Some common pharmacological inhibitors of VEGF pathway
Bevacizumab
Bevacizumab is the first drug to be applied in clinic to anti-angiogenesis in many solid tumors, which have been approved for first-line treatment in renal cancer.57 Bevacizumab is a humanized monoclonal antibody. The anti-angiogenesis mechanism of bevacizumab is that it neutralizes VEGF released by tumor cells, causing the inhibition of tumor angiogenesis and growth. Recently, it has been reported that bevacizumab can not only inhibit angiogenesis but also induce vessel normalization in preclinical and clinical trials. Some rectal cancer studies have shown that vessel normalization occurred when patients were treated with 5 mg/kg bevacizumab. The vessel normalization was evaluated by tumor physiology (blood perfusion, blood volume, permeability, MVD, perivascular coverage, and IFP), systemic response, and tumor response via biopsies, functional computed tomography scans and positron emission tomography scans. In a clinical trail, six patients with primary and locally advanced adenocarcinoma of the rectum had been treated with bevacizumab (5 mg/kg intravenously). Twelve days after bevacizumab infusion, a decrease in blood perfusion, vascular volume, MVD, IFP, and permeability surface product and an increase in drug delivery and the fraction of vessels with pericyte coverage were observed.58 Similarly Willett CG’s study observed the same result in rectal cancer patients with 5 mg/kg bevacizumab administration after 12 days.59 However, 12 days after the 10 mg/kg bevacizumab dose, MVD, blood flow, and IFP were reduced, but fluorodeoxyglucose uptake and permeability surface product did not change because a high dose of 10 mg/kg induced dose-limiting toxicities and excessive destruction on vessels.
DC101
DC101 is a rat monoclonal antibody against mouse VEGFR2, which is expressed highly in ECs of the tumor vasculature and mediates most of the angiogenic properties of VEGF. Specific VEGFR2 pathway blockade with DC101 provides an insight into normalization of tumor vessels. Studies have reported that DC101 therapy induced vessel normalization in mice bearing lung, breast, colorectal, and glioblastoma multiforme tumors.60,61 After DC101 administration in these model, the characteristics of vascular normalization was evaluated by intravital microscopy and dorsal skin fold chambers. The results showed the reduction of MVD and vessel diameter, an increase in PVC coverage, a decrease in IFP, and even a modification in vascular basement membrane.60 Additionally, another study used orthotopic mammary tumor mouse model to validate that normalizing the abnormal blood vessels cloud improve the delivery and effectiveness of chemotherapeutics.61 Vessel normalization by DC101 decreased IFP in tumor and reduced vessel wall pore sizes, allowing small nanoparticles to enter them more rapidly.
Ramucirumab
Ramucirumab is an entirely humanized monoclonal antibody directed against VEGFR2, which was approved for patients with advanced or metastatic gastric cancer or gastroesophageal junction carcinoma. Its role for metastatic breast cancer or advanced non-small-cell lung cancer is still debated.62 With a similar mechanism of anti-angiogenesis to DC101, ramucirumab is thought to have potential ability to normalize tumor vessels via blocking specific VEGFR2 pathway. One study demonstrated that it decreased vascularity and permeability in a liver metastasis after treatment with ramucirumab.63 However, this trial lacked further studies about the characteristics of tumor vascular normalization, which still needs more trials to validate it.
Sunitinib
VEGFR on ECs is tyrosine kinase-dependent receptor, and inhibiting tyrosine kinase allows anti-angiogenesis. It is one of the most representative tyrosine kinase inhibitors of VEGFRs. Studies showed that sunitinib treatment of gliomas reduced tumor IFP and increased the delivery of chemotherapy (temozolomide) to tumor cells.64,65 Temozolomide delivery to tumors directly correlated with the degree of PVC coverage and inversely correlated with the density of the abnormally thick collagen IV basement membrane. It suggested that sunitinib could induce vessel normalization in solid tumor.
