Back to Journals » OncoTargets and Therapy » Volume 10
Targeting the PD-1 pathway in pediatric solid tumors and brain tumors
Received 18 January 2017
Accepted for publication 8 March 2017
Published 12 April 2017 Volume 2017:10 Pages 2097—2106
DOI https://doi.org/10.2147/OTT.S124008
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yao Dai
Lars M Wagner,1 Val R Adams2
1Division of Pediatric Hematology/Oncology, 2Department of Pharmacy Practice and Science, University of Kentucky, Lexington, KY, USA
Abstract: While remarkable advances have been made in the treatment of pediatric leukemia over the past decades, new therapies are needed for children with advanced solid tumors and high-grade brain tumors who fail standard chemotherapy regimens. Immunotherapy with immune checkpoint inhibitors acting through the programmed cell death-1 (PD-1) pathway has shown efficacy in some chemotherapy-resistant adult cancers, generating interest that these agents may also be helpful to treat certain refractory pediatric malignancies. In this manuscript we review current strategies for targeting the PD-1 pathway, highlighting putative biomarkers and the rationale for investigation of these drugs to treat common pediatric tumors such as sarcoma, neuroblastoma, and high-grade glioma. We summarize the completed and ongoing clinical trial data available, and suggest potential applications for further study.
Keywords: PD-1, nivolumab, pembrolizumab, pediatric, sarcoma, neuroblastoma, glioma
Introduction
Although the majority of children with newly diagnosed cancer are expected to be long-term survivors, cure remains elusive for patients with certain tumor types. For example, patients with high-risk neuroblastoma, advanced sarcoma, or high-grade glioma who relapse after initial therapy are rarely cured, and these diseases collectively account for over half of pediatric cancer deaths.1 Given that further increases or modifications in existing cytotoxic therapies are unlikely to provide substantial benefit, there is great interest in pursuing alternative approaches for these refractory cancers.
Targeting the immune system as a method for controlling tumor growth is intuitively appealing, as it represents an enabling of the body’s defenses instead of the usual weakening that is brought about by toxic conventional therapy. There is growing evidence that the tumor microenvironment and immune effector cells may help control proliferation of a variety of different pediatric tumors, prompting the investigation of tumor vaccines or other immunomodulating strategies to eradicate malignant cells, or at least restrain tumor growth.2–4
The most effective immunotherapy to date for pediatric solid tumors has been the chimeric monoclonal antibody ch14.18 (dinutuximab; Unituxin®), which targets the ganglioside GD2 that is ubiquitously present on the surface of neuroblastoma cells. Use of dinutuximab following autologous stem-cell transplantation improved the 2-year event-free survival for high-risk neuroblastoma patients from 46% to 66%,5 leading to US Food and Drug Administration (FDA) approval of this agent. This breakthrough study convincingly showed that, in certain contexts, immunotherapy can indeed improve the outcome for particular pediatric tumors. Given this success, and the recent expansion of immunotherapies for adult cancers detailed below, attention is now turning to whether targeting the programmed cell death-1 (PD-1) pathway will be beneficial for refractory pediatric solid tumors and brain tumors. While responses have been seen in adult cancers once thought intractable, these agents have not been efficacious in all clinical scenarios, and patient selection will be critical for success. In this review, we discuss the mechanisms and potential biomarkers for PD-1-targeted therapy, and outline how this may potentially be applied to pediatric solid tumors.
Overview of PD-1 pathway
In the last three decades, cancer therapy has increasingly focused on immunotherapy, based in part on observations that cancer patients may have better outcomes when tumor-infiltrating lymphocytes (TIL) are present.6 The premise is that these TIL are capable of recognizing and killing tumor cells, thus leading to prolonged patient survival. Cytotoxic T-cells are a key mediator of this immune-mediated antitumor effect, and there is now great interest in understanding how these cells are not only activated but also regulated.
As tumor cells die, dendritic cells present tumor-specific antigens through major histocompatibility complex (MHC) to T-cells, and provide additional costimulatory signals through surface receptors like CD28 (Figure 1).7 Once the T-cell becomes activated, other costimulatory molecules are expressed, such as 4.1BB, OX40, and CD40L. Activated T-cells then undergo clonal expansion and migrate to the site of tumor-related antigens, where they infiltrate the tumor and kill malignant cells through perforin and granzyme B. Shortly after activation, mechanisms for turning off the cells also appear, namely the expression of the checkpoint receptors CTLA-4 and PD-1. Collectively, these receptors control the quality, intensity, and duration of the immune response.
