Back to Journals » Journal of Inflammation Research » Volume 19
Targeting mTOR in Systemic Lupus Erythematosus: From Immune Cell Dysfunction to Clinical Translation
Authors Jiang X, Sun L, Ji J, Zhao C, Wang Y, Zhang H, Wu Q, Su Q, Li F, Zhang S, Cheng T
Received 3 January 2026
Accepted for publication 20 March 2026
Published 8 June 2026 Volume 2026:19 593467
DOI https://doi.org/10.2147/JIR.S593467
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Adrian Lee
Xiaojing Jiang,1– 3 Lu Sun,2,3 Jiahui Ji,2,3 Chaoran Zhao,2,3 Yan Wang,2– 4 Han Zhang,2,3,5 Qi Wu,2,3 Qinyi Su,1– 3 Fang Li,1,2 Shengxiao Zhang,1– 3 Ting Cheng2,3,6
1Department of Rheumatology and Immunology, The Second Hospital of Shanxi Medical University, Taiyuan, Shanxi Province, People’s Republic of China; 2Shanxi Provincial Key Laboratory of Rheumatism Immune Microecology, Taiyuan, Shanxi Province, People’s Republic of China; 3Key Laboratory of Cellular Physiology at Shanxi Medical University, Ministry of Education, Taiyuan, Shanxi Province, People’s Republic of China; 4Department of Hepatobiliary, Surgery and Liver Transplantation Center, Shanxi Key Laboratory of Digestive Diseases and Organ Transplantation, The First Hospital of Shanxi Medical University, Taiyuan, Shanxi Province, People’s Republic of China; 5Department of Psychiatry, The Second Hospital of Shanxi Medical University, Taiyuan, People’s Republic of China; 6Department of Geriatric Medicine, Shanxi Bethune Hospital, Shanxi Academy of Medical Sciences, Third Hospital of Shanxi Medical University, Tongji Shanxi Hospital, Taiyuan, People’s Republic of China
Correspondence: Ting Cheng, Email [email protected]
Abstract: Systemic lupus erythematosus (SLE) is an autoimmune disease mainly characterized by the production of autoantibodies and the deposition of immune complexes, leading to damage to multiple organs and tissues. The mammalian target of rapamycin (mTOR), a serine/threonine kinase, regulates cell growth, proliferation, and survival. The dysfunction of immune cells, such as T cells, B cells, macrophages, and dendritic cells, plays a crucial role in the pathogenesis of systemic lupus erythematosus (SLE) by leading to the overproduction of autoantibodies, deposition of immune complexes, and subsequent tissue damage. The mTOR signaling plays an important role in SLE pathogenesis by affecting the proliferation and differentiation of immune cells and the secretion of inflammatory cytokines. Numerous studies have confirmed that mTOR inhibition can significantly relieve the clinical symptoms and delay disease progression in patients with SLE. Treatment with mTOR inhibitors effectively reduces disease activity and enables glucocorticoid-sparing. However, its therapeutic efficacy in lupus nephritis remains to be investigated. Furthermore, some studies have reported limited efficacy or adverse effects of mTOR inhibitors in certain SLE subsets. Given that mTOR-independent pathways (e.g. JAK-STAT, NF-κB) are also implicated in SLE pathogenesis, combination therapeutic strategies may be necessary. Consequently, further research into the role of mTOR in SLE and the potential of mTOR-targeted therapy is warranted. In this Review, we summarized the biological functions of mTOR, its effects on various immune cells populations, and the therapeutic effects of mTOR inhibitors in SLE, with the aim of providing a reference for the further exploration of therapeutic targets in SLE treatment.
Keywords: systemic lupus erythematosus, SLE, mTOR, treatment, immunometabolism
Introduction
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease characterized by widespread inflammation and tissue damage.1 It predominantly affects women, especially those of reproductive age.2 Clinical manifestations are highly variable, ranging from mild fatigue and joint pain to potentially severe, persistent organ damage, including skin rashes, joint pain, renal involvement, and neurological manifestations, all of which contributes to its severity and unpredictability.3
mTOR is a serine/threonine protein kinase belonging to the PI3K-related kinase family. It forms two functionally distinct complexes in cells—mTORC1 and mTORC2—and serves as a central regulatory hub for cell growth, metabolism, and immune responses. Its activity is precisely regulated by multiple upstream signals, including growth factors, energy status, oxygen levels, and amino acids. The physiological functions primarily mediated by mTOR include protein synthesis, cellular metabolism, autophagy regulation, and cytoskeleton remodeling.
The underlying pathogenesis of SLE is the breakdown of immune tolerance to self-antigens.4 Within this process, the mammalian target of rapamycin (mTOR) plays a key regulatory role in immune cell function. It influences the activation, differentiation, and survival of various immune cells, including T cells, B cells, and macrophages,5 thereby positioning it a central factor in immune responses and the development of autoimmune diseases such as SLE. Furthermore, dysregulated mTOR signaling can promote aberrations in multiple cellular processes, including growth factor receptor signaling, glycolytic and lipid metabolism, and autophagy.6
Understanding how mTOR dysregulation contributes to these immune and inflammatory processes is crucial for developing targeted therapies aimed at modulating its activity in SLE patients. Therefore, this review describes the structure and key processes of the mTOR pathway, highlighting its role in SLE pathogenesis and the mechanisms by which mTOR inhibitors may contribute to disease treatment. By synthesizing these findings, this review aims to explore the potential of harnessing mTOR-targeted therapies to improve clinical outcomes for patients with SLE, as well as to discuss the potential challenges in its clinical translation.
The Biology of mTOR
In 1975, Sehgal et al identified an antifungal macrolide antibiotic from soil samples on Easter Island, which was subsequently named Rapamycin according to its site of origin.7 Rapamycin associates with the intracellular receptor FK 506-binding protein of 12 kDa (FKBP12), and this complex then binds to and inhibits mTOR.8,9 mTOR is a member of an evolutionarily conserved family of serine/threonine kinases that serves as a central regulator of cellular growth, metabolism, and homeostasis by integrating various extracellular and intracellular signals.5 It is essential for numerous biological processes, including cellular metabolism, immune responses, autophagy, survival, proliferation, and migration.6,10
mTOR participates in the formation of two different functional complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2).11 These complexes process different upstream regulators and downstream effectors, thereby eliciting unique signaling events to govern specific cellular processes.
mTORC1 is composed of three core components and two inhibitory components. The core components include mTOR, mammalian lethal with Sec13 protein 8 (mLST8), and Raptor (regulatory associated protein of mTOR complex 1).12 mLST8 associates with the catalytic domain of mTORC1 and stabilizes its kinase domain.13 Raptor is vital for recruiting substrates to mTORC1 and ensuring its proper subcellular localization.14 The two inhibitory components of mTORC1 are DEP-domain-containing mTOR-interacting protein (DEPTOR) and proline-rich AKT substrate 40 kDa (PRAS40).15,16 The primary function of mTORC1 is to promote anabolic processes by phosphorylating substrates that drive the production of macromolecules, including proteins, lipids, and nucleotides, and by stimulating ATP production.17 Additionally, mTORC1 inhibits catabolic processes such as autophagy.18 In proliferating cells, mTORC1 drives the synthesis of macromolecules required for growth. In metabolic tissues such as the liver, mTORC1 enhances nutrient storage and inhibits catabolic metabolism.19 These functions collectively ensure an adequate supply of energy and building blocks for cell growth and proliferation.
The core components of mTORC2 also include mTOR and mLST8, both of which are essential for its stability and function.20 In contrast to mTORC1, mTORC2 consists of rapamycin-insensitive companion of mTOR (Rictor), which, together with DEPTOR and the regulatory subunits mSin1 and Protor1/2, forms the functional complex.17 The presence of Rictor and mSin1 physically blocks the binding of the FKBP12-rapamycin complex to mTOR, rendering mTORC2 acutely insensitive to rapamycin. Regarding its biological functions, mTORC2 phosphorylates members of the AGC kinases family including AKT, to modulate cell survival, proliferation, and cytoskeletal organization.19
Upstream Regulators of mTOR
mTORC1 activity is tightly regulated by various environmental cues, including growth factors, energy status, oxygen levels, and amino acid availability. Growth factors primarily signal through the phosphatidylinositol 3-kinase (PI3K) – phosphoinositide-dependent kinase-1 (PDK1) –protein kinase B (AKT) axis, while energy stress and hypoxia act through the LKB1 liver kinase B1 (AKT)/ AMP-activated protein kinase (AMPK) pathway.5,6
The PI3K/AKT/TSC signaling cascade constitutes the major regulatory mechanism for mTORC1 in response to growth factors. Ligand binding to cell surface receptors initiates the recruitment and phosphorylation of PI3K, which generates phosphatidylinositol 3,4,5-trisphosphate (PIP3). This facilitates the membrane recruitment and activation of PDK1 and AKT. Subsequently, activated AKT phosphorylates the tuberous sclerosis complex 1/2 (TSC1/TSC2) complex, thereby inhibiting its GAP (GTPase-activating protein) activity toward RHEB (Ras homolog enriched in brain).21 Maintaining RHEB in the GTP-bound active state enables the activated RHEB to directly bind to mTORC1 and activate it. Conversely, phosphatase and tensin homolog negatively (PTEN) regulates this pathway by dephosphorylating PIP3, thereby antagonizing PI3K signaling and suppressing mTORC1 activation.5
Under hypoxic conditions, regulated in development and DNA damage responses 1 (REDD1) is induced and promotes TSC2 function, leading to mTORC1 inhibition.5 DNA damage can also suppress mTORC1 through multiple mechanisms, including transcriptional upregulation of TSC2 and PTEN.5 Additionally, both hypoxia and energy depletion activate LKB1-dependent AMPK. Activated AMPK inhibits mTORC1 through two parallel pathways: one is to phosphorylate and activate TSC2, promoting the formation of the inhibitory TSC1/TSC2 complex; the other is to directly phosphorylate Raptor, leading to the conformational inhibition of mTORC1.22 This dual mechanism ensures robust suppression of mTORC1 under conditions of energetic stress.