Everolimus
The mammalian target of rapamycin (mTOR), a multifunctional signal transducing protein, which is not only a crucial regulator for cell growth and metabolism but also a promoter on angiogenesis. mTOR increases the translation of HIF-1/HIF-2, activating the expression of HIF-1α responsive genes, including VEGF, platelet-derived growth factor-β, transforming growth factor-α (TGF-α). It has been validated that inhibiting mTOR-HIF-1α-VEGF pathway contributes to vascular normalization in solid tumor. Everolimus is the primary inhibitor of this pathway, and it has been approved for clinical application.66
Everolimus has been shown to have significant effects including reduction in expression of HIF-1 and VEGF in cultured tumor cells, and reduction in microvessel area and blood vessel density in several different tumor model, such as renal cell cancer, ovarian cancer, and glioma xenografts.66–69 Moreover, it has been proved that everolimus has the ability of prolonging survival and increases radiosensitivity in solid tumor.68,70
Interestingly, Chen et al suggested that mTOR also promoted macrophage-induced angiogenesis through STAT3, and the effects of mTOR inhibitor were non-overlapping with anti-VEGF therapies.71 Based on this concept, it indicated that bevacizumab and everolimus in combination with chemotherapy would coordinately target parallel angiogenic signaling pathways and emerge a synergistic action to improve vessel normalization and suppress some potential resistance mechanisms.72
VEGF gene alternative splicing
VEGF is grouped into two protein families, which is resulted from different alternative selection of the pre-mRNA splicing site. Proximal splice-site selection in exon 8 (exon 8a) results in pro-angiogenic VEGFxxx isoforms, whereas distal splice-site selection in exon 8 (exon 8b) results in anti-angiogenic VEGFxxxb isoforms, where xxx means the number of amino acids on the polypeptide chain.73 The difference of two isoforms in structure is the six different amino acids in C-terminal region.74 VEGFxxx family members consist of VEGF189, VEGF165, VEGF121, and other variants. VEGFxxxb family is composed by VEGF121b, 145b, 165b, 183b, and VEGF189b.75–77
The selection of splicing suggests that control of splicing might be an important regulatory mechanism of VEGF expression. Thus, if we inhibit the pro-angiogenic splicing pathway, namely VEGFxxx selection splicing, the vessel growth will be suppressed, subsequently vasculature normalization will occur in tumor. Indeed many studies suggested that it significantly inhibited EC proliferation, angiogenesis, and tumor growth when the C-terminal region of VEGF was changed.78,79
VEGF165b isoform seems to be an endogenous anti-angiogenic agent, which could neutralize VEGF165 in vivo.80 Studies have shown that VEGF165b overexpression inhibits the growth of colon and prostate tumors, kidney cell carcinoma, Ewing sarcoma, and metastatic melanoma in animal models.80–82
Base on the selection of splicing site to VEGF isoforms, two possibilities of therapy were proposed. The first is specific antibody blocking for the exon 8a, resulting in the inhibition of angiogenic isoform. The second is using molecular inhibitors that could alter the splicing mechanism for the expression of VEGFxxxb isoform in vivo and resulting in vessel normalization.83 To date, documents have not shown much about the pathways that regulate alternative splicing of VEGF pre-mRNA in general and of the exon 8a/8b alternative 3′ splice site in particular. But some studies suggest that regulatory splicing factors (particularly SRp55) and growth factors (especially TGF-β1) can preferentially select the distal splice site.75 The regulatory splicing factors include the SR proteins (eg, 9G8, ASF/SF2, SRp40, SRp55, among others) which regulate pre-mRNA binding to enhancer and silencer.84
SiRNA implication in vessel normalization
siRNA is a <30 nucleotides long mRNA sequence, and siRNA recognizes that dsRNAs combine with the homogenous mRNA sequence and degrade it.85,86 It has been shown that this kind of gene silencing provided a new approach to study and cure some gene-associated diseases, such as oncogene, gene associated with anti-apoptosis, growth factors, and angiogenesis.87–89
Utilizing siRNA to inhibit angiogenesis in tumor is a novel but effective approach to treat solid tumors through normalizing tumor vasculature. In the retinoblastoma xenograft model, it was observed that siRNA targeting VEGF suppressed angiogenesis and made vessels normalized.90 In this study, the result showed that VEGF expression was strongly reduced by 87% and the tumor volume was reduced by 66% compared to controls. More crucial, the MVD (0.42±0.24) were significantly decreased in tumors treated by VEGF siRNA, compared to control (2.04±0.28). This was in line with the results of Zhang et al, who demonstrated that anti-HIF-1α reduced VEGF level and MVD in osteosarcoma xenograft model91 and reduced resistance against radiotherapy in lung cancer xenograft model.29
