Back to Journals » OncoTargets and Therapy » Volume 12
Targeting off-target effects: endoplasmic reticulum stress and autophagy as effective strategies to enhance temozolomide treatment
Authors He Y, Su J, Lan B, Gao Y, Zhao J
Received 15 November 2018
Accepted for publication 1 February 2019
Published 7 March 2019 Volume 2019:12 Pages 1857—1865
DOI https://doi.org/10.2147/OTT.S194770
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Geoffrey Pietersz
Yichun He,1 Jing Su,2 Beiwu Lan,1 Yufei Gao,1 Jingxia Zhao3
1Department of Neurosurgery, China-Japan Union Hospital, Jilin University, Changchun, Jilin, China; 2Department of Pathophysiology, Key Laboratory of Pathobiology, Ministry of Education, College of Basic Medical Sciences, Jilin University, Changchun, Jilin, China; 3Experimental Teaching Center, School of Nursing, Jilin University, Changchun, Jilin, China
Abstract: Glioblastoma multiforme (GBM) is the most common and aggressive adult primary central nervous system tumor. Unfortunately, GBM is resistant to the classic chemotherapy drug, temozolomide (TMZ). As well as its classic DNA-targeting effects, the off-target effects of TMZ can have pro-survival or pro-death roles and regulate GBM chemoradiation sensitivity. Endoplasmic reticulum (ER) stress is one of the most common off-target effects. ER stress and its downstream induction of autophagy, apoptosis, and other events have important roles in regulating TMZ sensitivity. Autophagy is an evolutionarily conserved cellular homeostasis mechanism that is closely associated with ER stress-induced apoptosis. Under ER stress, autophagy cannot only remove misfolded/unfolded proteins and damaged organelles and degrade and inhibit apoptosis-related caspase activation to reduce cell damage, but may also promote apoptosis dependent on ER stress intensity. Although some protein interactions between autophagy and apoptosis and common upstream signaling pathways have been found, the underlying regulatory mechanisms are still not fully understood. This review summarizes the possible mechanisms underlying the current known off-target roles of ER stress and downstream autophagy in the regulation of cell fate and evaluates their role in TMZ treatment and their potential as therapeutic targets.
Keywords: autophagy, apoptosis, chemotherapy resistance, temozolomide, glioma, endoplasmic reticulum stress
Introduction
Glioblastoma multiforme (GBM) is the most common and aggressive adult primary central nervous system tumor, with a 2-year survival rate of only 3%–5%.1 Currently, the standard treatment for GBM is a multimodal comprehensive approach that includes surgery, radiotherapy, and chemotherapy. Because complete surgical resection cannot be performed, and because of its multichemotherapy resistance, including the classic chemotherapeutic temozolomide (TMZ), recurrence is almost unavoidable, resulting in a very short median survival period of 12–15 months. To improve prognosis, research into the molecular mechanisms involved in resistance is imperative to increasing chemosensitivity.
TMZ, an oral alkylated chemotherapeutic drug, is a first-line drug for clinical glioma chemotherapy and can efficiently pass through the blood–brain barrier to reach the lesion. However, over time, GBM develops resistance to TMZ-induced damage, which is associated with several mechanisms such as DNA repair pathways (O6-methylguanine DNA methyltransferase [MGMT], DNA mismatch repair, base excision repair system), epidermal growth factor receptor (EGFR) and MDM2 expression, p53 mutation, and PTEN expression. In addition, changes in micro-RNA expression profiles are also associated with resistance development. These mechanisms are centered on DNA repair and mutation of endogenous tumor suppressor genes. However, in addition to the classic targeting of DNA damage, TMZ induces some off-target effects, which may promote survival or death, resulting in regulation of GBM chemoradiation sensitivity.
Among the off-target effects, endoplasmic reticulum (ER) stress is the most common. ER stress and its downstream signaling, such as autophagy and apoptosis, play an important role in regulating TMZ sensitivity. The ER is the intracellular organelle responsible for the synthesis of proteins and lipids. It can be disrupted by a variety of physiological or pathological stimuli (eg, withdrawal of nutrients, energy deprivation, hypoxia, oxidative stress, chemotherapeutic drugs) that can induce ER stress, leading to a decrease in ER protein folding ability and resulting in accumulation of misfolded or unfolded proteins, thereby activating the ER unfolded protein response (UPR). The UPR regulates autophagy and other degradation pathways that degrade misfolded proteins to restore homeostasis by limiting the rate of global protein synthesis, which finally determines cell fate. Autophagy, an important downstream target of ER stress, has been found to play a pro-survival or pro-apoptotic role at the cellular or organ level in response to different stresses.2 The exact mechanism by which autophagy performs two seemingly contradictory roles is still unclear. Current research shows that there is a complex interplay between autophagy and apoptosis. Therefore, during GBM chemotherapy, autophagy may be an important event resulting from off-target effects, leading to a change in TMZ sensitivity (Figure 1).3,4
 | Figure 1 Schematic representation of the functional relationship between unfolded protein response, autophagy, apoptosis, and glioma cell fate under TMZ treatment. |
Thus, studies of the off-target effects of ER stress will contribute to develop new therapeutic strategies that sensitize chemoresistant glioma and/or prevent glioma resistance.
ER stress in GBM
ER stress can be observed in a variety of solid tumor samples (such as glioma), and studies have shown that it is closely related to the regulation of tumor chemosensitivity. The expression of the ER stress marker, 78 kDa glucose-regulated protein (GRP78), is lower in normal brain tissue than in other normal tissues, but it is most significantly increased in glioblastoma compared with other tumors.5 Furthermore, GRP78 expression significantly increases with the pathological grade level in gliomas.6 Several studies in glioma and other cancer cell line models have demonstrated that ER stress-associated proteins, such as GRP78/Bip,6–9 ATF4,10 ATF6,11 and P4HB,12,13 have pro-survival roles and function as resistance-related factors. These evidences indicate that ER stress and TMZ sensitivity of gliomas are closely related.