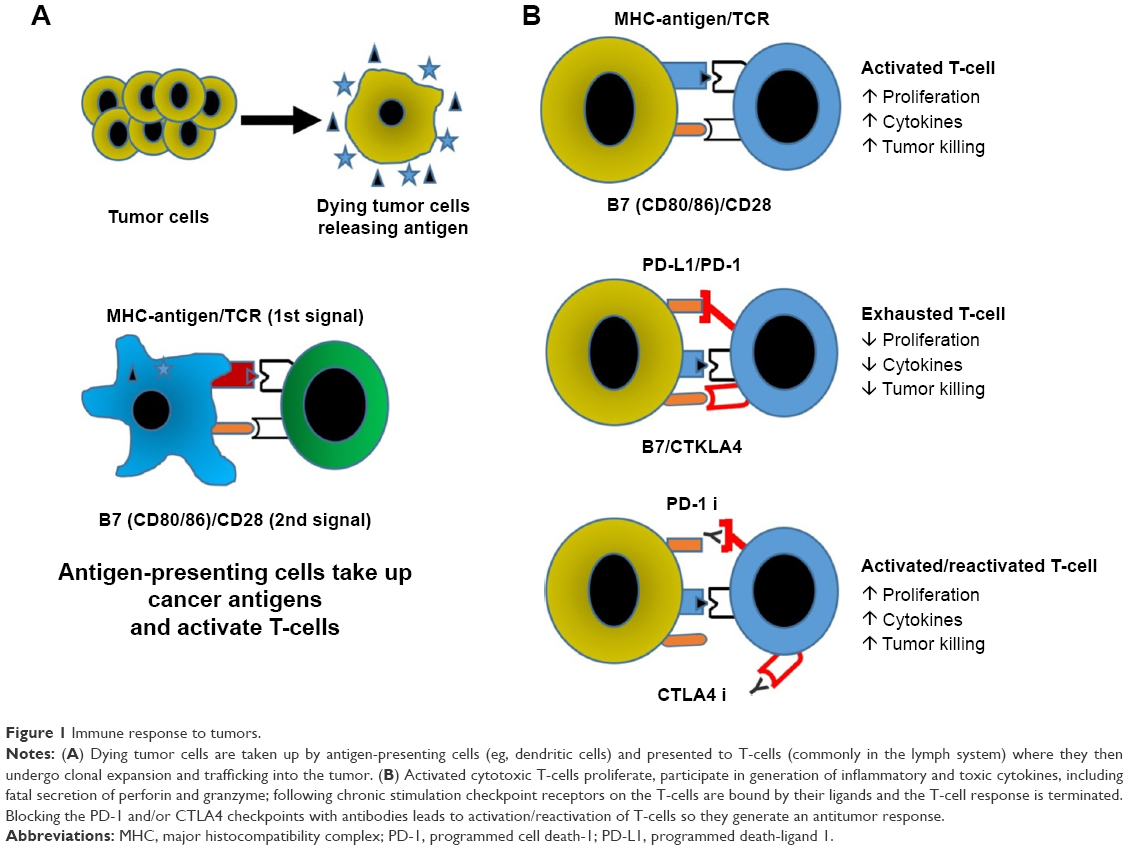 | Figure 1 Immune response to tumors. |
Augmenting clonal expansion of these cytotoxic T-cells has been accomplished with aldesleukin (interleukin 2), which is FDA approved for the treatment of metastatic renal cell carcinoma and melanoma.8,9 However, due to the high toxicity and relatively low response rates of aldesleukin, focus has shifted from augmenting expansion to prolonging the activation of cytotoxic T-cells by blocking checkpoint signals that can terminate the immune response. Although checkpoint signals are important to protect against autoimmune disease, they also protect the tumor from immune attack. The first target for checkpoint inhibition was CTLA-4, which competes with CD28 to bind the stimulatory molecules B7.1 and B7.2. CTLA-4 expression occurs upon T-cell activation, and once present CTLA-4 helps terminate T-cell activity. Ipilimumab is a monoclonal antibody against CTLA-4, and as such helps remove the brakes on the immune response and overcome tolerance by prolonging the period of T-cell activation. Ipilimumab has received FDA approval for the treatment of metastatic melanoma based on improved survival when compared to tumor vaccine;10 however, the overall response rate was only 11%. A pediatric phase I trial of 33 unselected patients with relapsed solid tumors treated with ipilimumab identified a maximum-tolerated dose of 5 mg/kg in patients <12 years old and 10 mg/kg in older patients.11 This compares to the FDA-approved dose of 3 mg/kg in adults. Unfortunately, no objective responses were seen, and focus has turned to second-generation checkpoint inhibitors targeting PD-1 with the hopes of further improving activity. In adult melanoma patients, pembrolizumab and nivolumab (PD-1 inhibitors) have proven to be superior to ipilimumab.12,13
PD-1 is another checkpoint that is important in terminating the response of activated T-cells, and is upregulated by chronically activated T-cells. PD-1 is expressed on activated effector T-cells and other TIL such as natural killer cells, and when activated causes the T-cells to become unresponsive. This phenotype of PD-1+ T-cells that is no longer cytotoxic is called T-cell exhaustion. Of note, mice deficient in PD-1 develop autoimmune diseases from unchecked immune activation, and blocking this pathway in PD-1-proficient mice with chronic viral infections restores antiviral immunity and reverses T-cell exhaustion.14 PD-1 signaling is induced following binding to either of its two ligands (PD-L1 or PD-L2), of which PD-L1 appears to be the more important in regulating T-cell antitumor function. PD-L1 is expressed in a variety of different tumors, and in dendritic cells, macrophages, and T-cells. Upregulation of PD-L1 expression by tumors is driven by inflammation when cytokines such as interferon gamma are expressed. Additionally, there is also a link between hypoxia, which can lead to infiltration of tumor-associated macrophages that express PD-L1. These immune-regulatory signals appear to be a mechanism tumors utilize to escape immune surveillance. Logically, it follows that tumors that express PD-L1 or contain macrophages that express PD-L1 might respond best to a PD-1 or PD-L1 inhibitor.15 This concept is depicted in Figure 2.
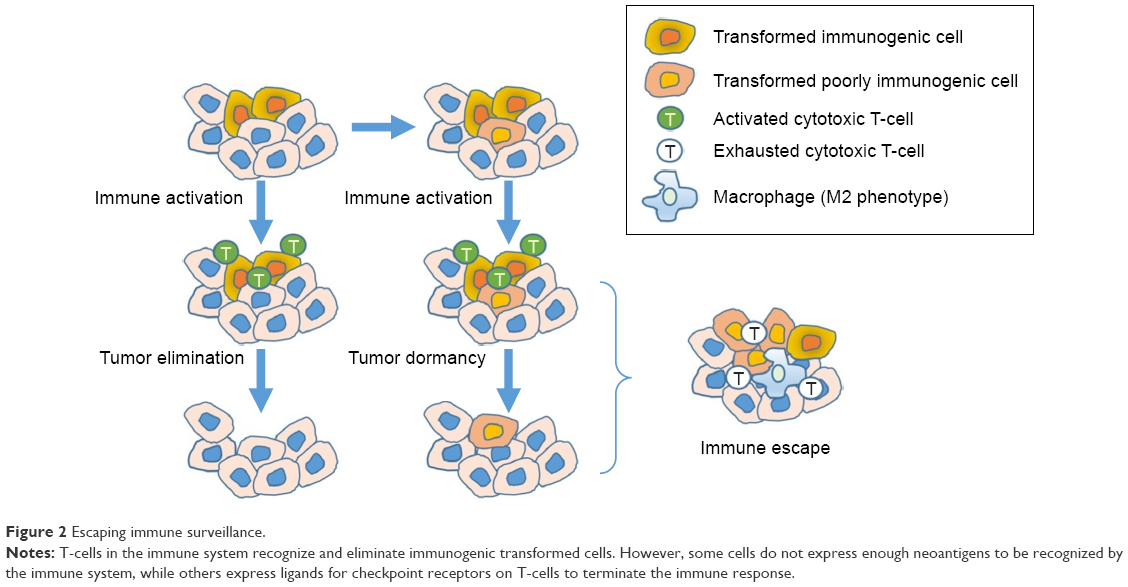 | Figure 2 Escaping immune surveillance. |
There are currently three FDA-approved antibodies targeting either PD-1 (nivolumab, pembrolizumab) or PD-L1 (aletuzumab), with several others in clinical trials. A summary is provided in Table 1. To date, there has been no published comparison of efficacy in patients treated with antibodies within a class (eg, nivolumab vs pembrolizumab), or between classes (PD-1 vs PD-L1). The toxicities of these compounds are nearly similar, with autoimmune adverse events being most common and concerning.16 Historically, immunotherapy was thought to only be efficacious in the setting of minimal residual disease;17 however, PD-1-targeted antibodies have routinely produced responses in patients with bulky disease.