Summary and perspective
Since Folkman first hypothesized the importance of angiogenesis in tumor progression, anti-angiogenesis therapy has become a novel strategy to cure solid tumor. Unlike conventional anti-cancer therapies designed to directly kill proliferating malignant cells, anti-angiogenesis therapy is to destroy tumor vasculature. In this concept, the regression of tumor vessels could make tumor be a tiny and dormant state and turn cancer from a killer into a chronic asymptomatic disease. In the past 40 years, this hypothesis has been evaluated in many preclinical studies. These data suggested that anti-angiogenesis therapy indeed eliminated tumor vessels and inhibited tumor growth to some degree, but could not keep tumor in a dormant state. Since the US Food and Drug Administration granted approval to the first anti-angiogenesis drug bevacizumab, anti-angiogenesis therapy began to be widely used in clinic to treat various tumors. Later, many other anti-angiogenic drugs have been approved in succession. Unfortunately, the benefits of these anti-angiogenesis agents proved to be modest in a large number of clinical trials.92,93 The disparity between the theory and medical reality anti-angiogenesis therapy might attribute to the difference of human cancers and xenograft model, for example, the growth rate of human tumor is slower, the time scale is longer, and each human patient with metastatic disease typically has multiple lesions. On the other hand, excessive vessel pruning leads to hypoxia and acidosis and even induces metastasis and relapse.94,95
Based on anti-angiogenesis theory, Jain hypothesized the theory of vasculature normalization and tried to make up the defects of anti-angiogenesis therapy. The hypothesis suggested that judicious dosing and administrative time of anti-angiogenic agents can restore the structure and function of vessels by decreasing hypoxia and vascular permeability and improving pericyte coverage, tumor perfusion, and blood flow, and synergize with immunotherapy and chemotherapy. Vascular normalization therapy exhibited benefits not only in preclinical trial but also in clinical trials. Although a large development was observed in patients with cancers, the concept of vascular normalization as a therapeutic strategy against tumors is relatively young and has some limitations. First, human tumors presented spatial and temporal heterogeneity, and the effects of vascular normalization maybe different from animal models. For instance, the fundamental effect of anti-angiogenesis therapy in many animal models (ie, tumor growth delay or shrinkage) is not generally seen in humans. Second, the state of vascular normalization is not permanent but transient, and the time window is difficult to confirm. Only in time window, the vessels present normal characteristics in structure and function. After time window, the tumor vessels return to abnormal, which is deemed to be relevant to activation of alternative pro-angiogenic signaling pathways. However, appearing point and period of time window rely on agent types and dose, tumor types, and locations. Therefore, the challenge is inducing time window by optimal dose and prolonging it.96
As the most important factor in angiogenesis, VEGF is a commendable target to make tumor vessel normalization. We summarize some key roles in VEGF expression, including NO gradient, upstream regulator like HIF-1, and transcription modulation like STAT3. We provide some common methods to normalize the tumor vasculature through targeting VEGF. It deserves to consider how to control its anti-angiogenensis effect and promote the normalization of tumor blood vessels. Optimal dose and duration of treatment control the effect anti-angiogenesis. As mentioned earlier, in Willett CG’s study, normalized vessels were observed in rectal cancer patients with 5 mg/kg bevacizumab administration after 12 days, but not in the group with 10 mg/kg bevacizumab.59 High dose of bevacizumab blocked VEGF completely and made excessive destruction on vessels. In this study, 5 mg/kg dose and 12 days of bevacizumab seemed like the optimal dose and duration in rectal patients. Due to tumor heterogeneity, the optimal dose and time are different and dubious in various tumors, which needs many trials to confirm. In addition, a long period of inhibiting VEGF might activate other potential alternative pathways for neovascularization, such as Ang-Tie2 axis.97 So in preclinic or clinic, only targeting VEGF might not acquire enough normalization, and multiple anti-angiogenesis approaches might make vessel more normal.