ER stress and UPR
Under ER stress, cells can activate a series of adaptive responses, such as the UPR. The UPR cannot only deal with stress by reducing new protein synthesis and increasing protein folding capacity, but can also reduce protein toxicity by reducing misfolded/unfolded protein accumulation through ER-associated protein degradation or autophagy. However, when stress persists or is too severe, the UPR initiates the apoptotic process to eliminate damaged cells. This indicates that the UPR determines cell survival and death depending on the stress intensity and duration.
The UPR consists of three classical pathways, PERK–eIF2α–ATF4, IRE1α–XBP1, and ATF6. PERK, IRE1, and ATF6 are three transmembrane proteins that can form complexes with the molecular chaperone, GRP78, and remain inactive under physiological conditions. When ER stress occurs, unfolded proteins accumulate in the ER and GRP78/BiP is released from the complex, leading to PERK, IRE1, and ATF6 activation.
PERK is a major protein that, under ER stress, reduces mRNA translation, inhibits global protein synthesis, and preferentially induces ATF4 expression by phosphorylating eIF2α.14 ATF4 is the core factor in the integration of the stress response, which can transcriptionally regulate autophagy, apoptosis, antioxidation, and other related proteins with its wide transcriptional capacity. ATF4 induces autophagy through transcriptional regulation of autophagy-related genes (eg, ATG7, ATG5, LC3B)15 and activates apoptosis by cooperating with upregulated CHOP and promoting XIAP degradation.16 CHOP initiates apoptosis by activating BIM and DR5 and inhibiting the antiapoptotic protein, BCL-2.17–19 DR5 is a death receptor that can be transcriptionally upregulated by CHOP and induces apoptosis by activating caspase-8.20 In addition, CHOP can induce apoptosis through the regulation of IP3R, which leads to changes in calcium homeostasis.21 Currently, PERK is the most studied branch of the UPR in the field of autophagy and apoptosis regulation by ER stress.
When the UPR is activated, the cytoplasmic region of IRE1α oligomerizes and autophosphorylates and is activated by forming a complex with tumor necrosis factor receptor-associated factor-2 (TRAF2) and apoptosis signal-regulating kinase-1 (ASK-1), which in turn actives Jun-N-terminal kinase (JNK) and induces autophagy or apoptosis.22 Moreover, IRE1α can activate the apoptotic process by its RNase activity through the cleavage of miRNAs that inhibit caspase-2, such as miR-17, miR-34a, miR-96, and miR-125b.23 IRE1α can also induce autophagy by regulating the expression of Beclin-1 via XBP1.24
ATF6 is a transcription factor activated under ER stress, whereupon it translocates to the Golgi and is cleaved by S1P and S2P to its short form, which translocates to the nucleus and binds to the ATF/CRE element or the ERSE element to initiate target gene transcription (eg, GRP78, CHOP, XBP1) and regulate autophagy and apoptosis through XBP1 and CHOP.25
ER stress: a double-edged sword in TMZ sensitivity adjustment
The protective roles of ER stress
ER stress plays an important role in the resistance of GBM to TMZ toxicity by enhancing the ability of the molecular chaperones’ protein folding capacity and upregulating the ER-associated protein degradation pathway, autophagy, and other cellular adaptive responses. Currently, studies on the link between ER stress and TMZ chemoresistance have primarily focused on cellular models and on multiple glioma cell models. However, the exact mechanism has not yet been clarified. These studies suggest that GRP78 is crucial for the changes in glioma chemosensitivity.26 In GBM cell lines, knockdown of GRP78 by siRNA induced cell death by inducing CHOP expression and caspase-7 activation, whereas overexpression of GRP78 significantly increased the resistance of GBM cells to chemotherapeutic agents such as TMZ.9 Epigallocatechin 3-gallate (EGCG) is a green tea extract that inhibits GRP78 by targeting the ATP-binding domains and has been demonstrated to enhance GBM sensitivity to TMZ in vitro. Furthermore, using two pairs of TMZ-sensitive/resistant cell lines (D54-S and D54-R; U87-S and U87-R), Sun et al discovered that the ER stress-induced protein, prolyl 4-hydroxylase subunit beta (P4HB), has an important role in glioma TMZ resistance. P4HB is an ER stress-inducible multifunctional protein with disulfide isomerase activity and is overexpressed in TMZ-resistant and recurrent GBM. P4HB overexpression or siRNA knockdown in GBM cell lines resulted in resistance or sensitivity to TMZ.12 In addition, it has been shown that miR-210 is involved in the regulation of P4HB expression and confers GBM cell resistance to TMZ.13 Thus far, there is little evidence linking ER stress molecules with TMZ-resistance; most studies have focused on the cellular adaptive response mediated by ER stress, such as autophagy (described in detail below). By analyzing cell models, xenograft tumor models, human GBM pathology samples, and databases, Epple et al found that the UPR may generate resistance via regulation of cell metabolism.27 Moreover, it has been demonstrated at the cellular level that the ER stress factor, ATF4, can regulate oxidative metabolism through xCT (SLC7a11) and increase the resistance of glioma cells to TMZ.10
Thus, under mild ER stress, the activated UPR can maintain cellular homeostasis via activating molecular chaperones, inhibiting global protein synthesis, increasing the degradation of misfolded or unfolded proteins, changing the oxidative metabolism, and then inhibiting cell death, resulting in reduced sensitivity of GBM to chemotherapy.