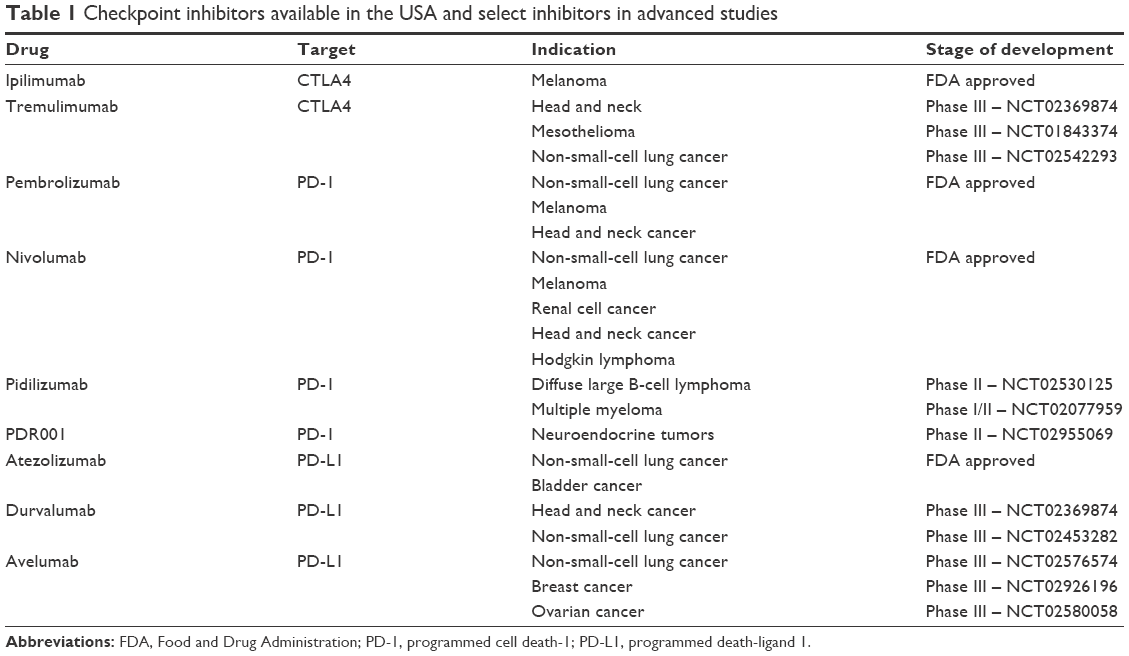 | Table 1 Checkpoint inhibitors available in the USA and select inhibitors in advanced studies |
Putative biomarkers and mechanisms of resistance for PD-1-targeted therapy
At present, there is no single biomarker that accurately predicts response to anti-PD-1 therapy. Based on the current understanding of the immune system’s antitumor response, there are several key elements that can explain how tumors escape immune surveillance and grow. Tumors that are not immunogenic will not be recognized by the immune system. Immunogenic tumors can avoid immune surveillance by preventing inflammation and lymphocyte penetration into the tumor, or they can avoid immune surveillance by terminating the immune response through expressed checkpoint ligands.
Intuitively, tumors that have escaped immune surveillance and express PD-L1 on the cell surface should benefit from either anti-PD-1 or anti-PD-L1 therapy. However, clinical trial experience shows that this is not always the case.18 Key areas of investigation include how expression of PD-L1 is assessed, what threshold is linked to clinical benefit from these agents, and whether PD-L1 expression is required for therapeutic success. As seen in Table 2, PD-L1 is most commonly assessed by immunohistochemistry (IHC), although a variety of different antibodies, different tissues (fresh, frozen, paraffin embedded, primary vs metastatic), and different thresholds for positivity have been used (reviewed in Hutarew).19 Despite these limitations, most trials involving lung cancer and melanoma patients show a general association between outcome and PD-L1 expression, although it is not necessarily required for therapeutic benefit.18 In contrast, PD-L1 expression in urothelial cancer or renal cell cancer seems even less predictive of response to nivolumab20,21 or pembrolizumab22, as some patients clearly benefit despite little or no expression. These results suggest that the utility of PD-L1 as a biomarker is limited and may not be broadly applicable across all tumor types.