Although vascular normalization of anti-VEGF therapeutic strategy is faced with several challenges, the development of theory and drugs would lead it toward a more efficient and successful clinical application in the future. Recently, nanoparticles (50–200 nm size) presented a favorable ability of anti-angiogenesis for they have greater permeability, better penetrance, and retention.98,99 The treatment might be greatly better when nanoparticles are implicated in vascular normalization of anti-VEGF therapy.
Acknowledgment
This work was supported by National Program on Key Research Project of China (2016YFC0902700), National Natural Science Foundation of China grants (Nos 81672672, 81572650, 81772891, and 81621062), and State Key Laboratory of Oral Diseases Special Funded Projects.
Disclosure
The authors report no conflicts of interest in this work.
References
Gerlowski LE, Jain RK. Microvascular permeability of normal and neoplastic tissues. Microvasc Res. 1986;31(3):288–305. | ||
Jain RK. Transport of molecules across tumor vasculature. Cancer Metastasis Rev. 1987;6(4):559–593. | ||
Hobbs SK, Monsky WL, Yuan F, et al. Regulation of transport pathways in tumor vessels: Role of tumor type and microenvironment. Proc Natl Acad Sci U S A. 1998;95(8):4607–4612. | ||
Stohrer M, Boucher Y, Stangassinger M, Jain RK. Oncotic pressure in solid tumors is elevated. Cancer Res. 2000;60:4251–4255. | ||
Jain RK. Determinants of tumor blood flow: a review. Cancer Res. 1988;48(10):2641–2658. | ||
Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307(5706):58–62. | ||
Jain RK. 1995 Whitaker Lecture: delivery of molecules, particles, and cells to solid tumors. Ann Biomed Eng. 1996;24(4):457–473. | ||
Facciabene A, Peng X, Hagemann IS, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and Treg cells. Nature. 2011;475(7355):226–230. | ||
Wang J, Zhu C. Anticoagulation in combination with antiangiogenesis and chemotherapy for cancer patients: evidence and hypothesis. Onco Targets Ther. 2016;9:4737–4746. | ||
Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285(21):1182–1186. | ||
Folkman J. Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov. 2007;6(4):273–286. | ||
Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8(8):592–603. | ||
Jain RK. Normalizing tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy. Nat Med. 2001;7(9):987–989. | ||
Goel S, Duda DG, Xu L, et al. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev. 2011;91(3):1071–1121. | ||
Dickson PV, Hamner JB, Sims TL, et al. Bevacizumab-induced transient remodeling of the vasculature in neuroblastoma xenografts results in improved delivery and efficacy of systemically administered chemotherapy. Clin Cancer Res. 2007;13(13):3942–3950. | ||
Leung D, Cachianes G, Kuang W, Goeddel D, Ferrara N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science. 1989;246(4935):1306–1309. | ||
Plouet J, Schilling J, Gospodarowicz D. Isolation and characterization of a newly identified endothelial-cell mitogen produced by Att-20 cells. EMBO J. 1989;8(12):3801–3806. | ||
Senger D, Galli S, Dvorak A, Perruzzi C, Harvey V, Dvorak H. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science. 1983;219(4587):983–985. | ||
Carmeliet P, Ferreira V, Breier G, et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380(6573):435–439. | ||
Kim KJ, Li B, Winer J, et al. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature. 1993;362(6423):841–844. | ||