Beyond growth factor and energy sensing, mTORC1 activation is uniquely dependent on amino acid availability, particularly leucine and arginine, and this signaling occurs at the lysosomal membrane surface.5 Unlike growth factor signaling, the amino acid sensing mechanism does not rely on the TSC1/2 complex but is mediated by a specific protein.5 The Rag family of GTPases serves as a critical relay for amino acid sufficiency. Mammalian cells express four Rag isoforms: RagA, RagB, RagC, and RagD. These proteins form obligate heterodimers consisting of either RagA or RagB with either RagC or RagD.23 In the presence of sufficient amino acids, RagA/B is loaded with GTP while RagC/D binds GDP. The Rag GTPases, together with their regulator, the Ragulator complex, are anchored to the lysosomal surface. By recruiting mTORC1 to the lysosomal membrane, they achieve amino acid-dependent activation of mTORC1.5 Specifically, sufficient amino acids promote the GTP-bound state of RagA/B, enabling the recruitment of mTORC1 via interaction with Raptor. This transfer brings mTORC1 close to the direct activator RHEB, which is also located on the lysosomal membrane, thereby completing the activation.24
The cells’ perception of sufficient amino acids depends on the accumulation of amino acids within the lysosomal cavity. This process requires the participation of vacuolar H+-adenosine triphosphatase (v-ATPase). This proton pump, situated on the lysosomal membrane, acidifies the lysosomal lumen and initiates a signaling cascade that promotes Rag GTPase interaction with RHEB, thereby facilitating mTORC1 activation.25 A feedback loop exists between mTORC1 and v-ATPase, allowing for coordinated regulation of lysosomal function and mTORC1 signaling25 (Figure 1).
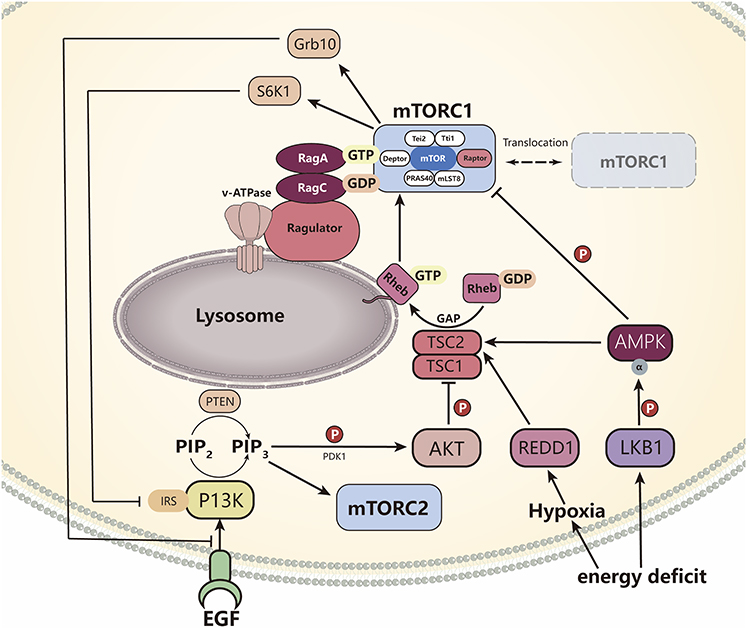 |
Figure 1 Upstream regulatory pathway of mTOR. Hypoxia and low energy can regulate the TSC1/TSC2 complex by activating the PI3K/Akt and LKB1-AMPK pathways, thereby inhibiting its GAP activity, activating RHEB, and promoting mTORC1 activity. AMPK can also directly phosphorylate Raptor to inhibit mTORC1. Hypoxia can also enhance TSC2 function by upregulating REDD1. Adequate amino acids promote the binding of Rag A/B-GTP to Rag C/D-GDP, guiding the translocation of mTORC1 to the surface of lysosomes and activating it through interaction with RHEB. V-ATPase enhances this process by adjusting the pH value inside the cavity. The increase in mTORC1 activity can be inhibited by S6K1 phosphorylation of IRS-1 and Grb10 to suppress mTORC2, forming a negative feedback regulation of insulin signaling. |
Compared to mTORC1, the regulation of mTORC2 is considerably less understood.5 Current evidence indicates that mTORC2 is primarily responsive to growth factor and hormone signaling, such as insulin, through the PI3K pathway.26,27 The PI3K product PIP3 promotes mTORC2 activation and facilitates the phosphorylation of AKT at Ser473, a residue commonly used as a marker of mTORC2 activity.5 PIP3 can also engage the PH domain of the mTORC2 subunit mSin1, relieving autoinhibition and promoting complex activation.5 Growth factor signaling enhances the association of mTORC2 with ribosomes, and insulin/PI3K signaling may promote AKT translation. Notably, there is a negative feedback loop between mTORC1 and mTORC2. Increased mTORC1 activity elevates ribosomal protein S6 kinase 1 (S6K1) function, which in turn phosphorylates insulin receptor substrate-1 (IRS-1) and growth factor receptor-bound protein 10 (Grb10). Finally, it inhibits the insulin/PI3K signaling pathway, thereby suppressing the activation of mTORC226 (Figure 1).
Downstream Targets of mTOR
Protein Synthesis
Ribosomal protein S6 kinase beta-1 (S6K1) and eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1) are two key downstream effectors through which mTORC1 regulates mRNA translation and protein synthesis. Upon activation, mTORC1 directly phosphorylates both S6K1 and 4E-BP1, thereby promoting cap-dependent translation initiation.6
4E-BP1 contains multiple phosphorylation sites, including Thr37, Thr46, Ser65, and Thr70. mTOR regulates 4E-BP1 phosphorylation through at least two mechanisms: first, the protein kinase activity of mTOR directly phosphorylates Thr37 and Thr46; second, mTOR inhibits phosphatases that would otherwise dephosphorylate Ser65 and Thr70.28 Phosphorylation of 4E-BP1 reduces its binding affinity for Eukaryotic Initiation Factor 4E (eIF4E), resulting in the release of this cap-binding protein. eIF4E subsequently participates in forming the eIF4F complex, a heterotrimer composed of eIF4E, the RNA helicase eIF4A, and a large scaffolding protein eIF4G. This complex recruits ribosomes to mRNA, thereby initiating the process of protein translation.29
In parallel, amino acid sufficiency activates mTORC1-S6K1 signaling pathway.30 mTORC1 directly phosphorylates S6K1 at the hydrophobic motif site Thr389, resulting in partial activation and exposing a docking site for PDK1. PDK1 then binds and phosphorylates S6K1 at the activation loop site Thr229, leading to full kinase activation.6 Once activated, S6K1 translocates to the cytoplasm, where it directly phosphorylates RPS6 (Ribosomal protein S6), a major component of the 40S ribosomal subunit.31 Phosphorylation of RPS6 enhances the translational efficiency of ribosomes, promoting the synthesis of proteins required for cell growth and proliferation.32
Beyond RPS6, S6K1 promotes protein translation by phosphorylating additional regulators of the translational machinery. These include Eukaryotic Initiation Factor 4B (eIF4B) and Programmed Cell Death Protein 4 (PDCD4).33 eIF4B acts as a positive regulator of the eIF4F complex; its phosphorylation by S6K1 enhances its interaction with eIF4A, facilitating mRNA unwinding and translation initiation. Conversely, PDCD4 functions as a negative regulator of eIF4Aby sequestering it and inhibiting its helicase activity. Through an indirect mechanism involving S6K1, PDCD4 is phosphorylated and subsequently targeted for degradation, thereby relieving this inhibition and further promoting translation initiation34 (Figure 2).
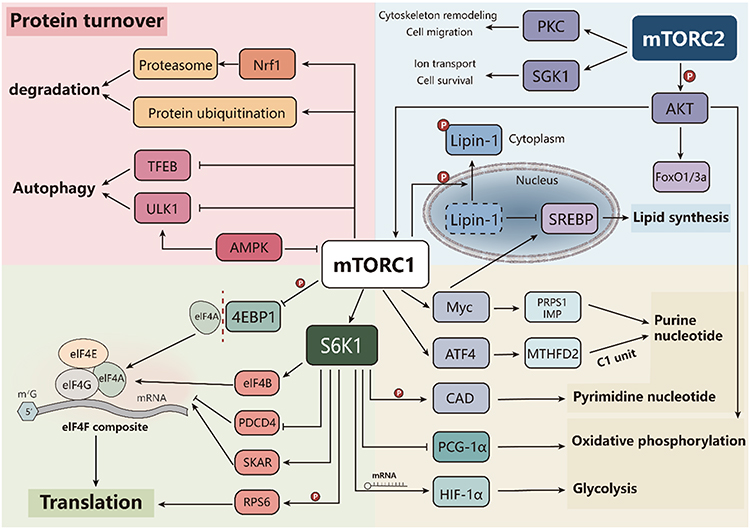 |
Figure 2 Downstream regulatory pathways of mTOR. Activated mTORC1 promotes protein translation by phosphorylating S6K1 and 4E-BP1, regulating the activity of eIF4E, eIF4B, and their related complexes. In nucleotide metabolism, the expression of MTHFD2 can be upregulated, and pyrimidine and purine synthesis can be promoted by activating CAD and transcription factor Myc. In addition, by enhancing the translation induced glycolysis of HIF-1 α and inhibiting the expression or activity of PGC-1 α, the oxidative phosphorylation level is reduced. In lipid metabolism, lipid synthesis is promoted by activating the SREBP pathway. MTORC2 participates in regulating cell metabolism, skeletal remodeling, and cell survival by phosphorylating Akt, PKC, and SGK1. In terms of protein turnover, mTORC1 inhibits the activity of ULK1 and TFEB, thereby negatively regulating autophagy. At the same time, upregulation of Nrf1 can enhance the expression of proteasome subunits and maintain protein homeostasis. |
Metabolism Regulation
mTORC1 coordinates multiple aspects of cellular metabolism, including lipid, glucose, and nucleotide metabolism, to support cell growth and proliferation.