ER stress acts as a pro-death mechanism
Under mild ER stress, a variety of cytoprotective effects can lead to glioma cell resistance to TMZ. However, under persistent and severe ER stress, cell homeostasis cannot be restored, resulting in the activation of various pathways. For instance, knockout of the ER stress-related factor mentioned above will transform protective ER stress into a fatal stress response. Hence, increasing the ER stress level has become a viable strategy for improved TMZ sensitivity. For example, sphingosine kinase inhibitors (SKIs) can increase TMZ toxicity through increasing ER stress, while the ER stress inhibitor 4-PBA reverses this sensitization effect.28 Fluoxetine, an antitumor-associated depression drug, works synergistically with TMZ through a CHOP-dependent apoptotic pathway mediated by ER stress. The new ER stress inducer, JLK1486, the systemic cancer chemotherapeutic drug, perillyl alcohol, and the novel trinorguaiane-type sesquiterpene, Radicol, can increase TMZ sensitization through CHOP by inducing lethal ER stress.29–31 Perillyl alcohol and TMZ have been conjugated to synthesize a new compound, NEO212, which shows stronger antiglioma properties, probably because of the enhanced apoptotic pathway of lethal ER stress.32 However, Xipell et al have found that ER stress-inducing agents can also increase the sensitivity of glioma cells to TMZ by downregulating MGMT, MPG, and Rad51.33
In conclusion, when ER stress is too severe, the UPR cannot restore homeostasis, and thus induces cell apoptosis through ATF4, CHOP, and other factors. Hence, increased ER stress may be an efficient target to enhance TMZ sensitivity. However, the specificity of its targets remains to be resolved.
ER stress-induced autophagy: the two faces of TMZ sensitivity regulation
At present, a variety of antitumor treatment methods (such as those that increase ER stress) cause autophagy in tumor cells. Autophagy is an important downstream ER stress event that has a double-edged sword role in ER stress largely through the regulation of apoptosis. Under ER stress, apoptosis occurs mainly through the mitochondrial pathways (endogenous) and death receptor pathways (exogenous). Numerous studies have shown that autophagy has a role in inhibiting or promoting apoptosis by participating in the critical steps of these two apoptosis pathways and in the regulation of crucial proteins.
Autophagy as a protective mechanism
Thus far, studies have shown that autophagy mainly acts as a pro-survival mechanism in the treatment of glioma with TMZ. Inhibition of autophagy can significantly increase TMZ-induced apoptosis at the clinical treatment concentration (≤100 μM), indicating that inhibition of autophagy is an effective strategy for improving the therapeutic effect of TMZ.34,35 Knockout of critical autophagy genes (eg, ATG5, ATG7, BECN1) or autophagy blockers (eg, chloroquine and its derivatives) can increase ER stress-related apoptosis, leading to increased CHOP expression and caspase-3 and PARP cleavage.36,37 This evidence suggests that autophagy has an important role in degrading unfolded proteins, reducing protein load, and promoting cell survival. In addition, ER stress can induce mitochondrial autophagy, degrade damaged mitochondria, and protect cells from death.38
Autophagy can protect cells from apoptosis by clearing unfolded proteins, reducing protein load, and eliminating damaged organelles (mainly mitochondria), as well as reduce apoptosis by regulating the activity of caspase family proteins. Studies have shown that inhibiting autophagy by knockdown of BECN1 (which encodes Beclin1) and VPS34 obviously increased the activation of caspase-8, the release of cytochrome C, and the proportion of Annexin V-positive cells in apoptosis-deficient cell lines HCT116 (BAX−/−) and MCF-7 (CASP3−/−) treated with the exogenous apoptosis pathway-inducing drug, TRAIL, and the classic chemotherapy drug, cisplatin. This effect was counteracted by caspase-8 inhibitors and caspase-8-targeting RNAi.39 These findings suggest that inhibiting autophagy mediated by ER stress and induced by the inhibition of the death receptor and chemotherapeutic drugs increases caspase-8 activation in apoptotic cell lines. Similarly, Amir et al have demonstrated that autophagy occurs during hepatocyte apoptosis induced by D-galactosamine (GalN) and lipopolysaccharide (LPS) in mice, and inhibiting autophagy by hepatic tissue-specific inhibition of ATG7 increased hepatocellular caspase-8 activity and apoptosis.40 Zhang et al have demonstrated that autophagy protected U87MG glioma cells from H2O2-induced oxidative stress by degrading caspase-8.41 Taken together, these findings suggest that autophagy contributes to avoidance of apoptosis by blocking caspase-8 activation.
It has been demonstrated that blocking autophagic flux can increase TMZ sensitization,36–38,42 and combination TMZ adjuvant therapy has entered Phase I/II clinical trials.43 In addition, several experiments have demonstrated that ataxia telangiectasia mutated (ATM) kinase inhibitors and plant extracts (eg, resveratrol) inhibited autophagy in both cellular models and mouse xenograft tumor models, and markedly enhanced TMZ cytotoxicity, reduced tumor volume, and increased mouse lifespan.44–48 However, these experiments did not involve analysis of caspase family proteins. Whether autophagy is involved in the regulation of caspase activity under the action of TMZ requires further study.
In conclusion, autophagy has a protective role in ER stress by clearing misfolded proteins and damaged organelles (mainly mitochondria) and by inhibiting caspase-8 activity.