Ferrara N. Vascular endothelial growth factor: basic science and clinical progress. Endocr Rev. 2004;25(4):581–611. | ||
Kashiwagi S, Izumi Y, Gohongi T, et al. NO mediates mural cell recruitment and vessel morphogenesis in murine melanomas and tissue-engineered blood vessels. J Clin Invest. 2005;115(7):1816–1827. | ||
Konno H, Yamamoto M, Ohta M. Recent concepts of antiangiogenic therapy. Surg Today. 2010;40(6):494–500. | ||
Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem. 1995;270(3):1230–1237. | ||
Mole DR, Maxwell PH, Pugh CW, Ratcliffe PJ. Regulation of HIF by the von Hippel-Lindau tumour suppressor: implications for cellular oxygen sensing. IUBMB Life. 2001;52(1–2):43–47. | ||
Ema M. Taya S, Yokotani N, Youichimatsuda KS, Fujii-kuriyama Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1α regulates the VEGF expression and is potentially involved in lung and vascular development. Proc Natl Acad Sci U S A. 1997;94:4273–4278. | ||
Azoitei N, Becher A, Steinestel K, et al. PKM2 promotes tumor angiogenesis by regulating HIF-1α through NF-κB activation. Mol Cancer. 2016;15(1):3. | ||
Yang Y, Sun M, Wang L, Jiao B, Hifs JBH. HIFs, angiogenesis, and cancer. J Cell Biochem. 2013;114(5):967–974. | ||
Zhu H, Zhang S. Hypoxia inducible factor-1α/vascular endothelial growth factor signaling activation correlates with response to radiotherapy and its inhibition reduces hypoxia-induced angiogenesis in lung cancer. J Cell Biochem. 2018 Epub Jun 15:1–12. | ||
Lee K, Zhang H, Qian DZ, Rey S, Liu JO, Semenza GL. Acriflavine inhibits HIF-1 dimerization, tumor growth, and vascularization. Proc Natl Acad Sci U S A. 2009;106(42):17910–17915. | ||
Yu J, Demuinck ED, Zhuang Z, et al. Endothelial nitric oxide synthase is critical for ischemic remodeling, mural cell recruitment, and blood flow reserve. Proc Natl Acad Sci U S A. 2005;102(31):10999–11004. | ||
Dulak J, Jozkowicz A, Dembinska-Kiec A, et al. Nitric oxide induces the synthesis of vascular endothelial growth factor by rat vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2000;20(3):659–666. | ||
Chin K, Kurashima Y, Ogura T, Tajiri H, Yoshida S, Esumi H. Induction of vascular endothelial growth factor by nitric oxide in human glioblastoma and hepatocellular carcinoma cells. Oncogene. 1997;15(4):437–442. | ||
Ziche M, Parenti A, Ledda F, et al. Nitric oxide promotes proliferation and plasminogen activator production by coronary venular endothelium through endogenous bFGF. Circ Res. 1997;80(6):845–852. | ||
Kimura H, Weisz A, Kurashima Y, et al. Hypoxia response element of the human vascular endothelial growth factor gene mediates transcriptional regulation by nitric oxide: control of hypoxia-inducible factor-1 activity by nitric oxide. Blood. 2000;95(1):189–197. | ||
Kashiwagi S, Tsukada K, Xu L, et al. Perivascular nitric oxide gradients normalize tumor vasculature. Nat Med. 2008;14(3):255–257. | ||
Mukherjee T, Hombría JC-G, Zeidler MP. Opposing roles for Drosophila JAK/STAT signalling during cellular proliferation. Oncogene. 2005;24(15):2503–2511. | ||
Grandis JR, Drenning SD, Zeng Q, et al. Constitutive activation of Stat3 signaling abrogates apoptosis in squamous cell carcinogenesis in vivo. Proc Natl Acad Sci U S A. 2000;97(8):4227–4232. | ||
Wang S-C, Zhang L, Hortobagyi GN, Hung M-C. Targeting HER2: Recent developments and future directions for breast cancer patients. Semin Oncol. 2001;28(6 Suppl 18):21–29. | ||