In lipid metabolism, mTORC1 promotes lipogenesis by regulating the activity of sterol regulatory element-binding proteins (SREBPs), master transcription factors for fatty acid and cholesterol synthesis. mTORC1 phosphorylates lipin-1, a phosphatidic acid phosphatase that functions as a transcriptional coactivator, causing its nuclear exclusion and thereby alleviating its inhibitory effect on SREBP activity.3 Additionally, mTORC1 activates SREBP1 through an S6K1-dependent mechanism. However, the underlying mechanism remains unclear.35
Regarding glucose metabolism, mTORC1 promotes a metabolic shift from oxidative phosphorylation to aerobic glycolysis, a phenomenon known as the Warburg effect.3 As previously described, mTORC1 enhances HIF-1α mRNA translation through activation of the 4E-BP1 pathways. HIF-1α subsequently induces the expression of glycolytic enzymes and lactate dehydrogenase, thereby increasing glycolytic flux.35 Concurrently, mTORC1 signaling through inhibiting the expression and activity of peroxisome proliferator-activated receptor gamma coactivator 1α (PGC-1α), a key regulator of mitochondrial biogenesis and oxidative phosphorylation, further reinforcing the shift toward glycolysis.36
As for nucleotide metabolism, mTORC1 regulates nucleotide metabolism through both substrate supply and biosynthetic enzyme regulation. To supply one-carbon units for purine synthesis, mTORC1 increases ATF4-dependent expression of methylenetetrahydrofolate dehydrogenase 2 (MTHFD2), a critical enzyme in the mitochondrial tetrahydrofolate cycle.37 For pyrimidine synthesis, mTORC1 enhances the activity of carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase (CAD), the rate-limiting enzyme complex in de novo pyrimidine biosynthesis that catalyzes the first three steps from glutamine to dihydroorotate. This activation occurs through S6K1-mediated direct phosphorylation of CAD, which increases its enzymatic activity.38 Furthermore, mTORC1 indirectly promotes purine nucleotide synthesis by activating transcription factors such as Myc, which upregulate the expression of key purine synthesis enzymes, including phosphoribosyl pyrophosphate synthetase 1 (PRPS1) and IMP dehydrogenase.39
mTORC2 primarily regulates cellular metabolism and survival through its downstream kinase targets. One of the most critical functions of mTORC2 is the phosphorylation and activation of AKT at Ser473. Fully activated AKT promotes cell survival, proliferation, and growth by inhibiting pro-apoptotic factors such as FoxO1 and FoxO3a, enhancing glucose metabolism through increased glucose transporter expression and glycolytic enzyme activity, and activating mTORC1.40,41 mTORC2 also phosphorylates members of the protein kinase C (PKC) family, which modulates cytoskeletal remodeling, cell polarity, and cell migration.42–45 Additionally, mTORC2 phosphorylates and activates serum/glucocorticoid regulated kinase 1 (SGK1), which regulates ion transport, cell survival, and growth46 (Figure 2).
Protein Turnover
In addition to the various anabolic processes mentioned above, mTORC1 also promotes cell growth by suppressing catabolic pathways, particularly autophagy.47 The Unc-51 Like Autophagy Activating Kinase 1 (ULK1) complex, which comprises ULK1, ATG13, FIP200, and ATG101, serves as a critical initiator of autophagosome formation. mTORC1 directly phosphorylates ULK1, thereby preventing its activation by AMPK, a critical regulator of autophagy. Under conditions of energy depletion, activated AMPK can directly phosphorylate ULK1 at alternative sites, initiating autophagy even in the presence of residual mTORC1 activity. Therefore, the relative activities of mTORC1 and AMPK coordinately determine the extent of autophagy induction.47,48 Additionally, mTORC1 phosphorylates and inhibits Unc-51 Like Autophagy Activating Kinase 1 (TFEB), a master regulator of autophagy and lysosomal gene expression, thereby indirectly suppressing autophagic flux.34
The ubiquitin-proteasome system (UPS) represents the second major pathway for intracellular protein degradation. The relationship between mTORC1 signaling and proteasomal activity is complex and context-dependent. Acute mTORC1 inhibition has been shown to robustly promote protein ubiquitination, suggesting that active mTORC1 signaling normally suppresses this degradation pathway.1,49,50 Conversely, sustained mTORC1 activation upregulates proteasome subunit expression via the transcription factor Nrf1, thereby enhancing proteasomal activity to maintain protein homeostasis under conditions of elevated protein synthesis demands51 (Figure 2).
The Role of mTOR in SLE
The pathogenesis of SLE involves three major immunological disturbances: activation of the innate immune system, production of self-reactive antibodies, and emergence of autoreactive T cells.52 As a key driver of metabolic stress and inflammatory lineage development, mTOR is widely implicated in the metabolic reprogramming and functional alterations of immune cells in SLE, playing an important role in both innate and adaptive immunity.53 The mTOR signaling pathway influences immune cells proliferation, differentiation, and inflammatory cytokines secretion through its regulation of anabolism, energy metabolism, and autophagy, which is essential to SLE pathogenesis.54 Notably, studies have demonstrated that in SLE patients, mTORC1 is activated while mTORC2 is inhibited, suggesting a complex and differential involvement of the two mTOR complexes in disease mechanisms.55,56
Regulation of Macrophages by mTOR
Macrophages are indispensable components of innate immune systems and serve as a critical link between innate and adaptive immune through antigen presentation. Macrophage abnormalities represent a vital factor in the occurrence and development of SLE.57 Macrophage infiltration has been reported in association with lupus nephritis, indicating their direct involvement in organ damage.58 Depending on environmental cues, macrophages can polarize into two distinct phenotypes: classically activated M1 macrophages, which primarily exert pro-inflammatory activity, and alternatively activated M2 macrophages, which predominantly possess anti-inflammatory properties.59 An imbalance in macrophage polarization is also a typical characteristic of SLE. Abnormally activated M1 macrophages promote the release of inflammatory cytokines, exacerbating tissue damage.60 In contrast, M2 macrophages participate in immune regulation and tissue repair by releasing anti-inflammatory factors and suppressing pro-inflammatory cytokines expression.61 Adoptive transfer of M2 macrophages or induction of M2 polarization macrophages has been shown to alleviate SLE manifestations, whereas M1 macrophage transfer exacerbates disease progression.62,63 Collectively, impaired phagocytic ability and the dysregulation of M1/M2 macrophages balance contribute significantly to SLE pathogenesis.
mTOR is an important regulatory factor for macrophage activation and metabolism, with numerous studies demonstrating its essential role in macrophage polarization.19 However, the underlying mechanisms are complex, and conflicting findings regarding mTOR-mediated macrophage activation have been reported. Myeloid cell-specific deletion of PTEN in mice leads to constitutively enhanced PI3K and mTOR signaling, resulting in increased expression of M2 macrophage markers and enhanced activation of the Signal transducer and activator of transcription 6 (STAT6), a transcription factor important for M2 macrophage polarization signaling pathway, which is critical for M2 polarization.64 Conversely, inhibition of mTORC1 with rapamycin stimulates M1 macrophage polarization.65
Studies involving TSC1 deletion have yielded particularly complex results. Ablation of TSC1, which leads to increased mTORC1 activation and decreased mTORC2 activation, has been shown to promote M1 macrophage polarization while reducing M2 polarization. This effect occurs through two distinct mechanisms: first, TSC1 deletion promotes M1 polarization independently of STAT6 and Peroxisome Proliferator-Activated Receptor Gamma (PPARγ) by activating a GTPase–Rapidly Accelerated Fibrosarcoma 1 (RAF1)–Mitogen-Activated Protein Kinase Kinase (MEK)–Extracellular Signal-Regulated Kinase (ERK) pathway that operates independently of mTORC1; second, it reduces M2 polarization by activating mTORC1, which in turn suppresses the expression of CCAAT/Enhancer-Binding Protein Beta (CEBPβ).66 In contrast, another study found that TSC1 deletion promotes both M1 and M2 macrophage polarization.67 This discrepancy may be attributed to differences in experimental design, with one study examining functional responses upon inflammatory stimulation and the other accessing steady-state responses under basal conditions. AKT, a core component of the PI3K/AKT/mTOR signaling pathway, also plays a critical role in macrophage polarization, with different isoforms exerting opposing effects. Ablation of AKT1 promotes M1 macrophages polarization, whereas deletion of AKT2 favors M2 polarization.68 Consistent with these findings, AMPK deletion enhances M1 macrophage polarization by reducing AKT activation. Collectively, these results demonstrate that PI3K-AKT-mTOR pathway controls macrophage polarization, although the precise functions and interactions of each pathway member require further investigation. Impaired macrophage phagocytic ability, along with dysregulated autophagy and apoptosis, represents an essential link in SLE pathogenesis.69,70 Accumulation of apoptotic cells promotes the formation of autoimmune antigens and antibodies, thereby aggravating inflammatory response. Given that mTORC1 inhibits autophagy by phosphorylating ULK1, mTORC1 inhibition promotes autophagic progression, which may influence the clearance of apoptotic debris71 (Figure 3). In summary, the effects of mTOR on macrophage function are multifaceted and complex. Further research is needed to elucidate how modulation of macrophage polarization and autophagy through the mTOR pathway might be harnessed to improve clinical outcomes in SLE patients.