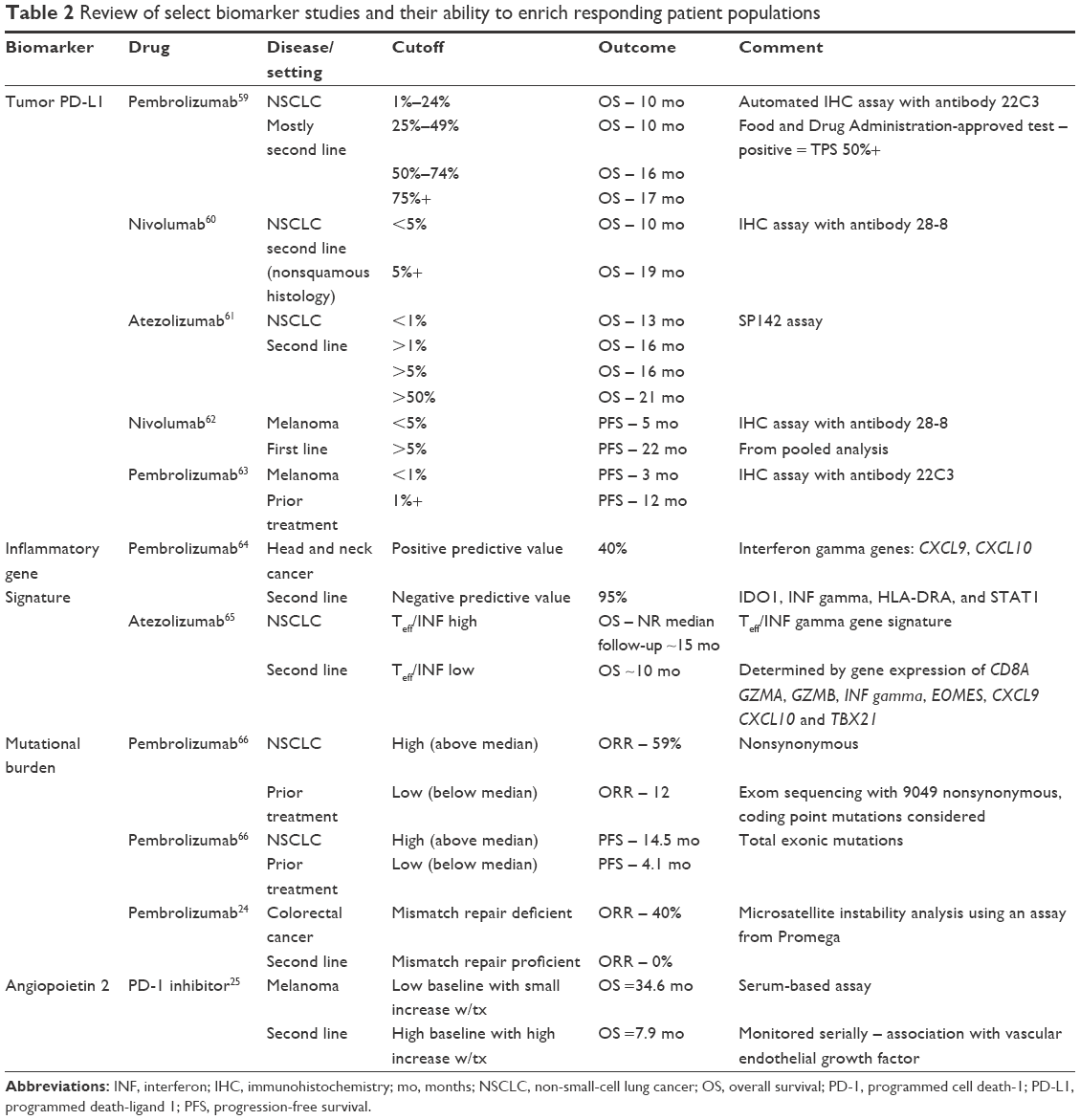 | Table 2 Review of select biomarker studies and their ability to enrich responding patient populations |
PD-L1 expression is induced by inflammatory cytokines such as interferon gamma, and the expression of this cytokine may mark the mechanism the tumor has utilized to escape immune surveillance and consequently be a biomarker for PD-1 inhibitor activity. For example, in a preliminary report describing the treatment of patients with non-small-cell lung cancer with the anti-PD-L1 antibody durvalumab, patients whose tumors coexpressed interferon gamma and PD-L1 had improved survival (hazard ratio [HR] 0.4, P=0.016) compared with those expressing PD-L1 alone (HR 0.64, P=0.18).23 These findings suggest that some type of panel of biomarkers may be most efficacious for patient selection.
Another putative biomarker for response to anti-PD-1 therapy is the total mutational burden, with studies showing improved response rates in patients with >100 mutations in tumor tissue.18 Somatic mutations have the potential to encode “non-self” immunogenic antigens (also called neoantigens), which may make the tumor more visible to the immune system. Mutations may be induced by exposure, such as sunlight or cigarette smoke, and perhaps this may be one reason why PD-1 agents have been successful in melanoma and lung cancer. Alternatively, mutations also may be dramatically increased in tumors demonstrating mismatch repair (MMR) deficiency, and this can result in microsatellite instability (MSI) and sensitivity to PD-1 inhibition. For example, patients treated with pembrolizumab for colon cancer showed response rates as high as 40% in patients whose tumors showed MMR deficiency, compared with 0% in MMR-proficient tumors.24 Whole-exome sequencing demonstrated a mean of 1,782 mutations per tumor in the MMR-deficient cohort, compared to 73 in the proficient group. Assessing for mutational burden and MSI can now be done by commercial assays, and may be another potentially important factor to consider regarding patient selection.
Finally, there is some recent information suggesting that angiopoietin-2 (Ang2) may also be a biomarker for PD-1-targeted therapy, and importantly one that can be assessed by peripheral blood at various time points. Ang2 is a critical regulator of tumor-associated blood vessel maturation, and is involved with the recruitment of monocytes/macrophages into the tumor microenvironment and induction of PD-L1 expression in M2-polarized macrophages. Wu et al found that for 43 melanoma patients treated with PD-1 blockade, those with high circulating levels of Ang2 (defined as >3,175 pg/mL) or rising levels during treatment had reduced overall survival,25 suggesting that Ang2 may mediate resistance to checkpoint inhibitors, and could be used to select and follow patients. Although pediatric data are limited, one study of 35 various pediatric solid tumor or brain tumor patients showed that the median circulating Ang2 level at diagnosis would be considered low at 2,482 pg/mL, suggesting potential sensitivity to PD-1 inhibition according to this parameter.26 A list of biomarkers used in key clinical trials is provided in Table 2.