Grivennikov SI, Karin M. Dangerous liaisons: STAT3 and NF-κB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010;21(1):11–19. | ||
Niu G, Heller R, Catlett-Falcone R, et al. Gene therapy with dominant-negative Stat3 suppresses growth of the murine melanoma B16 tumor in vivo. Cancer Res. 1999;59(20):5059–5063. | ||
Niu G, Wright KL, Huang M, et al. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene. 2002;21(13):2000–2008. | ||
Choudhry H, Harris AL, Mcintyre A. The tumour hypoxia induced non-coding transcriptome. Mol Aspects Med. 2016;47–48:3535–3553. | ||
Camps C, Saini HK, Mole DR, et al. Integrated analysis of microRNA and mRNA expression and association with HIF binding reveals the complexity of microRNA expression regulation under hypoxia. Mol Cancer. 2014;13(1):28. | ||
Wang S, Olson EN. AngiomiRs – Key regulators of angiogenesis. Curr Opin Genet Dev. 2009;19(3):205–211. | ||
Anand S, Cheresh DA. MicroRNA-mediated regulation of the angiogenic switch. Curr Opin Hematol. 2011;18(3):171–176. | ||
Matejuk A, Collet G, Nadim M, Grillon C, Kieda C. MicroRNAs and tumor vasculature normalization: impact on anti-tumor immune response. Arch Immunol Ther Exp. 2013;61(4):285–299. | ||
Collet G, Skrzypek K, Grillon C, et al. Hypoxia control to normalize pathologic angiogenesis: Potential role for endothelial precursor cells and miRNAs regulation. Vascul Pharmacol. 2012;56(5–6):252–261. | ||
Wang S, Aurora AB, Johnson BA, et al. The Endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev Cell. 2008;15(2):261–271. | ||
Fish JE, Santoro MM, Morton SU, et al. miR-126 regulates angiogenic signaling and vascular integrity. Dev Cell. 2008;15(2):272–284. | ||
van Solingen C, Seghers L, Bijkerk R, et al. Antagomir-mediated silencing of endothelial cell specific microRNA-126 impairs ischemia-induced angiogenesis. J Cell Mol Med. 2009;13(8a):1577–1585. | ||
Hua Z, Lv Q, Ye W, et al. MiRNA-directed regulation of VEGF and other angiogenic factors under hypoxia. PLoS One. 2006;1(1):e116. | ||
Cascio S, D’Andrea A, Ferla R, et al. miR-20b modulates VEGF expression by targeting HIF-1 alpha and STAT3 in MCF-7 breast cancer cells. J Cell Physiol. 2010;224(1):242–249. | ||
Fang L, Deng Z, Shatseva T, et al. MicroRNA miR-93 promotes tumor growth and angiogenesis by targeting integrin-β8. Oncogene. 2011;30(7):806–821. | ||
Yamakuchi M, Lotterman CD, Bao C, et al. P53-induced microRNA-107 inhibits HIF-1 and tumor angiogenesis. Proc Natl Acad Sci U S A. 2010;107(14):6334–6339. | ||
Zhang X, Tang J, Zhi X, et al. miR-874 functions as a tumor suppressor by inhibiting angiogenesis through STAT3/VEGF-A pathway in gastric cancer. Oncotarget. 2015;6(3):1605–1617. | ||
Inglis DJ, Lavranos TC, Beaumont DM, et al. The vascular disrupting agent BNC105 potentiates the efficacy of VEGF and mTOR inhibitors in renal and breast cancer. Cancer Biol Ther. 2014;15(11):1552–1560. | ||
Willett CG, Boucher Y, di Tomaso E, et al. Direct evidence that the VEGF-specific antibody bevacizumab has antivascular effects in human rectal cancer. Nat Med. 2004;10(2):145–147. | ||
Willett CG, Boucher Y, Duda DG, et al. Surrogate markers for antiangiogenic therapy and dose-limiting toxicities for bevacizumab with radiation and chemotherapy: continued experience of a phase I trial in rectal cancer patients. J Clin Oncol. 2005;23(31):8136–8139. | ||
Tong RT, Boucher Y, Kozin SV, Winkler F, Hicklin DJ, Jain RK. Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res. 2004;64(11):3731–3736. | ||