Autophagy as a pro-death mechanism
Although autophagy under ER stress can degrade misfolded/unfolded proteins to maintain cellular homeostasis and inhibit cell death, it can also participate in ER stress-induced apoptosis.49 Autophagy-associated cell death is currently thought to result from complete degradation of cellular components caused by excessive autophagy. In T98G glioma cells, silencing of the critical autophagy genes BECN1 and ATG7 by siRNA partially decreased the cytotoxicity of TMZ.50 In addition, drugs that promote autophagy-mediated apoptosis such as Δ9-tetrahydrocannabinol51 and oncolytic adenovirus CRAd–Surivin–pk7,52 in combination with TMZ, significantly reduced the volume of xenograft tumors. Furthermore, dasatinib,53 rapamycin,50 the dual class IA inhibitor of PI3K/mTOR, PI-103,54 pan BCL-2 inhibitors,55 and VPA56 improved the TMZ effect through autophagy in different GBM cell lines.
Studies on ER stress have indicated that the autophagosomal membrane serves as a platform for intracellular death-inducing signaling complex (iDISC), which mediates caspase-8 activation and apoptosis. In 1998, Martin et al found that forced membrane localization and self-association of caspase-8 promoted apoptosis.57 Furthermore, it has been demonstrated that caspase-8 can be recruited to the ATG12–ATG5 complex by FADD and that the autophagic machinery has an important role in caspase-8 activation.58–60 Moreover, Young et al found that the caspase-8–FADD complex colocalized with ATG5 in ATG16- and LC3-positive structures upon treatment with SKI-I (a pan-sphingosine kinase inhibitor) and bortezomib (a proteasome inhibitor), indicating that the autophagosomal membrane serves as a platform for this complex and demonstrated that the recruitment of caspase-8 to this complex depends on p62/SQSTM1.61 Inhibition of the early stage of autophagy through knockdown of ATG5 or ATG3 significantly reduced caspase-8 activation and apoptosis, whereas inhibiting late-stage autophagic flux with bafilomycin A1 increased caspase-dependent cell death.61 Furthermore, using HAMLET treatment, Zhang et al demonstrated that p62/SQSTM1 is involved in the regulation of apoptosis by regulating caspase-8 activation in U87MG glioma cells and that the p62/SQSTM1 UBA domain is required for it to activate caspase-8.62 And another study showed that E3 ubiquitin ligase TRIM13 could regulate caspase-8 ubiquitination and translocation to autophagosomal membranes under ER stress.63 And in mitochondrial apoptotic pathway-deficient cell lines, ER stress inducers tunicamycin and thapsigargin promoted the formation of ATG5–FADD–caspase-8 complexes and caspase-8 activation.39 Furthermore, Tang et al found that caspase-8 dimerization and activation based on iDISC formation occur on accumulated immature autophagosomal membranes and require the LC3 conjugation machinery.64 Inhibiting the initial stages of autophagy by knocking out ATG5 and ATG7 decreased caspase-8-dependent apoptosis.39,65 Conversely, increased induction of autophagy would make the cell sensitive to autophagy blockers such as chloroquine and bafilomycin A1 through p62/SQSTM1-dependent caspase-8 activation.66–68 However, autophagy could also play a cytoprotective role by degrading caspase-8 or blocking intrinsic or extrinsic apoptosis pathways, and inhibiting autophagy initiation by siRNA or inhibitors enhances the activation of caspase-8 and apoptosis.40,41,69 However, the factors that determine the completion of the autophagy process or that serve as a platform for caspase-8 activation and iDISC formation remain unknown.
In addition to the regulation of apoptosis through the interaction with caspases, autophagy may promote apoptosis by degrading endogenous antiapoptotic factors. Inhibitors of apoptosis (IAPs) are an evolutionarily conserved protein family that inhibit apoptosis by inactivating caspases.70 Hou et al found that ATG1 and ATG7 mutations caused a decrease in DNA fragmentation, but did not affect nuclear condensation in degenerating mid-stage egg chambers, indicating that autophagy is partially involved in the regulation of cell death. During this stage, the effector caspase, DCP-1, and the IAP protein, Bruce, closely interact with autophagy.71 Subsequently, Nezis et al revealed that inhibiting autophagy by knocking out ATG1, ATG13, and type III PI3K (VPS34) blocks developmental apoptosis in the late-stage nurse cells of Drosophila melanogaster oogenesis. Furthermore, it has been shown that dBruce colocalized with the autophagy marker, ATG8-GFP, and accumulated in autophagy-mutated cells.72,73 Xu et al demonstrated that CHAL-24 induced the degradation of c-FLIP (L) and c-IAP via autophagy, thereby significantly enhancing the antitumor activity of TRAIL.74 Additionally, it has been reported that the PERK–eIF2α–ATF4 pathway of the UPR is involved in the degradation of the antiapoptotic protein, XIAP.16 Although the PERK axis is closely related to autophagy, whether autophagy is directly involved in the degradation of XIAP protein still lacks direct experimental evidence. Taken together, this evidence demonstrates that autophagy can promote apoptosis by autophagic degradation of certain types of IAPs.
In summary, autophagy undertakes a cytoprotective role by eliminating misfolded/unfolded proteins and damaged organelles and inhibiting the activation of caspases under ER stress. It also serves as a platform for the activation of caspase-8 and degradation of IAPs, promoting ER stress-induced apoptotic death.