There are several proposed mechanisms of resistance to PD-1 blockade, and include interruptions of any step along the pathway of T-cell cytotoxicity. Some tumors such as prostate cancer have very little PD-L1 expression despite the presence of TIL, and so may be resistant for this reason.27 Mutations in tumor can account for downregulation of MHC and decreased immunogenicity,28 or altered dendritic cell migration.29 In addition, involvement of other PD-1-independent pathways may help tumor cells evade T-cell destruction. Some tumor cells are induced by interferon gamma to express indoleamine 2,3-dioxygenase, which can render experimental models of melanoma resistant to PD-1 blockade.30 Other complex mechanisms such as extreme T-cell exhaustion or impaired development of effective T-cell memory may also contribute to resistance (reviewed in O’Donnell et al).31
Preclinical testing in pediatric cancers
In considering whether PD-1 targeting would be appropriate for pediatric cancers, a key issue is whether potential biomarkers suggested by adult studies are present in childhood tumors as well. Although limited, there is a growing body of evidence about PD-L1 expression, mutational burden, and MSI in important childhood cancers such as bone and soft tissue sarcoma, neuroblastoma, and high-grade glioma.
Pediatric sarcoma
In two of the larger studies to date of osteosarcoma, IHC analysis showed PD-L1 expression in 25%–47% of primary tumor samples, and tumors expressing PD-L1 were significantly more likely to have infiltrating immune cells, which often expressed PD-1.32,33 Importantly, PD-L1 expression correlated with worse event-free survival, and these findings are consistent with the concept that PD-L1 in tumor cells may interact with PD-1-expressing cells in the microenvironment to help evade immune rejection by the host. Interestingly, PD-L1 expression has been reported to be more common in metastatic lesions than in primary tumors,34,35 which is relevant considering that metastases are more likely than primary tumors to cause death in this disease. Further, osteosarcoma has a greater inherent genomic instability and higher mutational burden than many other pediatric solid tumors,36 which may also predispose to benefit from PD-1-targeted therapy. Finally, use of the immune adjuvant mifamurtide has previously shown benefit in osteosarcoma37 and is approved by the European Medicines Agency, further demonstrating the potential role of immunotherapy for this disease.
PD-L1 expression has been less well studied in Ewing sarcoma, another important pediatric sarcoma that may originate either in bone or soft tissue. One study demonstrated expression in 8 (57%) of 14 primary bone tumor samples,32 while 8 (38%) of 21 soft tissue primaries have reported PD-L1 expression in other studies.38,39 For rhabdomyosarcoma, the most common pediatric soft tissue sarcoma, data are limited but Chowdhury et al have reported expression in 13 (86%) of 15 alveolar rhabdomyosarcoma and 8 (50%) of 16 embryonal rhabdomyosarcoma patient samples.32 More robust analysis of the tumor microenvironment is not yet available, although infiltrating PD-1 CD8 lymphocytes are present in a subset of these tumors.
Neuroblastoma
Neuroblastoma is the most common extracranial solid tumor in children, and high-risk disease accounts for up to 15% of pediatric cancer deaths. An early study did not identify PD-L1 expression in any of the 18 tested samples from neuroblastoma patients.40 In contrast, Chowdhury et al reported PD-L1 surface expression in 31 (72%) of 43 patient tumor samples,32 with lower survival in patients expressing PD-L1. Dondero et al confirmed that PD-1 expression is present in lymphocytes obtained from metastatic bone marrow samples, and that PD-L1 expression in tumor cells is inducible with interferon gamma.41 Interestingly, the production of cytokines such as interferon is particularly high in patients following treatment with the recently approved monoclonal antibody dinutuximab, suggesting potential benefit for combination therapy.
High-grade glioma
PD-L1 expression has been identified in 6 (30%) of 20 pediatric high-grade glioma samples,42 and larger studies in adults showed expression in 68% of 62 glioblastoma samples, with a strong trend for worse outcome in these patients.43 In addition, a subset of pediatric high-grade glioma tumors has evidence of MSI, which reflects the increased mutational status of the tumor and the potential greater number of neoantigens that could induce a host immune response. Viana-Pereira et al have reported MSI to be present in 14 (19%) of 71 high-grade glioma samples, significantly higher than that seen with adult tumors.44 While some of those patients may have had the rare congenital MMR deficiency syndrome (MMRDS, previously called Turcot syndrome),45 MSI may also arise independently outside of this defined cancer predisposition. The potential benefit for using anti-PD-1 antibodies to treat these hypermutated tumors was shown by Bouffet et al, who reported sustained responses in two siblings with pediatric glioblastoma and MMRDS who were treated with single-agent nivolumab.46 Taken together, these findings support further investigation of PD-1-targeted drugs for this disease.
Completed and upcoming clinical trials with PD-1 agents to treat pediatric cancers
To date, there have been only a few reports of treatment of pediatric cancer patients with agents targeting the PD-1 pathway. Apart from the glioma patients with MMR deficiency treated with pembrolizumab as above,46 all other reports involve genetically unselected patients. Blumenthal et al retrospectively described outcomes in 22 patients with recurrent primary brain tumors treated with pembrolizumab, of whom five were children with pontine glioma (2), glioblastoma (1), atypical teratoid/rhabdoid tumor (1), and medulloblastoma (1).47 These children were ages 3–7 years, and received an adjusted flat dose of 50 mg every 3 weeks for a median of four infusions (range 2–10). Disappointingly, no responses were seen in either pediatric or adult patients.
Although preclinical data would suggest that osteosarcoma would be sensitive to agents targeting PD-1, this has not been the case in the limited data available to date. For example, in the SARC028 multi-institutional phase II trial of pembrolizumab, only one of 19 patients with recurrent osteosarcoma had an objective imaging response.48 In addition, no responses were seen in 13 patients treated with recurrent Ewing sarcoma, although there has been one anecdotal case report of a heavily pretreated patient with relapsed disease who had a sustained response to pembrolizumab.49 Interestingly, the SARC028 study did show activity for a subset of adult soft tissue sarcoma, namely undifferentiated pleomorphic sarcoma, in which four of 10 patients experienced a partial response. This sarcoma histiotype is somewhat heterogeneous, and it is not known whether PD-1 agents will have similar efficacy in the soft tissue sarcoma subtypes more commonly seen in pediatrics such as rhabdomyosarcoma.