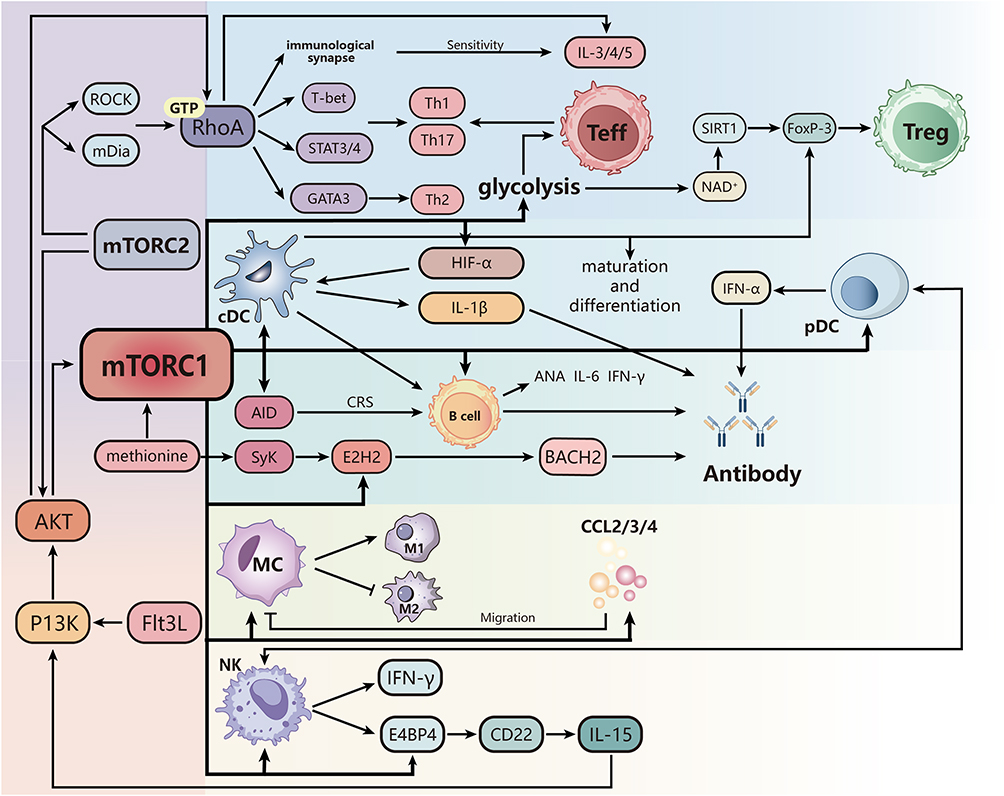 |
Figure 3 The role of mTOR in SLE. MTORC1 drives glycolysis in T cells, enhances effector T cell differentiation, and inhibits SIRT1 activity through an imbalance of NAD+/NADH ratio, thereby reducing Foxp3 expression and weakening Treg homeostasis. MTORC2 promotes the differentiation of Th1, Th2, and Th17 cells and enhances the production of IL-3/4/5 by activating the RhoA/Akt pathway. In dendritic cells, mTORC1 promotes cDC maturation and IL-1 β release, enhances pDC production of IFN-α, and interferes with antigen presentation by upregulating antigen uptake receptors and co stimulatory molecules; In addition, mTOR signaling drives DC induced B cells to promote IL-6, IFN-γ, and ANA production, while inhibiting Treg function through FoxP-3. MTORC1 upregulates AID in B cells, promotes antibody class switching, and induces EZH2 expression in synergy with Syk, inhibits BACH2, and promotes plasma cell differentiation. In macrophages, mTORC1 promotes M1 polarization and inhibits M2 differentiation, while downregulating CCL2, CCL3, and CCL4, limiting their chemotactic migration. IL-15 regulates NK cell development through the PI3K/AKT/mTORC1 axis and induces E4BP4 upregulation of CD22 to form a positive feedback; MTORC1 also enhances the synergy between NK and pDC, promotes the secretion of IFN-α and IFN-γ, and exacerbates SLE related inflammatory responses. |
Regulation of Dendritic Cells by mTOR
Dendritic cells (DCs) are the most powerful and important antigen-presenting cells in the immune system, capable of initiating primary immune responses. DCs can both activate effective immune responses and maintain immune tolerance to prevent autoimmunity through the secretion of various cytokines.72 In SLE, DCs secrete cytokines that promote DC maturation, B cell differentiation, Th cell polarization, and cytotoxic T cell activation, thereby amplifying subsequent immune responses. Dysregulation of DC function has been implicated in SLE pathogenesis, and alterations in the mTOR pathway may represent an upstream regulatory mechanism contributing to DC dysfunction in this disease.73
The mTOR pathway regulates multiple aspects of DC differentiation and function. Fms-related tyrosine kinase 3 ligand (Flt3L) is a growth factor essential for DC development and differentiation, and the mTOR pathway regulates Flt3L-mediated activation of resting dendritic cells through Toll-like receptors (TLR) via the PI3K/AKT/TSC/mTOR axis, driving the differentiation of resting DCs into mature DCs.53 Abnormally activated mTOR also affects DC differentiation through the IFN-α pathway. Specifically, enhanced mTOR signaling promotes plasmacytoid DCs (pDCs) to secrete large amounts of IFN-α, driving myeloid DCs (MDCs) to release IL‑12. These cytokines initiate a cascade of immune activation with dual consequences. First, IFN‑α and IL‑12 synergistically induce Th1 cell polarization, promote abnormal T cell proliferation, and stimulate B cell activation and proliferation, leading B cells to secrete antinuclear antibodies (ANA) and other autoantibodies.72,74 Second, mature DCs highly express the costimulatory molecules B7-1 (CD80) and B7-2 (CD86), which bind to costimulatory receptors on the surface of T cells, strongly activating autoreactive T cells and further amplifying the autoimmune response.73 On the contrary, accumulating evidence indicates that abnormal mTOR activation disrupts the regulatory balance between DCs and regulatory T cells (Treg). Dysregulated mTOR signaling inhibits the differentiation and function of FOXP3+ Treg cells, thereby weakening Treg-mediated immunosuppressive effect of DCs, and releasing constraints on DC immunogenicity. In turn, elevated cytokines within the SLE microenvironment including IFN‑α, IL‑6, and IL‑18, further exacerbate mTOR-mediated DC dysfunction. This creates a self-perpetuating cycle in which abnormal mTOR signaling leads to excessive DC maturation, enhanced antigen presentation, massive release of pro-inflammatory cytokines (IFN‑α, IL‑12), impaired Treg suppressive function, activation of autoreactive lymphocytes, and abundant autoantibody production.75,76 This cascade continuously triggers autoimmune inflammatory damage, ultimately promoting the onset and progression of SLE. The detailed molecular mechanism underlying these processes require further investigation (Figure 3).
Regulation of Natural Killer Cells by mTOR
Natural killer (NK) cells participate in both protective and pathogenic immunity, rapidly mediating killing of intracellular pathogens and malignant cells, although their self-directed cytotoxicity can also exacerbate tissue pathology. Additionally, NK cells secrete cytokines and chemokines such as IFN-γ, enabling coordination with other immune cells. A hallmark of SLE is activation of type I interferon system, triggered by nucleic acid-containing immune complexes, leading to tissue inflammation and damage. In SLE pathogenesis, NK cells interact with pDCs to strongly promote pDC-derived IFN-α production. Conversely, pDC-derived IFN-α promotes NK cell development, maturation, and IFN-γ production, establishing a reciprocal NK-pDC crosstalk that contributes to SLE pathogenesis.77
mTOR is involved throughout NK cell development, and its dysregulation directly links NK cell dysfunction to SLE pathogenesis.
NK cell development is triggered by IL-15, which activates the PI3K/AKT/TSC1/mTORC1 axis to regulate early cell development. In turn, NK cells induce the NK cell-specific transcription factor E4BP4 through this pathway, which also upregulates CD22, thereby amplifying IL-15 activity by positive feedback. Notably, TSC1 functions as a negative regulatory factor that modulates the duration of mTORC1, although it is not indispensable for the final maturation, survival, or function of NK cells.78 During immune responses, activated NK cells rely on mTORC1-dependent aerobic glycolysis to achieve their effector functions. In SLE patients, overactivation of mTORC1 in immune cells disrupts the delicate balance of NK cell development and metabolism This dysregulation can lead to two distinct but equally detrimental outcomes: either NK cells become overactivated, enhancing pathogenic NK-pDC crosstalk and promoting excessive IFN-α production, or they develop functional defects that impair immune homeostasis. Both situations can lead to breakdown of immune tolerance and drive SLE progression53 (Figure 3).