However, the role of autophagy in the treatment of GBM with TMZ remains controversial, and the mechanism by which autophagy switches between a pro-survival and a pro-death role remains unclear. Combining the close relationship between ER stress and autophagy, it is likely that ER stress determines the role of autophagy in the regulation of cell survival and death. Hence, we propose the following model: in mild or moderate ER stress, the activated UPR maintains cellular homeostasis by inhibiting new protein synthesis and increasing protein folding capacity, in which autophagy has an important role by eliminating misfolded/unfolded proteins and inhibiting or degrading caspases. In severe ER stress, the UPR activates the apoptotic process by CHOP and other proteins, while autophagy serves as a platform for the activation of caspase-8 to promote apoptosis. This hypothetical model considers that autophagy specifically determines the fate of cells under ER stress, and that ER function is mainly mediated through the modulation of autophagy function (Figure 2).
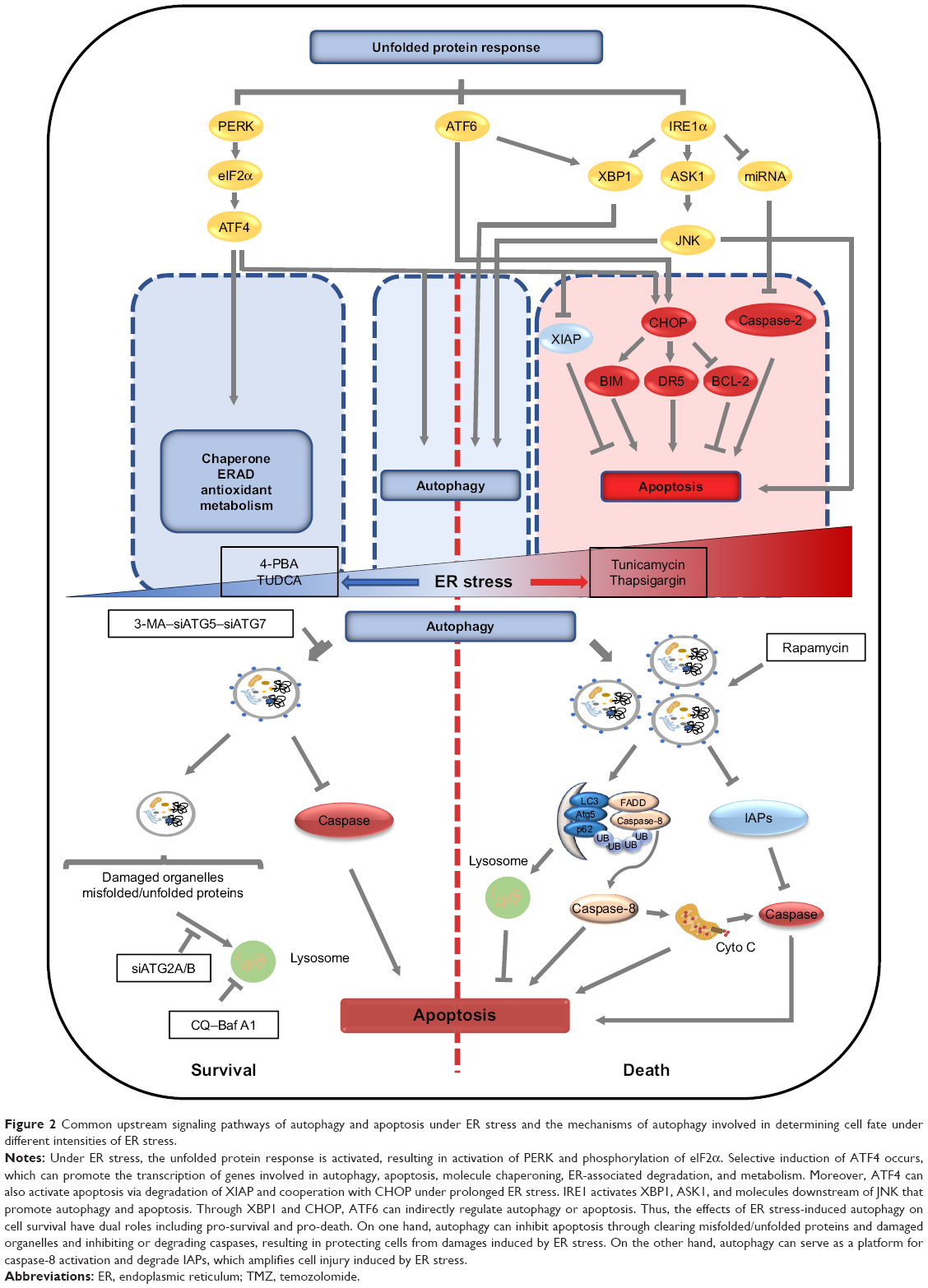 | Figure 2 Common upstream signaling pathways of autophagy and apoptosis under ER stress and the mechanisms of autophagy involved in determining cell fate under different intensities of ER stress. |
Conclusion and prospect
The resistance of GBM to TMZ treatment is the bottleneck in the clinical treatment of this disease. The off-target effects of ER stress have various and intricate outcomes on the sensitivity of GBM to chemotherapy (mainly TMZ). Undoubtedly, ER stress and autophagy are crucial regulators of TMZ sensitivity but may be more likely to function in a cell-specific manner. Finally, the survival and death of GBM cells theoretically depend on the dose and duration of TMZ treatment, the DNA repair capacity of cells, the rate of proliferation, and the critical proteins that activate DNA repair, such as ATM. However, it is unclear to how the mechanism of ER stress and autophagy transition between pro-survival and pro-apoptosis roles to ultimately determine cell fate. Under low doses and short-term TMZ treatment, ER stress and autophagy may have a cytoprotective role; however, it is unknown if they have a role in promoting cell death. If this hypothesis is validated, the dose and duration of TMZ treatment will become an important factor affecting prognosis after chemotherapy. The switch between the pro-survival and pro-death effects induced by ER stress and its downstream autophagy in different therapeutic environments requires further exploration. It is of great importance to develop more combinatorial solutions that will increase glioblastoma sensitivity to TMZ and reduce off-target effects.