Table 3 lists several key ongoing clinical trials in pediatric patients, which are expected to better establish the potential role of anti-PD-1 antibodies and hopefully identify biomarkers that can be used for patient selection. The Children’s Oncology Group is performing an important phase I/II study that will determine both the recommended phase II doses of single-agent nivolumab and the combination of nivolumab with the CTLA-4 inhibitor ipilimumab in patients with unselected recurrent solid tumors or lymphoma (ClinicalTrials.gov identifier NCT02304458). In the phase II expansion portion of the study, activity of nivolumab will be assessed in at least 10 patients in each of the following cohorts: neuroblastoma, osteosarcoma, rhabdomyosarcoma, Ewing sarcoma, Hodgkin lymphoma, non-Hodgkin lymphoma, and melanoma. If sufficient activity is not seen in a cohort treated with nivolumab, then the activity of the combination of nivolumab and ipilimumab will be assessed for that tumor type. This expansive trial includes many correlative studies and should help better define the role of PD-1 inhibition in treating pediatric tumors.
 | Table 3 Ongoing trials of agents targeting the PD-1 or PD-L1 pathway for treatment of pediatric cancer |
Other ongoing studies include a Pediatric Brain Tumor Consortium phase I/II study in which pembrolizumab is being evaluated for treatment of recurrent high-grade glioma or other hypermutated brain tumors (NCT02359565). Pembrolizumab is also being studied in a pediatric phase I/II trial as single-agent treatment for recurrent melanoma or PD-L1-positive tumors (NCT02332668). In addition, anti-PD-1 antibodies are also being included in so-called “basket trials” for recurrent pediatric solid tumors, in which patients are stratified based on molecular characteristics of the tumor rather than the histiotype (NCT02813135). In regard to PD-L1 antibodies, a phase I/II study of atezolizumab is now open for patients <30 years with recurrent solid tumors (NCT02541604), and a phase II study of avelumab is open for patients with recurrent osteosarcoma (NCT03006848).
Safety and response considerations
As opposed to the myelosuppression usually seen with conventional chemotherapy, the toxicity of anti-PD-1 antibodies has often been immune-related such as pneumonitis, colitis, hepatitis, hypophysitis, and thyroiditis.16 In general, the occurrence of these toxicities has correlated with antitumor effects, consistent with the mechanism of action of these agents. In the limited number of pediatric patients studied to date, the severity and scope of adverse events seem similar to adults. The development of pneumonitis and pleural effusions is of particular concern in pediatric sarcoma patients, who commonly have bulky lung or pleural metastases that may be associated with effusions. Such patients being treated with anti-PD-1 antibodies should be monitored carefully, and be considered for drainage or other medical management that could include steroids.50
Another characteristic of PD-1-targeted therapy is the potential for the temporary increase in size of lesions as a consequence of therapy-related inflammation. This effect is termed pseudoprogression, and has led to the development of a modified response assessment system called immune-related response criteria that are now used in trials of these agents.51 This revised system allows for more interval growth of tumor to occur before the determination of progressive disease is made, based on past observations of patients going on to have prolonged disease stability despite some initial growth of tumor. The concept of tumor pseudoprogression may have the greatest clinical implications in the treatment of brain tumors, as at least one death has now been reported in a pediatric patient with recurrent glioblastoma who was treated with nivolumab and developed fatal cerebral edema despite operative intervention.52
Combination therapies
Given the common tendency of cancers to develop resistance to monotherapies, it is likely that some patients may benefit only when PD-1 agents are combined with other drugs that may be synergistic. The only FDA-approved combination is nivolumab together with the CTLA-4 inhibitor ipilimumab for the treatment of metastatic melanoma. While this intuitively appealing strategy of double checkpoint inhibition increases the response rate and overall survival, the rate of grade 3–4 toxicity is also increased when compared to nivolumab alone,53 emphasizing that toxicity considerations will be important when planning combination therapies.
Some conventional chemotherapy drugs such as cyclophosphamide, platinum analogs, and taxanes can elicit immunogenicity by recruiting immune cells to the microenvironment, stimulating natural killer-dependent antitumor immunity and T-cell responses, and disrupting immune suppressor mechanisms by depleting regulatory T-cells and myeloid-derived suppressor cells.54 These immunomodulatory effects may be optimized by using lower or more protracted dosing,55 and this strategy is being utilized in an ongoing pediatric study combining pembrolizumab with low-dose oral cyclophosphamide (NCT02813135).
The ability of radiotherapy to induce immune responses is only now beginning to be understood. Preclinical studies suggest that the combination of a PD-1 antibody with radiation can be synergistic,56 and combination phase III studies are now underway for glioblastoma (NCT02617589) and lung cancer (NCT027768558). If this strategy is successful in adults, it is likely that pediatric trials combining radiotherapy and anti-PD-1 antibodies will soon follow. Other possible combinations include targeted agents such as dabrafenib or trametinib together with anti-PD-1 therapy,57 or even tumor vaccines58 or oncolytic viruses such as tailmogene laherparepvecm, which produces GM-CSF and is being evaluated together with pembrolizumab in a phase III trial for melanoma patients (NCT02263508).
Conclusions and future directions
The use of PD-1-targeted agents has revolutionized the treatment of certain adult cancers, generating excitement in part because some of the most responsive tumors have been previously refractory to conventional therapies. Still emerging is the identification of a reliable biomarker or panel of characteristics that can allow appropriate patient selection. Available information to date suggests that PD-L1 expression on tumor cells, a strong lymphocytic presence in the tumor microenvironment, and an increased mutational burden are important but not yet definitive markers that suggest efficacy. These features are variably present in some high-risk pediatric tumors, and support further investigation of PD-1-targeted therapy. However, data from adult studies clearly show that the presence or absence of these putative biomarkers does not guarantee the success or failure of this therapeutic strategy. Although off-label use is tempting for patients with multiply recurrent tumors, caution should be taken in light of the still unproven benefit of these agents for pediatric cancers, and the potential for organ toxicity related to immune effects. Trials are underway, which will better define the role these drugs may play for treating pediatric cancers, either as single agents or in combination with chemotherapy, radiation, or other molecularly targeted or biologic therapies.