Regulation of T Cells by mTOR
T lymphocytes play a crucial role in the pathogenesis of SLE. Effector T cells (Teff cells), particularly Th1, Th2, and Th17 cells, represent important pro-inflammatory T cell populations that activate other immune cells by secretion of inflammatory cytokines such as IL-17, IL-22, and IL-6, leading to enhanced inflammatory responses. In contrast, Regulatory T cells (Treg cells) are responsible for maintaining immune tolerance and preventing excessive immune responses and the development of autoimmune diseases. Dysfunction of Treg cells impairs the immune system’s ability to control the activity of Teff cells, further promoting the progression of SLE.79 Therefore, the overactivation of Teff cells and the dysfunction of Treg cells are major causes of immune dysregulation in SLE patients.80 Among these processes, mTOR signaling plays an important regulatory role in the proliferation and differentiation of T cell lineages, with mTORC1 and mTORC2 contributing in different ways.81
mTORC1 Regulates T Cell Differentiation
mTORC1 promotes the conversion of oxidative phosphorylation to glycolysis through HIF-1α, facilitating the differentiation of T cells into effector T cells (especially Th1 and Th17 cells).82 Glycolysis, as a rapid energy-producing pathway, better meets the energy and metabolite demands required for the rapid proliferation of Th1 and Th17 cells compared to oxidative phosphorylation, and its ability to function without relying on oxygen is crucial for the quick response and adaptability of T cells under various conditions.83,84 In contrast, the increase in glycolysis induced by mTORC1 and HIF-1α activation may inhibit the differentiation and function of Treg cells for two reasons: First, unlike effector T cells (such as Th1 and Th17 cells), Treg cells generally need to remain in the periphery for long periods after an immune response to maintain immune tolerance.34,85 Therefore, they require a stable energy supply, and oxidative phosphorylation provides sufficient energy support for long-term survival. Excessive glycolysis may disrupt Treg cell function.86 Second, glycolysis not only suppresses the expression of Treg-specific transcription factor (Foxp3) but its byproducts may also affect Foxp3 stability. NAD+ is a substrate for deacetylases such as sirtuin 1 (SIRT1), which are crucial for the deacetylation and stability of Foxp3.87 Changes in the NAD+/NADH ratio during glycolysis lead to the accumulation of NADH, inhibiting the regeneration of NAD+, reducing SIRT1 activity, and subsequently affecting the deacetylation and expression of Foxp3, further leading to the loss of Treg cell function.
PTEN, as a tumor suppressor gene, can inhibit the activity of mTORC1 by dephosphorylating key molecules in the PI3K/Akt signaling pathway, thereby indirectly regulating the differentiation of Treg cells.88 Notably, (Indoleamine 2,3-dioxygenase) upregulates PTEN expression, which in turn inhibits the mTORC1 signaling pathway and promotes the differentiation of Treg cells.89 Additionally, the deficiency of amino acids such as leucine and serine also suppresses mTORC1 activity, limiting the differentiation of T cells into effector T cells, further demonstrating the crucial role of mTORC1 in the differentiation process of immune cells90 (Figure 3).
mTORC2 Regulates T-Cell Differentiation
mTORC2 regulates T cell differentiation through upstream activation of RhoA, a small GTPase, via pathways such as Rho-associated kinases (ROCKs) or Mammalian Diaphanous-related formin 1 (mDia1), which directly modulate RhoA.91 Additionally, mTORC2 can indirectly enhance RhoA activity by activating Akt, an important downstream effector of mTORC2, which regulates molecules associated with RhoA activation. This process promotes the differentiation and function of Th2 cells.92 Studies have shown that T cells with mTORC1 defects cannot differentiate into Th1 or Th17 cells, whereas T cells with mTORC2 defects cannot differentiate into Th2 cells.93,94 Furthermore, mTORC2 also exerts a negative regulatory effect on Treg cells.95
Regulation of B Cells by mTOR
B cells play an important role in SLE pathogenesis through antibody production, cytokine production, and antigen presentation.96 It has been reported that the Dysregulated balance of B cell subsets, including dysfunctional regulatory B cells (Bregs), expanded T-bet+ CD11c+ B cells, and increased plasma cells, is closely associated with SLE disease activity, autoantibody generation, and organ injury.97–99 And mTOR signaling pathway is essential for regulating the development, metabolism, and function of B cells.
Studies have found that the mTORC1 signaling pathway is significantly activated in early B cell development.100 Meanwhile, mice with knockout of mTOR or Raptor gene exhibit a development blockade in early B cells.100,101 Besides mTORC1 signaling pathway plays an important role in the humoral immune response of B cells by regulating antibody production, somatic hypermutation, and class switch recombination.102
It has been reported that the mTOR pathway in B cells from SLE patients is activated, and mTOR can induce plasmablast differentiation through metabolic and transcription factor regulation. Two of the pathways that have been extensively studied are the PI3K-Akt-mTOR pathway and the essential amino acids-mTOR pathway.103 Researchers have found enhanced PI3K-Akt signaling is critical for B-cell proliferation and survival.104 Hyperactivity of mTORC1 promotes the differentiation of plasma cells.105 Meanwhile, mTORC1 plays a critical role in B-cell antibody class switching.106 Conversely, mTORC2, independent of mTORC1, inhibits antibody class switching by regulating AKT.107 Splenic tyrosine kinase (Syk) is vital for the effective induction of CD40 and TLR signaling by BCR, and the increase of sky phosphorylation in B cells is positively correlated with disease activity.108,109 Methionine, which can regulate syk and mTORC1 signals, is important for plasmablast differentiation.110 In the presence of methionine, Syk and mTORC1 signals can synergistically induce EZH2 (an enzyme highly correlated with the activity of SLE) expression, which suppresses BACH2 expression through epigenomic modification, and promotes plasmablast differentiation in SLE.110
Recently, researchers found that glutaminolysis is abnormally enhanced in lymphocytes from SLE patients, which is related to mTOR/P70S6K/4EBP1 and NLRP3/caspase-1/IL-1β pathways.111 Besides Wu et al reported that the mTORC1 pathway in atypical memory B cells (ie, CD11c+ T-bet+ B cells) is abnormally activated112 (Figure 3).
mTOR-Targeted Therapy
mTOR Inhibitors
Rapamycin, an allosteric inhibitor of mTOR, has been shown to control antigen-induced autoimmunity, T cell hyperreactivity, and titers of antinuclear antibodies (ANA) and anti-dsDNA antibodies, thereby inhibiting SLE development.11
Sirolimus has been successfully used in clinical trials for the treatment of SLE, with both efficacy and safety profiles assessed.113 One study demonstrated that systemic administration of rapamycin in SLE patients effectively inhibited overactivated T cells, B cells, and hepatocytes.114 In an open-label Phase 1/2 trial, sirolimus treatment inhibited antigen-induced T-cell proliferation, reversed the deficiency of circulating regulatory T cells, and significantly improved skin, mucosal, and musculoskeletal manifestations in participating. Moreover, rapamycin reduces high levels of IL-4 and IL-17, further contributing to symptomatic improvement.54 Notably, most sirolimus-associated side effects were mild, with dyslipidemia being the most common adverse reaction, which can be managed with statins. Its use in combination can reduce the dose of other disease-mod antirheumatic drugs required during the treatment of SLE.115,116 However, high doses of everolimus, one of the most commonly used sirolimus analogues, are associated with proteinuria.116 Therefore, the therapeutic efficacy of sirolimus in LN remains unclear, and further experiments are required to determine the optimal dosage for its use in LN treatment.
Compared with the first-generation mTOR inhibitors, second-generation mTOR inhibitors, namely Dual PI3K/mTOR inhibitors can produce better biological responses, enhance therapeutic potential, and minimize resistance.117 A study demonstrated that INK128 effectively suppresses disease activity in lupus mice by targeting the mTOR signaling pathway to regulate the proportion of CD11b⁺Gr1⁺ cells (MDSCs), a cell type that plays a critical role in the early stages of SLE pathogenesis.118 In summary, mTOR inhibitor is effective in treating patients with clinically active SLE, especially those with musculoskeletal and mucocutaneous manifestations, through various means. However, further studies are required to determine its optimal regimen and dosage.119 Importantly, not all studies have shown consistent benefits. A cohort study reported that sirolimus treatment was associated with notable tolerability issues, leading to discontinuation in 5 out of 16 patients, primarily due to drug-related adverse effects. Additionally, although renal function remained stable in most patients, one individual with pre-existing stage 4 chronic kidney disease progressed to end-stage renal failure, and another experienced a renal flare despite treatment. Worsening dyslipidemia was also observed in a subset of patients, underscoring the need for careful metabolic monitoring.120 These observations suggest that patient stratification and combination strategies may be necessary to mitigate risks and optimize long-term outcomes.
N-Acetylcysteine
The therapeutic action of NAC is mainly to increase the level of glutathione (GSH, one of the most critical intracellular antioxidants) in the cell, which affects the mitochondrial membrane potential (Δψm) and oxidative stress processes, further preventing mTORC1 activation and regulating T cell differentiation.121 Moreover, GSH directly scavenges and neutralizes reactive oxygen species (ROS), and by reducing ROS, GSH can inhibit this signal and prevent mTOR overactivation. With the blockage of its mTORC1 activity, the prevention and treatment of SLE are achieved.122 In summary, through its antioxidant effects, NAC can modulate mitochondrial membrane potential and oxidative stress to indirectly influence the mTOR pathway.123 Thus, the inhibitory effect of NAC on mTOR is non-targeted. NAC is considered an oxidation-reduction sensitive driving factor in the pathogenesis of lupus.124
Partial preclinical studies have demonstrated that NAC can reduce autoantibody levels and ameliorate organ damage in SLE mouse models.125–127 Clinical studies have shown that N-acetylcysteine (NAC) reduces oxidative stress in lymphocytes and inhibits mTORC1 activity in patients with SLE.56,124,128 Another clinical trial indicated that NAC treatment significantly alleviated attention deficit hyperactivity disorder (ADHD) symptoms in patients with SLE.129 A recent randomized double-blind clinical trial found that NAC therapy significantly reduces disease activity and complication rates in SLE patients.130 Therefore, NAC holds significant promise as an adjunctive therapy for SLE.