Chauhan VP, Stylianopoulos T, Martin JD, et al. Normalization of tumour blood vessels improves the delivery of nanomedicines in a size-dependent manner. Nat Nanotechnol. 2012;7(6):383–388. | ||
Aprile G, Fontanella C, Rihawi K, Grossi F, Rijavec E. Ramucirumab: preclinical research and clinical development. Onco Targets Ther. 2014;7:1997–2006. | ||
Spratlin JL, Cohen RB, Eadens M, et al. Phase I Pharmacologic and biologic study of ramucirumab (IMC-1121B), a fully human immunoglobulin G1 monoclonal antibody targeting the vascular endothelial growth factor receptor-2. J Clin Oncol. 2010;28(5):780–787. | ||
Zhou Q, Gallo JM. Differential effect of sunitinib on the distribution of temozolomide in an orthotopic glioma model. Neuro Oncol. 2009;11(3):301–310. | ||
Zhou Q, Guo P, Gallo JM. Impact of angiogenesis inhibition by sunitinib on tumor distribution of temozolomide. Clin Cancer Res. 2008;14(5):1540–1549. | ||
Atkins MB, Hidalgo M, Stadler WM, et al. Randomized phase II study of multiple dose levels of CCI-779, a novel mammalian target of rapamycin kinase inhibitor, in patients with advanced refractory renal cell carcinoma. J Clin Oncol. 2004;22(5):909–918. | ||
Mabuchi S, Altomare DA, Connolly DC, et al. RAD001 (everolimus) delays tumor onset and progression in a transgenic mouse model of ovarian cancer. Cancer Res. 2007;67(6):2408–2413. | ||
Mabuchi S, Altomare DA, Cheung M, et al. RAD001 inhibits human ovarian cancer cell proliferation, enhances cisplatin-induced apoptosis, and prolongs survival in an ovarian cancer model. Clin Cancer Res. 2007;13(14):4261–4270. | ||
Shinohara ET, Cao C, Niermann K, et al. Enhanced radiation damage of tumor vasculature by mTOR inhibitors. Oncogene. 2005;24(35):5414–5422. | ||
Manegold PC, Paringer C, Kulka U, et al. Antiangiogenic therapy with mammalian target of rapamycin inhibitor RAD001 (everolimus) increases radiosensitivity in solid cancer. Clin Cancer Res. 2008;14(3):892–900. | ||
Chen W, Ma T, Shen X-N, et al. Macrophage-induced tumor angiogenesis is regulated by the TSC2-mTOR pathway. Cancer Res. 2012;72(6):1363–1372. | ||
Rangwala F, Bendell JC, Kozloff MF, et al. Phase I study of capecitabine, oxaliplatin, bevacizumab, and everolimus in advanced solid tumors. Invest New Drugs. 2014;32(4):700–709. | ||
Biselli-Chicote PM, Oliveira ARCP, Pavarino EC, Goloni-Bertollo EM. VEGF gene alternative splicing: pro- and anti-angiogenic isoforms in cancer. J Cancer Res Clin Oncol. 2012;138(3):363–370. | ||
Ladomery MR, Harper SJ, Bates DO. Alternative splicing in angiogenesis: The vascular endothelial growth factor paradigm. Cancer Lett. 2007;249(2):133–142. | ||
Nowak DG, Woolard J, Amin EM, et al. Expression of pro- and anti-angiogenic isoforms of VEGF is differentially regulated by splicing and growth factors. J Cell Sci. 2008;121(20):3487–3495. | ||
Harper SJ, Bates DO. VEGF-A splicing: the key to anti-angiogenic therapeutics? Nat Rev Cancer. 2008;8(11):880–887. | ||
Rosales-Torres AM, Alonso I, Vergara M, et al. Vascular endothelial growth factor isoforms 120, 164 and 205 are reduced with atresia in ovarian follicles of sheep. Anim Reprod Sci. 2010;122(1–2):111–117. | ||
Bates DO, Cui TG, Doughty JM, et al. VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, is down-regulated in renal cell carcinoma. Cancer Res. 2002;62(14):4123–4131. | ||