Disclosure
The authors report no conflicts of interest in this work.
References
Ward E, DeSantis C, Robbins A, Kohler B, Jemal A. Childhood and adolescent cancer statistics, 2014. CA A Cancer J Clin. 2014;64(2):83–103. | ||
Roberts SS, Chou AJ, Cheung NK. Immunotherapy of childhood sarcomas. Front Oncol. 2015;5:181. | ||
Eyrich M, Rachor J, Schreiber SC, Wolfl M, Schlegel PG. Dendritic cell vaccination in pediatric gliomas: lessons learnt and future perspectives. Front Pediatr. 2013;1:12. | ||
Croce M, Corrias MV, Rigo V, Ferrini S. New immunotherapeutic strategies for the treatment of neuroblastoma. Immunotherapy. 2015;7(3):285–300. | ||
Yu AL, Gilman AL, Ozkaynak MF, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med. 2010;363(14):1324–1334. | ||
Galon J, Angell HK, Bedognetti D, Marincola FM. The continuum of cancer immunosurveillance: prognostic, predictive, and mechanistic signatures. Immunity. 2013;39(1):11–26. | ||
Daud A. Current and emerging perspectives on immunotherapy for melanoma. Semin Oncol. 2015;42 (Suppl 3):S3–S11. | ||
Schmidinger M, Hejna M, Zielinski CC. Aldesleukin in advanced renal cell carcinoma. Expert Rev Anticancer Ther. 2004;4(6):957–980. | ||
Amaria RN, Reuben A, Cooper ZA, Wargo JA. Update on use of aldesleukin for treatment of high-risk metastatic melanoma. Immunotargets Ther. 2015;4:79–89. | ||
Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. | ||
Merchant MS, Wright M, Baird K, et al. Phase I clinical trial of ipilimumab in pediatric patients with advanced solid tumors. Clin Cancer Res. 2016;22(6):1364–1370. | ||
Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372(26):2521–2532. | ||
Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373(1):23–34. | ||
Barber DL, Wherry EJ, Masopust D, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439(7077):682–687. | ||
Boussiotis VA. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N Engl J Med. 2016;375(18):1767–1778. | ||
Abdel-Wahab N, Shah M, Suarez-Almazor ME. Adverse events Associated with immune checkpoint blockade in patients with cancer: a systematic review of case reports. PLoS One. 2016;11(7):e0160221. | ||
Parish CR. Cancer immunotherapy: the past, the present and the future. Immunol Cell Biol. 2003;81(2):106–113. | ||
Gibney GT, Weiner LM, Atkins MB. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016;17(12):e542–e551. | ||
Hutarew G. PD-L1 testing, fit for routine evaluation? From a pathologist’s point of view. Memo. 2016;9(4):201–206. | ||
Motzer RJ, Escudier B, McDermott DF, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373(19):1803–1813. | ||
Sharma P, Retz M, Siefker-Radtke A, et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): a multicentre, single-arm, phase 2 trial. Lancet Oncol. 2017;pii:S1470–S2045(17)30065–30067. | ||
Bellmunt J, de Wit R, Vaughn DJ, et al. Pembrolizumab as second-line therapy for advanced urothelial carcinoma. N Engl J Med. 2017;376:1015–1026. | ||
Higgs BW, Morehouse C, Streicher K, et al. Relationship of baseline tumoral IFNγ mRNA and PD-L1 protein expression to overall survival in durvalumab-treated NSCLC patients. J Clin Oncol. 2016;34:(Suppl; abstr 3036). | ||
Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509–2520. | ||
Wu X, Giobbie-Hurder A, Liao X, et al. Angiopoietin-2 as a biomarker and target for immune checkpoint therapy. Cancer Immunol Res. 2017;5(1):17–28. | ||
Hintsala E, Bono P, Andersson S, Kivivuori SM. Quantification of plasma and bone marrow VEGF and angiopoietin-2 levels in pediatric malignancies. J Pediatr Hematol Oncol. 2012;34(7):503–510. | ||
Martin AM, Nirschl TR, Nirschl CJ, et al. Paucity of PD-L1 expression in prostate cancer: innate and adaptive immune resistance. Prostate Cancer Prostatic Dis. 2015;18(4):325–332. | ||
Korkolopoulou P, Kaklamanis L, Pezzella F, Harris AL, Gatter KC. Loss of antigen-presenting molecules (MHC class I and TAP-1) in lung cancer. Br J Cancer. 1996;73(2):148–153. | ||
Spranger S, Gajewski TF. A new paradigm for tumor immune escape: beta-catenin-driven immune exclusion. J Immunother Cancer. 2015;3:43. | ||
Holmgaard RB, Zamarin D, Munn DH, Wolchok JD, Allison JP. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J Exp Med. 2013;210(7):1389–1402. | ||
O’Donnell JS, Long GV, Scolyer RA, Teng MW, Smyth MJ. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat Rev. 2017;52:71–81. | ||
Chowdhury F, Dunn S, Mitchell S, Mellows T, Ashton-Key M, Gray JC. PD-L1 and CD8+PD1+ lymphocytes exist as targets in the pediatric tumor microenvironment for immunomodulatory therapy. OncoImmunology. 2015;4(10):e1029701. | ||
Koirala P, Roth ME, Gill J, et al. Immune infiltration and PD-L1 expression in the tumor microenvironment are prognostic in osteosarcoma. Sci Rep. 2016;6:30093. | ||
Lussier DM, O’Neill L, Nieves LM, et al. Enhanced T-cell immunity to osteosarcoma through antibody blockade of PD-1/PD-L1 interactions. J Immunother. 2015;38(3):96–106. | ||
Shen JK, Cote GM, Choy E, et al. Programmed cell death ligand 1 expression in osteosarcoma. Cancer Immunol Res. 2014;2(7):690–698. | ||
Kansara M, Teng MW, Smyth MJ, Thomas DM. Translational biology of osteosarcoma. Nat Rev Cancer. 2014;14(11):722–735. | ||