Acknowledgments
This work was supported in part by the Norman Bethune Program of Jilin University (2015218), the Lateral Research Funds of Jilin University (2015377), the Development and Reform Commission of Jilin Province (2015Y032), the Education Office of Jilin Province (2015509), the Scientific Research Foundation for the Returned Overseas Chinese Scholars (2012–2014), the Human Resources and Social Security Department of Jilin Province (#2012–2014), and the International Cooperation Project funded by the Department of Science and Technology of Jilin Province (20160414007 GH). We thank Nikki March, PhD, from Liwen Bianji, Edanz Editing China, for editing the English text of a draft of this manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Fitzmaurice C, Dicker D, Pain A, et al; Global Burden of Disease Cancer Collaboration. The global burden of cancer 2013. JAMA Oncol. 2015;1(4):505–527. | ||
Fulda S, Kögel D. Cell death by autophagy: emerging molecular mechanisms and implications for cancer therapy. Oncogene. 2015;34(40):5105–5113. | ||
Sui X, Chen R, Wang Z, et al. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis. 2013;4(10):e838. | ||
Li S, Jiang T, Li G, Wang Z. Impact of p53 status to response of temozolomide in low MGMT expression glioblastomas: preliminary results. Neurol Res. 2008;30(6):567–570. | ||
Kang BR, Yang SH, Chung BR, Kim W, Kim Y. Cell surface GRP78 as a biomarker and target for suppressing glioma cells. Sci Rep. 2016;6(1):34922. | ||
Lee HK, Xiang C, Cazacu S, et al. GRP78 is overexpressed in glioblastomas and regulates glioma cell growth and apoptosis. Neuro Oncol. 2008;10(3):236–243. | ||
Virrey JJ, Dong D, Stiles C, et al. Stress chaperone GRP78/BIP confers chemoresistance to tumor-associated endothelial cells. Mol Cancer Res. 2008;6(8):1268–1275. | ||
Dong D, Ni M, Li J, et al. Critical role of the stress chaperone GRP78/BiP in tumor proliferation, survival, and tumor angiogenesis in transgene-induced mammary tumor development. Cancer Res. 2008;68(2):498–505. | ||
Pyrko P, Schönthal AH, Hofman FM, Chen TC, Lee AS. The unfolded protein response regulator GRP78/BiP as a novel target for increasing chemosensitivity in malignant gliomas. Cancer Res. 2007;67(20):9809–9816. | ||
Chen D, Rauh M, Buchfelder M, Eyupoglu IY, Savaskan N. The oxido-metabolic driver ATF4 enhances temozolamide chemo-resistance in human gliomas. Oncotarget. 2017;8(31):51164–51176. | ||
Dadey DY, Kapoor V, Khudanyan A, et al. The ATF6 pathway of the ER stress response contributes to enhanced viability in glioblastoma. Oncotarget. 2016;7(2):2080–2092. | ||
Sun S, Lee D, Ho AS, et al. Inhibition of prolyl 4-hydroxylase, beta polypeptide (P4HB) attenuates temozolomide resistance in malignant glioma via the endoplasmic reticulum stress response (ERSR) pathways. Neuro Oncol. 2013;15(5):562–577. | ||
Lee D, Sun S, Zhang XQ, et al. MicroRNA-210 and endoplasmic reticulum chaperones in the regulation of chemoresistance in glioblastoma. J Cancer. 2015;6(3):227–232. | ||
Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13(2):89–102. | ||
B’chir W, Maurin AC, Carraro V, et al. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013;41(16):7683–7699. | ||
Hiramatsu N, Messah C, Han J, LaVail MM, Kaufman RJ, Lin JH. Translational and posttranslational regulation of XIAP by eIF2α and ATF4 promotes ER stress–induced cell death during the unfolded protein response. Mol Biol Cell. 2014;25(9):1411–1420. | ||
McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. GADD153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001;21(4):1249–1259. | ||
Puthalakath H, O’Reilly LA, Gunn P, et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 2007;129(7):1337–1349. | ||
Yamaguchi H, Wang HG. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J Biol Chem. 2004;279(44):45495–45502. | ||
Chen P, Hu T, Liang Y, et al. Neddylation inhibition activates the extrinsic apoptosis pathway through ATF4-CHOP-DR5 axis in human esophageal cancer cells. Clin Cancer Res. 2016;22(16):4145–4157. | ||
Chao CC, Huang CC, Lu DY, et al. Ca2+ store depletion and endoplasmic reticulum stress are involved in P2X7 receptor-mediated neurotoxicity in differentiated NG108-15 cells. J Cell Biochem. 2012;113(4):1377–1385. | ||
Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta. 1833;2013(12):3460–3470. | ||
Maurel M, Chevet E. Endoplasmic reticulum stress signaling: the microRNA connection. Am J Physiol Cell Physiol. 2013;304(12):C1117–C1126. | ||
Margariti A, Li H, Chen T, et al. XBP1 mRNA splicing triggers an autophagic response in endothelial cells through BECLIN-1 transcriptional activation. J Biol Chem. 2013;288(2):859–872. | ||