Woolard J, Wang WY, Bevan HS, et al. VEGF165b, an inhibitory vascular endothelial growth factor splice variant: mechanism of action, in vivo effect on angiogenesis and endogenous protein expression. Cancer Res. 2004;64(21):7822–7835. | ||
Woolard J, Wang WY, Bevan HS, et al. VEGF165b, an inhibitory vascular endothelial growth factor splice variant: mechanism of action, in vivo effect on angiogenesis and endogenous protein expression. Cancer Res. 2004;64(21):7822–7835. | ||
Rennel ES, Waine E, Guan H, et al. The endogenous anti-angiogenic VEGF isoform, VEGF165b inhibits human tumour growth in mice. Br J Cancer. 2008;98(7):1250–1257. | ||
Varey AHR, Rennel ES, Qiu Y, et al. VEGF165b, an antiangiogenic VEGF-A isoform, binds and inhibits bevacizumab treatment in experimental colorectal carcinoma: balance of pro- and antiangiogenic VEGF-A isoforms has implications for therapy. Br J Cancer. 2008;98(8):1366–1379. | ||
Rennel ES, Harper SJ, Bates DO. Therapeutic potential of manipulating VEGF splice isoforms in oncology. Future Oncol. 2009;5(5):703–712. | ||
Cáceres JF, Kornblihtt AR. Alternative splicing: multiple control mechanisms and involvement in human disease. Trends Genet. 2002;18(4):186–193. | ||
Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391(6669):806–811. | ||
Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411(6836):494–498. | ||
Cioca DP, Aoki Y, Kiyosawa K. RNA interference is a functional pathway with therapeutic potential in human myeloid leukemia cell lines. Cancer Gene Ther. 2003;10(2):125–133. | ||
Holle L, Hicks L, Song W, Holle E, Wagner T, Yu X. Bcl-2 targeting siRNA expressed by a T7 vector system inhibits human tumor cell growth in vitro. Int J Oncol. 2004;24(3):615–621. | ||
Zhang L, Yang N, Mohamed-Hadley A, Rubin SC, Coukos G. Vector-based RNAi, a novel tool for isoform-specific knock-down of VEGF and anti-angiogenesis gene therapy of cancer. Biochem Biophys Res Commun. 2003;303(4):1169–1178. | ||
Jia RB, Zhang P, Zhou YX, et al. VEGF-targeted RNA interference suppresses angiogenesis and tumor growth of retinoblastoma. Ophthalmic Res. 2007;39(2):108–115. | ||
Zhang XD, Wu Q, Yang S. Effects of siRNA-mediated HIF-1α gene silencing on angiogenesis in osteosarcoma. Pak J Med Sci. 2017;33(2):341–346. | ||
Shojaei F. Anti-angiogenesis therapy in cancer: current challenges and future perspectives. Cancer Lett. 2012;320(2):130–137. | ||
Bellou S, Pentheroudakis G, Murphy C, Fotsis T. Anti-angiogenesis in cancer therapy: hercules and hydra. Cancer Lett. 2013;338(2):219–228. | ||
Ye W. The complexity of translating anti-angiogenesis therapy from basic science to the clinic. Dev Cell. 2016;37(2):114–125. | ||
Ramjiawan RR, Griffioen AW, Duda DG. Anti-angiogenesis for cancer revisited: is there a role for combinations with immunotherapy? Angiogenesis. 2017;20(2):185–204. | ||
Goel S, Wong AH-K, Jain RK. Vascular normalization as a therapeutic strategy for malignant and nonmalignant disease. Cold Spring Harb Perspect Med. 2012;2(3):a006486. | ||
Bottsford-Miller JN, Coleman RL, Sood AK. Resistance and escape from antiangiogenesis therapy: clinical implications and future strategies. J Clin Oncol. 2012;30(32):4026–4034. | ||
Yoncheva K, Momekov G. Antiangiogenic anticancer strategy based on nanoparticulate systems. Expert Opin Drug Deliv. 2011;8(8):1041–1056. | ||
Mukherjee S, Patra CR. Therapeutic application of anti-angiogenic nanomaterials in cancers. Nanoscale. 2016;8(25):12444–12470. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.