Meyers PA. Muramyl tripeptide (mifamurtide) for the treatment of osteosarcoma. Expert Rev Anticancer Ther. 2009;9(8):1035–1049. | ||
Paydas S, Bagir EK, Deveci MA, Gonlusen G. Clinical and prognostic significance of PD-1 and PD-L1 expression in sarcomas. Med Oncol. 2016;33(8):93. | ||
Kim C, Kim EK, Jung H, et al. Prognostic implications of PD-L1 expression in patients with soft tissue sarcoma. BMC Cancer. 2016;16:434. | ||
Aoki T, Hino M, Koh K, et al. Low frequency of programmed death ligand 1 expression in pediatric cancers. Pediatr Blood Cancer. 2016;63(8):1461–1464. | ||
Dondero A, Pastorino F, Della Chiesa M, et al. PD-L1 expression in metastatic neuroblastoma as an additional mechanism for limiting immune surveillance. Oncoimmunology. 2016;5(1):e1064578. | ||
Majzner RG, Martinez D, Pawel B, et al. Assessment of PD-L1 expression and tumor associated immune cells in pediatric cancer tissues. J Clin Oncol. 2016;34:(Suppl; abstr 11542). | ||
Zeng J, Zhang XK, Chen HD, Zhong ZH, Wu QL, Lin SX. Expression of programmed cell death-ligand 1 and its correlation with clinical outcomes in gliomas. Oncotarget. 2016;7(8):8944–8955. | ||
Viana-Pereira M, Lee A, Popov S, et al. Microsatellite instability in pediatric high grade glioma is associated with genomic profile and differential target gene inactivation. PLoS One. 2011;6(5):e20588. | ||
Wimmer K, Rosenbaum T, Messiaen L. Connections between constitutional mismatch repair deficiency syndrome and neurofibromatosis type 1. Clin Genet. Epub 2016 Oct 25. | ||
Bouffet E, Larouche V, Campbell BB, et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J Clin Oncol. 2016;34(19):2206–2211. | ||
Blumenthal DT, Yalon M, Vainer GW, et al. Pembrolizumab: first experience with recurrent primary central nervous system (CNS) tumors. J Neurooncol. 2016;129(3):453–460. | ||
Burgess MA, Crowley J, Reinke DK, et al. SARC 028: A phase II study of the anti-PD1 antibody pembrolizumab (P) in patients (Pts) with advanced sarcomas. J Clin Oncol. 2015;33:(suppl; abstr TPS10578). | ||
McCaughan GJ, Fulham MJ, Mahar A, et al. Programmed cell death-1 blockade in recurrent disseminated Ewing sarcoma. J Hematol Oncol. 2016;9(1):48. | ||
Weber JS, Postow M, Lao CD, Schadendorf D. Management of adverse events following treatment with anti-programmed death-1 agents. Oncologist. 2016;21(10):1230–1240. | ||
Nishino M. Immune-related response evaluations during immune-checkpoint inhibitor therapy: establishing a “common language” for the new arena of cancer treatment. J Immunother Cancer. 2016;4:30. | ||
Zhu X, McDowell MM, Newman WC, Mason GE, Greene S, Tamber MS. Severe cerebral edema following nivolumab treatment for pediatric glioblastoma: case report. J Neurosurg Pediatr. 2016;19:1–5. | ||
D’Angelo SP, Larkin J, Sosman JA, et al. Efficacy and safety of nivolumab alone or in combination with ipilimumab in patients with mucosal melanoma: a pooled analysis. J Clin Oncol. 2017;35(2):226–235. | ||
Bracci L, Schiavoni G, Sistigu A, Belardelli F. Immune-based mechanisms of cytotoxic chemotherapy: implications for the design of novel and rationale-based combined treatments against cancer. Cell Death Differ. 2014;21(1):15–25. | ||
Chang CL, Hsu YT, Wu CC, et al. Dose-dense chemotherapy improves mechanisms of antitumor immune response. Cancer Res. 2013;73(1):119–127. | ||
Kang J, Demaria S, Formenti S. Current clinical trials testing the combination of immunotherapy with radiotherapy. J Immunother Cancer. 2016;4:51. | ||
Hu-Lieskovan S, Mok S, Homet Moreno B, et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF(V600E) melanoma. Sci Transl Med. 2015;7(279):279ra241. | ||
Karaki S, Anson M, Tran T, et al. Is there still room for cancer vaccines at the era of checkpoint inhibitors. Vaccines (Basel). 2016;4(4):37. | ||
Baas P, Garon EB, Herbst RS, et al. Relationship between level of PD-L1 expression and outcomes in the KEYNOTE-010 study of pembrolizumab versus docetaxel for previously treated, PD-L1-positive NSCLC. J Clin Oncol. 2016;34(Suppl; abstr 9015). | ||
Borghaei H, Brahmer J. Nivolumab in nonsquamous non-small-cell lung cancer. N Engl J Med. 2016;374(5):493–494. | ||
Rittmeyer A, Barlesi F, Waterkamp D, et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet. 2017;386(10066):255–265. | ||
Long GV, Larkin J, Ascierto PA, et al. PD-L1 expression as a biomarker for nivolumab (NIVO) plus ipilimumab (IPI) and NIVO alone in advanced melanoma (MEL): a pooled analysis. ESMO 2016 Congress in Copenhagen, Denmark. 2016:(Suppl; Abstr 1112PD). | ||
Kefford R, Ribas A, Hamid O, et al. Clinical efficacy and correlation with tumor PD-L1 expression in patients (pts) with melanoma (MEL) treated with the anti-PD-1 monoclonal antibody MK-3475. J Clin Oncol. 2014;32:(Suppl; abstr 3005). | ||
Seiwert TY, Burtness B, Mehra R, et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. Lancet Oncol. 2016;17(7):956–965. | ||
Fehrenbacher L, Spira A, Ballinger M, et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet. 2016;387(10030):1837–1846. | ||
Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124–128. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.