Adachi Y, Yamamoto K, Okada T, Yoshida H, Harada A, Mori K. ATF6 is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell Struct Funct. 2008;33(1):75–89. | ||
Chen TC. GRP78/BiP modulation of GRP78/BiP in altering sensitivity to chemotherapy. Methods Enzymol. 2011;491:25–36. | ||
Epple LM, Dodd RD, Merz AL, et al. Induction of the unfolded protein response drives enhanced metabolism and chemoresistance in glioma cells. PLoS One. 2013;8(8):e73267. | ||
Noack J, Choi J, Richter K, et al. A sphingosine kinase inhibitor combined with temozolomide induces glioblastoma cell death through accumulation of dihydrosphingosine and dihydroceramide, endoplasmic reticulum stress and autophagy. Cell Death Dis. 2014;5(9):e1425. | ||
Weatherbee JL, Kraus JL, Ross AH. ER stress in temozolomide-treated glioblastomas interferes with DNA repair and induces apoptosis. Oncotarget. 2016;7(28):43820–43834. | ||
Cho HY, Wang W, Jhaveri N, et al. Perillyl alcohol for the treatment of temozolomide-resistant gliomas. Mol Cancer Ther. 2012;11(11):2462–2472. | ||
Li ZY, Zhang C, Chen L, et al. Radicol, a novel trinorguaiane-type sesquiterpene, induces temozolomide-resistant glioma cell apoptosis via ER stress and Akt/mTOR pathway blockade. Phytother Res. 2017;31(5):729–739. | ||
Cho HY, Wang W, Jhaveri N, et al. NEO212, temozolomide conjugated to perillyl alcohol, is a novel drug for effective treatment of a broad range of temozolomide-resistant gliomas. Mol Cancer Ther. 2014;13(8):2004–2017. | ||
Xipell E, Aragón T, Martínez-Velez N, et al. Endoplasmic reticulum stress-inducing drugs sensitize glioma cells to temozolomide through downregulation of MGMT, MPG, and Rad51. Neuro Oncol. 2016;18(8):1109–1119. | ||
Kanzawa T, Germano IM, Komata T, et al. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004;11(4):448–457. | ||
Nanegrungsunk D, Onchan W, Chattipakorn N, Chattipakorn SC. Current evidence of temozolomide and bevacizumab in treatment of gliomas. Neurol Res. 2015;37(2):167–183. | ||
Golden EB, Cho HY, Jahanian A, et al. Chloroquine enhances temozolomide cytotoxicity in malignant gliomas by blocking autophagy. Neurosurg Focus. 2014;37(6):E12. | ||
Zanotto-Filho A, Braganhol E, Klafke K, et al. Autophagy inhibition improves the efficacy of curcumin/temozolomide combination therapy in glioblastomas. Cancer Lett. 2015;358(2):220–231. | ||
Hori YS, Hosoda R, Akiyama Y, et al. Chloroquine potentiates temozolomide cytotoxicity by inhibiting mitochondrial autophagy in glioma cells. J Neurooncol. 2015;122(1):11–20. | ||
Deegan S, Saveljeva S, Logue SE, et al. Deficiency in the mitochondrial apoptotic pathway reveals the toxic potential of autophagy under ER stress conditions. Autophagy. 2014;10(11):1921–1936. | ||
Amir M, Zhao E, Fontana L, et al. Inhibition of hepatocyte autophagy increases tumor necrosis factor-dependent liver injury by promoting caspase-8 activation. Cell Death Differ. 2013;20(7):878–887. | ||
Zhang YB, Zhao W, Zeng RX. Autophagic degradation of caspase-8 protects U87mg cells against H2O2-induced oxidative stress. Asian Pac J Cancer Prev. 2013;14(7):4095–4099. | ||
Lee SW, Kim HK, Lee NH, et al. The synergistic effect of combination temozolomide and chloroquine treatment is dependent on autophagy formation and p53 status in glioma cells. Cancer Lett. 2015;360(2):195–204. | ||
Rosenfeld MR, Ye X, Supko JG, et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy. 2014;10(8):1359–1368. | ||
Lin CJ, Lee CC, Shih YL, et al. Resveratrol enhances the therapeutic effect of temozolomide against malignant glioma in vitro and in vivo by inhibiting autophagy. Free Radic Biol Med. 2012;52(2):377–391. | ||
Agnihotri S, Burrell K, Buczkowicz P, et al. ATM regulates 3-methylpurine-DNA glycosylase and promotes therapeutic resistance to alkylating agents. Cancer Discov. 2014;4(10):1198–1213. | ||
Zou Y, Wang Q, Wang W. MutL homolog 1 contributes to temozolomide-induced autophagy via ataxia-telangiectasia mutated in glioma. Mol Med Rep. 2015;11(6):4591–4596. | ||
Zou Y, Wang Q, Li B, Xie B, Wang W. Temozolomide induces autophagy via ATM-AMPK-ULK1 pathways in glioma. Mol Med Rep. 2014;10(1):411–416. | ||
Liao CL, Chen CM, Chang YZ, et al. Pine (Pinus morrisonicola Hayata) needle extracts sensitize GBM8901 human glioblastoma cells to temozolomide by downregulating autophagy and O(6)-methylguanine-DNA methyltransferase expression. J Agric Food Chem. 2014;62(43):10458–10467. | ||
Zhang J, Hummersone M, Matthews CS, Stevens MFG, Bradshaw TD, et al. N3-substituted temozolomide analogs overcome methylguanine-DNA methyltransferase and mismatch repair precipitating apoptotic and autophagic cancer cell death. Oncology. 2015;88(1):28–48. | ||
Palumbo S, Pirtoli L, Tini P, et al. Different involvement of autophagy in human malignant glioma cell lines undergoing irradiation and temozolomide combined treatments. J Cell Biochem. 2012;113(7):2308–2318. | ||