Metformin (Met)
Metformin is a commonly used drug for treating type 2 diabetes, but it also activates AMPK and inhibits mTORC1 to exert immune regulatory effects.131 In autoimmune diseases such as systemic lupus erythematosus, CD4+ T cells often show reduced IL-2 secretion or functional abnormalities, leading to immune system dysregulation.132 By inhibiting mTORC1 activation, metformin reduces cellular metabolism and ROS production, decreases oxidative stress, and further improves CD4+ T cell function, thereby restoring IL-2 secretion.133 Additionally, metformin suppressed the germinal center response and modulated T cell differentiation by reducing follicular helper T cells (Tfh) and Th17 cells while increasing Treg cells.3
In experiments assessing the therapeutic potential of metformin in a SLE mouse model, MET inhibited B cell differentiation into plasma cells and germinal center formation through the AMPK/mTOR/STAT3 signaling pathway, thereby reducing the production of autoantibodies and inflammation.134 Furthermore, the study demonstrated that metformin treatment significantly reduced the risk of disease flares in patients with SLE and subsequently allowed for a reduction in glucocorticoid dosage.135 A recent study demonstrated that metformin-treated patients exhibited significantly lower risks of lupus nephritis (LN), chronic kidney disease (CKD), and major adverse cardiovascular events (MACE).136
Other mTOR Related Drugs
The PI3K–AKT–mTOR pathway is activated in B cells, CD4+ T cells, and macrophages from SLE patients.103,137 Blockade of PI3Kγ with AS605240 can reduce the excessive number of lymphocytes, autoantibody production and the number of macrophages infiltrating the kidney, which reduces disease symptoms and prolongs life span in mouse models of lupus.138,139 Mesenchymal stem cells (MSCs) exhibit enhanced senescence in SLE patients, while broad PI3K inhibitor LY294002 can improve this situation.140 Therefore PI3K is a promising target for SLE.
Fingolimod, a receptor modulator targeting the S1P receptor which plays a role upstream of mTORC1, has been used to treat multiple sclerosis.141 However, fingolimod can block the expansion of DN T cells and prevent nephritis, which effectively inhibits the development of autoimmunity and prolongs the survival of MRL/lpr mice.142,143 Based on these research findings, fingolimod may be a promising drug adjuvant therapy or treatment of SLE.
In addition, calcium/calmodulin-dependent protein kinase IV (CaMK4) targeted therapy for SLE is also promising. Researchers found that overexpression of CaMK4 in T cells from lupus-prone mice affected the balance between Th17 cells and Tregs mainly by AKT/ mTOR pathway and increasing the binding of cAMP response element modulator α (CREM-α) to the IL17 genes.144 Inhibition of CaMK4 with KN-93 inhibits Th17 differentiation and averts IL–17–instigated inflammation.145 As for B cells, KN-93 can also inhibit the development of a lupus-related disease by decreasing the expression of CD86 and CD80 on B cells.146
Conclusion
The mTOR signaling pathway exerts multifaceted effects on SLE pathogenesis through its modulation of diverse immune cell populations. In innate immunity, mTOR influences disease onset and progression by affecting macrophage polarization and autophagy, as well as dendritic cell maturation and function. In adaptive immunity, mTOR induces the differentiation of T cells into pro-inflammatory subsets via metabolic reprogramming while concurrently promoting B cell differentiation and affecting antibody production. The pathway also modulates NK cell development and the critical regulatory balance between effector and regulatory T cells. The use of immune suppressants, while they can prevent or treat SLE, inevitably damages the body’s normal immune and immune surveillance, thus increasing the probability of developing tumors and infections. Current research shows that it is feasible to treat SLE by targeting the mTOR pathway, especially those with musculoskeletal and mucocutaneous manifestations. However, given the heterogeneity of SLE and the existence of mTOR-independent immune dysregulation, mTOR-targeted therapy may not be universally effective. Patients with certain disease manifestations (eg., LN) or genetic backgrounds may require combination approaches that simultaneously target complementary pathways such as JAK-STAT or NF-κB. Future studies should focus on biomarker-driven patient selection to maximize therapeutic benefit. Treatment with mTOR inhibitors can effectively reduce disease activity and reduce the dosage of glucocorticoid. However, non-selective inhibition of mTOR may affect the normal physiological functions of the organism, such as dyslipidemia. Moreover, the therapeutic efficacy of mTOR-targeted therapy for LN remains uncertain and require further investigation. Therefore, further exploration is necessary to determine the applicability, optimal regimen, dosage, and potential adverse effects of mTOR-targeted therapy in SLE patients with different disease backgrounds.
Data Sharing Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Author Contributions
X.J. Conceptualization, Formal analysis, Visualization, Writing – original draft, Writing – review and editing; L.S. Formal analysis, Conceptualization, Writing – original draft, Writing – review and editing; J.J. Formal analysis, Writing – original draft, Writing – review and editing; C.Z. Formal analysis, Visualization, Writing – original draft; Y.W. Conceptualization, Resources, Supervision, Writing – review and editing; H.Z. Conceptualization, Resources, Supervision, Writing – review and editing; Q.W. Conceptualization, Writing – original draft, Writing – review and editing; Q.S. Conceptualization, Writing – review and editing; F.L. Conceptualization, Funding acquisition, Writing – review and editing; S.Z. Conceptualization, Resources, Writing – review and editing; T.C. Conceptualization, Funding acquisition, Writing – review and editing.All authors gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This project was supported by grants from the Qingmiao Medical Research Project of Shanxi Provincial Health Commission (2025QM011) and the National Natural Science Foundation of China(No. 82001740).
Disclosure
The authors declare no conflicts of interest.
References
1. Morita M, Mizui M, Masuyama S, Tsokos GC, Isaka Y. Reduction of Cell Surface T-Cell Receptor by Non-Mitogenic CD3 Antibody to Mitigate Murine Lupus. Front Immunol. 2022;13:855812. doi:10.3389/fimmu.2022.855812
2. Arnaud L, Mathian A, Boddaert J, Amoura Z. Late-onset systemic lupus erythematosus: epidemiology, diagnosis and treatment. Drugs Aging. 2012;29(3):181–17. doi:10.2165/11598550-000000000-00000
3. Zhao X, Wang S, Wang S, Xie J, Cui D. mTOR signaling: a pivotal player in Treg cell dysfunction in systemic lupus erythematosus. Clin Immunol. 2022;245:109153. doi:10.1016/j.clim.2022.109153
4. Garcia-Romo GS, Caielli S, Vega B, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med. 2011;3(73):73ra20. doi:10.1126/scitranslmed.3001201
5. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–293. doi:10.1016/j.cell.2012.03.017
6. Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017;168(6):960–976. doi:10.1016/j.cell.2017.02.004
7. Vezina C, Kudelski A, Sehgal SN. Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J Antibiot. 1975;28(10):721–726. doi:10.7164/antibiotics.28.721
8. Aghdasi B, Ye K, Resnick A, et al. FKBP12, the 12-kDa FK506-binding protein, is a physiologic regulator of the cell cycle. Proc Natl Acad Sci U S A. 2001;98(5):2425–2430. doi:10.1073/pnas.041614198
9. Sabers CJ, Martin MM, Brunn GJ, et al. Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J Biol Chem. 1995;270(2):815–822. doi:10.1074/jbc.270.2.815
10. Panwar V, Singh A, Bhatt M, et al. Multifaceted role of mTOR (mammalian target of rapamycin) signaling pathway in human health and disease. Signal Transduct Target Ther. 2023;8(1):375. doi:10.1038/s41392-023-01608-z
11. Perl A. Activation of mTOR (mechanistic target of rapamycin) in rheumatic diseases. Nat Rev Rheumatol. 2016;12(3):169–182. doi:10.1038/nrrheum.2015.172
12. Hara K, Maruki Y, Long X, et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell. 2002;110(2):177–189. doi:10.1016/s0092-8674(02)00833-4
13. Yang H, Rudge DG, Koos JD, Vaidialingam B, Yang HJ, Pavletich NP. mTOR kinase structure, mechanism and regulation. Nature. 2013;497(7448):217–223. doi:10.1038/nature12122
14. Nojima H, Tokunaga C, Eguchi S, et al. The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J Biol Chem. 2003;278(18):15461–15464. doi:10.1074/jbc.C200665200
15. Peterson TR, Laplante M, Thoreen CC, et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009;137(5):873–886. doi:10.1016/j.cell.2009.03.046
16. Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol. 2007;9(3):316–323. doi:10.1038/ncb1547
17. Liu GY, Sabatini DM. mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol. 2020;21(4):183–203. doi:10.1038/s41580-019-0199-y
18. Laplante M, Sabatini DM. Regulation of mTORC1 and its impact on gene expression at a glance. J Cell Sci. 2013;126(Pt 8):1713–1719. doi:10.1242/jcs.125773
19. Covarrubias AJ, Aksoylar HI, Horng T. Control of macrophage metabolism and activation by mTOR and Akt signaling. Semin Immunol. 2015;27(4):286–296. doi:10.1016/j.smim.2015.08.001
20. Hwang Y, Kim LC, Song W, Edwards DN, Cook RS, Chen J. Disruption of the Scaffolding Function of mLST8 Selectively Inhibits mTORC2 Assembly and Function and Suppresses mTORC2-Dependent Tumor Growth In Vivo. Cancer Res. 2019;79(13):3178–3184. doi:10.1158/0008-5472.CAN-18-3658
21. Inoki K, Li Y, Xu T, Guan K-L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17(15):1829–1834. doi:10.1101/gad.1110003
22. Shaw RJ. LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol. 2009;196(1):65–80. doi:10.1111/j.1748-1716.2009.01972.x
23. Sancak Y, Peterson TR, Shaul YD, et al. The Rag GTPases Bind Raptor and Mediate Amino Acid Signaling to mTORC1. Science. 2008;320(5882):1496–1501. doi:10.1126/science.1157535
24. Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag Complex Targets mTORC1 to the Lysosomal Surface and Is Necessary for Its Activation by Amino Acids. Cell. 2010;141(2):290–303. doi:10.1016/j.cell.2010.02.024
25. Peña-Llopis S, Vega-rubin-de-celis S, Schwartz JC, et al. Regulation of TFEB and V-ATPases by mTORC1. EMBO J. 2011;30(16):3242–3258. doi:10.1038/emboj.2011.257
26. Szwed A, Kim E, Jacinto E. Regulation and metabolic functions of mTORC1 and mTORC2. Physiol Rev. 2021;101(3):1371–1426. doi:10.1152/physrev.00026.2020
27. Ragupathi A, Kim C, Jacinto E. The mTORC2 signaling network: targets and cross-talks. Biochem J. 2024;481(2):45–91. doi:10.1042/BCJ20220325
28. Katsara O, Attur M, Ruoff R, Abramson SB, Kolupaeva V. Increased Activity of the Chondrocyte Translational Apparatus Accompanies Osteoarthritic Changes in Human and Rodent Knee Cartilage. Arthritis Rheumatol. 2017;69(3):586–597. doi:10.1002/art.39947
29. Gingras AC, Raught B, Sonenberg N. mTOR signaling to translation. Curr Top Microbiol Immunol. 2004;279:169–197. doi:10.1007/978-3-642-18930-2_11
30. Willinger T, Staron M, Ferguson SM, De Camilli P, Flavell RA. Dynamin 2-dependent endocytosis sustains T-cell receptor signaling and drives metabolic reprogramming in T lymphocytes. Proc Natl Acad Sci U S A. 2015;112(14):4423–4428. doi:10.1073/pnas.1504279112
31. Nakhjavani M, Smith E, Palethorpe HM, et al. Anti-Cancer Effects of an Optimised Combination of Ginsenoside Rg3 Epimers on Triple Negative Breast Cancer Models. Pharmaceuticals. 2021;14(7):633. doi:10.3390/ph14070633
32. Bohlen J, Roiuk M, Teleman AA. Phosphorylation of ribosomal protein S6 differentially affects mRNA translation based on ORF length. Nucleic Acids Res. 2021;49(22):13062–13074. doi:10.1093/nar/gkab1157
33. Nakagawa T, Araki T, Nakagawa M, Hirao A, Unno M, Nakayama K. S6 Kinase- and beta-TrCP2-Dependent Degradation of p19Arf Is Required for Cell Proliferation. Mol Cell Biol. 2015;35(20):3517–3527. doi:10.1128/MCB.00343-15
34. Zhang F, Cheng T, Zhang SX. Mechanistic target of rapamycin (mTOR): a potential new therapeutic target for rheumatoid arthritis. Arthritis Res Ther. 2023;25(1):187. doi:10.1186/s13075-023-03181-w
35. Düvel K, Yecies JL, Menon S, et al. Activation of a Metabolic Gene Regulatory Network Downstream of mTOR Complex 1. Molecular Cell. 2010;39(2):171–183. doi:10.1016/j.molcel.2010.06.022
36. Cunningham JT, Rodgers JT, Arlow DH, Vazquez F, Mootha VK, Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature. 2007;450(7170):736–740. doi:10.1038/nature06322
37. Ben-Sahra I, Howell JJ, Asara JM, Manning BD. Stimulation of de Novo Pyrimidine Synthesis by Growth Signaling Through mTOR and S6K1. Science. 2013;339(6125):1323–1328. doi:10.1126/science.1228792
38. Tait-Mulder J, Hodge K, Sumpton D, Zanivan S, Vazquez A. The conversion of formate into purines stimulates mTORC1 leading to CAD-dependent activation of pyrimidine synthesis. Cancer Metab. 2020;8:20. doi:10.1186/s40170-020-00228-3
39. Huang F, Huffman KE, Wang Z, et al. Guanosine triphosphate links MYC-dependent metabolic and ribosome programs in small-cell lung cancer. J Clin Investig. 2021;131(1). doi:10.1172/jci139929
40. Guertin DA, Stevens DM, Thoreen CC, et al. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell. 2006;11(6):859–871. doi:10.1016/j.devcel.2006.10.007
41. Jacinto E, Facchinetti V, Liu D, et al. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127(1):125–137. doi:10.1016/j.cell.2006.08.033
42. Gan X, Wang J, Wang C, et al. PRR5L degradation promotes mTORC2-mediated PKC-delta phosphorylation and cell migration downstream of Galpha12. Nat Cell Biol. 2012;14(7):686–696. doi:10.1038/ncb2507
43. Jacinto E, Loewith R, Schmidt A, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6(11):1122–1128. doi:10.1038/ncb1183
44. Li X, Gao T. mTORC2 phosphorylates protein kinase Czeta to regulate its stability and activity. EMBO Rep. 2014;15(2):191–198. doi:10.1002/embr.201338119
45. Thomanetz V, Angliker N, Cloetta D, et al. Ablation of the mTORC2 component rictor in brain or Purkinje cells affects size and neuron morphology. J Cell Biol. 2013;201(2):293–308. doi:10.1083/jcb.201205030
46. Garcia-Martinez JM, Alessi DR. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem J. 2008;416(3):375–385. doi:10.1042/BJ20081668
47. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–141. doi:10.1038/ncb2152
48. Yarotskyy V, Gao G, Du L, Ganapathi SB, Peterson BZ, Elmslie KS. Roscovitine binds to novel L-channel (CaV1.2) sites that separately affect activation and inactivation. J Biol Chem. 2010;285(1):43–53. doi:10.1074/jbc.M109.076448
49. Rousseau A, Bertolotti A. An evolutionarily conserved pathway controls proteasome homeostasis. Nature. 2016;536:7615):184–9. doi:10.1038/nature18943
50. Zhao J, Zhai B, Gygi SP, Goldberg AL. mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proc Natl Acad Sci U S A. 2015;112(52):15790–15797. doi:10.1073/pnas.1521919112
51. Zhang Y, Nicholatos J, Dreier JR, et al. Coordinated regulation of protein synthesis and degradation by mTORC1. Nature. 2014;513:7518):440–3. doi:10.1038/nature13492
52. Bentham J, Morris DL, Graham DSC, et al. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat Genet. 2015;47(12):1457–1464. doi:10.1038/ng.3434
53. Furment MM, Perl A. Immmunometabolism of systemic lupus erythematosus. Clin Immunol. 2024;261:109939. doi:10.1016/j.clim.2024.109939
54. He J, Ma J, Ren B, Liu A. Advances in systemic lupus erythematosus pathogenesis via mTOR signaling pathway. Semin Arthritis Rheum. 2020;50(2):314–320. doi:10.1016/j.semarthrit.2019.09.022
55. Kato H, Perl A. Mechanistic target of rapamycin complex 1 expands Th17 and IL-4+ CD4-CD8- double-negative T cells and contracts regulatory T cells in systemic lupus erythematosus. J Immunol. 2014;192(9):4134–4144. doi:10.4049/jimmunol.1301859
56. Lai ZW, Hanczko R, Bonilla E, et al. N-acetylcysteine reduces disease activity by blocking mammalian target of rapamycin in T cells from systemic lupus erythematosus patients: a randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2012;64(9):2937–2946. doi:10.1002/art.34502
57. Li Y, Lee PY, Reeves WH. Monocyte and macrophage abnormalities in systemic lupus erythematosus. Arch Immunol Ther Exp. 2010;58(5):355–364. doi:10.1007/s00005-010-0093-y
58. Hill GS, Delahousse M, Nochy D, et al. Predictive power of the second renal biopsy in lupus nephritis: significance of macrophages. Kidney Int. 2001;59(1):304–316. doi:10.1046/j.1523-1755.2001.00492.x
59. Murray PJ. Macrophage Polarization. Annu Rev Physiol. 2017;79:541–566. doi:10.1146/annurev-physiol-022516-034339
60. Ahamada MM, Jia Y, Wu X. Macrophage Polarization and Plasticity in Systemic Lupus Erythematosus. Front Immunol. 2021;12:734008. doi:10.3389/fimmu.2021.734008
61. Mohammadi S, Saghaeian-Jazi M, Sedighi S, Memarian A. Immunomodulation in systemic lupus erythematosus: induction of M2 population in monocyte-derived macrophages by pioglitazone. Lupus. 2017;26(12):1318–1327. doi:10.1177/0961203317701842
62. Horuluoglu B, Bayik D, Kayraklioglu N, Goguet E, Kaplan MJ, Klinman DM. PAM3 supports the generation of M2-like macrophages from lupus patient monocytes and improves disease outcome in murine lupus. J Autoimmun. 2019;99:24–32. doi:10.1016/j.jaut.2019.01.004
63. Li F, Yang Y, Zhu X, Huang L, Xu J. Macrophage Polarization Modulates Development of Systemic Lupus Erythematosus. Cell Physiol Biochem. 2015;37(4):1279–1288. doi:10.1159/000430251
64. Yue S, Rao J, Zhu J, et al. Myeloid PTEN Deficiency Protects Livers from Ischemia Reperfusion Injury by Facilitating M2 Macrophage Differentiation. J Immunol. 2014;192(11):5343–5353. doi:10.4049/jimmunol.1400280
65. Mercalli A, Calavita I, Dugnani E, et al. Rapamycin unbalances the polarization of human macrophages to M1. Immunology. 2013;140(2):179–190. doi:10.1111/imm.12126
66. Byles V, Covarrubias AJ, Ben-Sahra I, et al. The TSC-mTOR pathway regulates macrophage polarization. Nat Commun. 2013;4:2834. doi:10.1038/ncomms3834
67. Fang C, Yu J, Luo Y, et al. Tsc1 is a Critical Regulator of Macrophage Survival and Function. Cell Physiol Biochem. 2015;36(4):1406–1418. doi:10.1159/000430306
68. Arranz A, Doxaki C, Vergadi E, et al. Akt1 and Akt2 protein kinases differentially contribute to macrophage polarization. Proc Natl Acad Sci U S A. 2012;109(24):9517–9522. doi:10.1073/pnas.1119038109
69. Denny MF, Chandaroy P, Killen PD, et al. Accelerated macrophage apoptosis induces autoantibody formation and organ damage in systemic lupus erythematosus. J Immunol. 2006;176(4):2095–2104. doi:10.4049/jimmunol.176.4.2095
70. Li H, Wu Q, Li J, et al. Cutting Edge: defective follicular exclusion of apoptotic antigens due to marginal zone macrophage defects in autoimmune BXD2 mice. J Immunol. 2013;190(9):4465–4469. doi:10.4049/jimmunol.1300041
71. Deretic V. Autophagy as an innate immunity paradigm: expanding the scope and repertoire of pattern recognition receptors. Curr Opin Immunol. 2012;24(1):21–31. doi:10.1016/j.coi.2011.10.006
72. Mackern-Oberti JP, Llanos C, Riedel CA, Bueno SM, Kalergis AM. Contribution of dendritic cells to the autoimmune pathology of systemic lupus erythematosus. Immunology. 2015;146(4):497–507. doi:10.1111/imm.12504
73. Hui Q, Xuelu X, Dongzhou L, et al. Relationship between interleukin-12 and interferon-α expression in dendritic cells of patients with systemic lupus erythematosus and their subtypes. Chinese J. 2004;2004(12):712–715.