Torres S, Lorente M, Rodríguez-Fornés F, et al. A combined preclinical therapy of cannabinoids and temozolomide against glioma. Mol Cancer Ther. 2011;10(1):90–103. | ||
Ulasov IV, Sonabend AM, Nandi S, et al. Combination of adenoviral virotherapy and temozolomide chemotherapy eradicates malignant glioma through autophagic and apoptotic cell death in vivo. Br J Cancer. 2009;100(7):1154–1164. | ||
Milano V, Piao Y, Lafortune T, de Groot J. Dasatinib-induced autophagy is enhanced in combination with temozolomide in glioma. Mol Cancer Ther. 2009;8(2):394–406. | ||
Choi EJ, Cho BJ, Lee DJ, et al. Enhanced cytotoxic effect of radiation and temozolomide in malignant glioma cells: targeting PI3K-AKT-mTOR signaling, Hsp90 and histone deacetylases. BMC Cancer. 2014;14(1):17. | ||
Voss V, Senft C, Lang V, et al. The pan-Bcl-2 inhibitor (−)-gossypol triggers autophagic cell death in malignant glioma. Mol Cancer Res. 2010;8(7):1002–1016. | ||
Cui-jie S, Ming-wei W, Fu-rong C, et al. Histone deacetylase inhibitor, 2-propylpentanoic acid, increases the chemosensitivity and radiosensitivity of human glioma cell lines in vitro. Chin Med J. 2012;125(24):4338–4343. | ||
Martin DA, Siegel RM, Zheng L, Lenardo MJ. Membrane oligomerization and cleavage activates the caspase-8 (FLICE/MACHalpha1) death signal. J Biol Chem. 1998;273(8):4345–4349. | ||
Laussmann MA, Passante E, Düssmann H, et al. Proteasome inhibition can induce an autophagy-dependent apical activation of caspase-8. Cell Death Differ. 2011;18(10):1584–1597. | ||
Bell BD, Leverrier S, Weist BM, et al. FADD and caspase-8 control the outcome of autophagic signaling in proliferating T cells. Proc Natl Acad Sci U S A. 2008;105(43):16677–16682. | ||
Jiang H, White EJ, Ríos-Vicil CI, Xu J, Gomez-Manzano C, Fueyo J. Human adenovirus type 5 induces cell lysis through autophagy and autophagy-triggered caspase activity. J Virol. 2011;85(10):4720–4729. | ||
Young MM, Takahashi Y, Khan O, et al. Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. J Biol Chem. 2012;287(15):12455–12468. | ||
Zhang YB, Gong JL, Xing TY, Zheng SP, Ding W. Autophagy protein p62/SQSTM1 is involved in HAMLET-induced cell death by modulating apotosis in U87MG cells. Cell Death Dis. 2013;4(3):e550. | ||
Tomar D, Prajapati P, Sripada L, et al. TRIM13 regulates caspase-8 ubiquitination, translocation to autophagosomes and activation during ER stress induced cell death. Biochim Biophys Acta. 1833;2013(12):3134–3144. | ||
Tang Z, Takahashi Y, Chen C, et al. Atg2A/B deficiency switches cytoprotective autophagy to non-canonical caspase-8 activation and apoptosis. Cell Death Differ. 2017;24(12):2127–2138. | ||
So KY, Lee BH, Oh SH. The critical role of autophagy in cadmium-induced immunosuppression regulated by endoplasmic reticulum stress-mediated calpain activation in RAW264.7 mouse monocytes. Toxicology. 2018;393:15–25. | ||
Nàger M, Sallán MC, Visa A, et al. Inhibition of WNT-CTNNB1 signaling upregulates SQSTM1 and sensitizes glioblastoma cells to autophagy blockers. Autophagy. 2018;14(4):619–636. | ||
Villar VH, Nguyen TL, Delcroix V, et al. MTORC1 inhibition in cancer cells protects from glutaminolysis-mediated apoptosis during nutrient limitation. Nat Commun. 2017;8:14124. | ||
Fu W, Li X, Lu X, et al. A novel acridine derivative, LS-1-10 inhibits autophagic degradation and triggers apoptosis in colon cancer cells. Cell Death Dis. 2017;8(10):e3086. | ||
Yao CJ, Chow JM, Chuang SE, et al. Induction of forkhead class box O3a and apoptosis by a standardized ginsenoside formulation, KG-135, is potentiated by autophagy blockade in A549 human lung cancer cells. J Ginseng Res. 2017;41(3):247–256. | ||
Salvesen GS, Duckett CS. IAP proteins: blocking the road to death’s door. Nat Rev Mol Cell Biol. 2002;3(6):401–410. | ||
Hou YC, Chittaranjan S, Barbosa SG, McCall K, Gorski SM. Effector caspase Dcp-1 and IAP protein Bruce regulate starvation-induced autophagy during Drosophila melanogaster oogenesis. J Cell Biol. 2008;182(6):1127–1139. | ||
Nezis IP, Shravage BV, Sagona AP, et al. Autophagic degradation of dBruce controls DNA fragmentation in nurse cells during late Drosophila melanogaster oogenesis. J Cell Biol. 2010;190(4):523–531. | ||
Nezis IP, Shravage BV, Sagona AP, et al. Autophagy as a trigger for cell death: autophagic degradation of inhibitor of apoptosis dBruce controls DNA fragmentation during late oogenesis in Drosophila. Autophagy. 2010;6(8):1214–1215. | ||
Xu J, Xu X, Shi S, et al. Autophagy-mediated degradation of IAPs and c-FLIP(L) potentiates apoptosis induced by combination of TRAIL and Chal-24. J Cell Biochem. 2016;117(5):1136–1144. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.