74. Rama I, Bruene B, Torras J, et al. Hypoxia stimulus: an adaptive immune response during dendritic cell maturation. Kidney Int. 2008;73(7):816–825. doi:10.1038/sj.ki.5002792
75. Liu Y, Gu Y, Cao X. The exosomes in tumor immunity. Oncoimmunology. 2015;4(9):e1027472. doi:10.1080/2162402X.2015.1027472
76. Xie Y, Zhang X, Zhao T, Li W, Xiang J. Natural CD8(+)25(+) regulatory T cell-secreted exosomes capable of suppressing cytotoxic T lymphocyte-mediated immunity against B16 melanoma. Biochem Biophys Res Commun. 2013;438(1):152–155. doi:10.1016/j.bbrc.2013.07.044
77. Yang Y, Day J, Souza-Fonseca Guimaraes F, Wicks IP, Louis C. Natural killer cells in inflammatory autoimmune diseases. Clin Transl Immunology. 2021;10(2):e1250. doi:10.1002/cti2.1250
78. Yang M, Chen S, Du J, et al. NK cell development requires Tsc1-dependent negative regulation of IL-15-triggered mTORC1 activation. Nat Commun. 2016;7:12730. doi:10.1038/ncomms12730
79. Kumar BV, Connors TJ, Farber DL. Human T Cell Development, Localization, and Function throughout Life. Immunity. 2018;48(2):202–213. doi:10.1016/j.immuni.2018.01.007
80. Kosmaczewska A, Swierkot J, Ciszak L, Wiland P. The role of Th1, Th17, and Treg cells in the pathogenesis of rheumatoid arthritis including anti-inflammatory action of Th1 cytokines. Postepy Hig Med Dosw. 2011;65:397–403. doi:10.5604/17322693.948971
81. Zeng H, Chi H. mTOR and lymphocyte metabolism. Curr Opin Immunol. 2013;25(3):347–355. doi:10.1016/j.coi.2013.05.002
82. Guma M, Tiziani S, Firestein GS. Metabolomics in rheumatic diseases: desperately seeking biomarkers. Nat Rev Rheumatol. 2016;12(5):269–281. doi:10.1038/nrrheum.2016.1
83. Peng HY, Lucavs J, Ballard D, et al. Metabolic Reprogramming and Reactive Oxygen Species in T Cell Immunity. Front Immunol. 2021;12:652687. doi:10.3389/fimmu.2021.652687
84. Qiu R, Yu X, Wang L, et al. Inhibition of Glycolysis in Pathogenic T(H)17 Cells through Targeting a miR −21-Peli1-c-Rel Pathway Prevents Autoimmunity. J Immunol. 2020;204(12):3160–3170. doi:10.4049/jimmunol.2000060
85. Cheru N, Hafler DA, Sumida TS. Regulatory T cells in peripheral tissue tolerance and diseases. Front Immunol. 2023;14:1154575. doi:10.3389/fimmu.2023.1154575
86. Yu X, Teng XL, Wang F, et al. Metabolic control of regulatory T cell stability and function by TRAF3IP3 at the lysosome. J Exp Med. 2018;215(9):2463–2476. doi:10.1084/jem.20180397
87. Kwon HS, Lim HW, Wu J, Schnolzer M, Verdin E, Ott M. Three novel acetylation sites in the Foxp3 transcription factor regulate the suppressive activity of regulatory T cells. J Immunol. 2012;188(6):2712–2721. doi:10.4049/jimmunol.1100903
88. Yang C, Chen C, Xiao Q, et al. Relationship Between PTEN and Angiogenesis of Esophageal Squamous Cell Carcinoma and the Underlying Mechanism. Front Oncol. 2021;11:739297. doi:10.3389/fonc.2021.739297
89. Gerriets VA, Kishton RJ, Johnson MO, et al. Foxp3 and Toll-like receptor signaling balance T(reg) cell anabolic metabolism for suppression. Nat Immunol. 2016;17(12):1459–1466. doi:10.1038/ni.3577
90. Sundrud MS, Koralov SB, Feuerer M, et al. Halofuginone inhibits TH17 cell differentiation by activating the amino acid starvation response. Science. 2009;324(5932):1334–1338. doi:10.1126/science.1172638
91. Thompson WR, Guilluy C, Xie Z, et al. Mechanically activated Fyn utilizes mTORC2 to regulate RhoA and adipogenesis in mesenchymal stem cells. Stem Cells. 2013;31(11):2528–2537. doi:10.1002/stem.1476
92. Gedaly R. mTOR Signaling in Regulatory T Cell Differentiation and Expansion. SOJ Immunology. 2015;3(1):122. doi:10.15226/soji/3/1/00122
93. Delgoffe GM, Pollizzi KN, Waickman AT, et al. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol. 2011;12(4):295–303. doi:10.1038/ni.2005
94. Lee K, Gudapati P, Dragovic S, et al. Mammalian target of rapamycin protein complex 2 regulates differentiation of Th1 and Th2 cell subsets via distinct signaling pathways. Immunity. 2010;32(6):743–753. doi:10.1016/j.immuni.2010.06.002
95. Delgoffe GM, Kole TP, Zheng Y, et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. 2009;30(6):832–844. doi:10.1016/j.immuni.2009.04.014
96. Franks SE, Getahun A, Hogarth PM, Cambier JC. Targeting B cells in treatment of autoimmunity. Curr Opin Immunol. 2016;43:39–45. doi:10.1016/j.coi.2016.09.003
97. Blair PA, Norena LY, Flores-Borja F, et al. CD19(+)CD24(hi)CD38(hi) B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic Lupus Erythematosus patients. Immunity. 2010;32(1):129–140. doi:10.1016/j.immuni.2009.11.009
98. Menon M, Blair PA, Isenberg DA, Mauri C. A Regulatory Feedback between Plasmacytoid Dendritic Cells and Regulatory B Cells Is Aberrant in Systemic Lupus Erythematosus. Immunity. 2016;44(3):683–697. doi:10.1016/j.immuni.2016.02.012
99. Pernis AB, Ivashkiv LB. ‘-Omics’ shed light on B cells in lupus. Nat Immunol. 2019;20(8):946–948. doi:10.1038/s41590-019-0446-6
100. Iwata TN, Ramirez JA, Tsang M, et al. Conditional Disruption of Raptor Reveals an Essential Role for mTORC1 in B Cell Development, Survival, and Metabolism. J Immunol. 2016;197(6):2250–2260. doi:10.4049/jimmunol.1600492
101. Yang K, Neale G, Green DR, He W, Chi H. The tumor suppressor Tsc1 enforces quiescence of naive T cells to promote immune homeostasis and function. Nat Immunol. 2011;12(9):888–897. doi:10.1038/ni.2068
102. Zhang S, Pruitt M, Tran D, et al. B cell-specific deficiencies in mTOR limit humoral immune responses. J Immunol. 2013;191(4):1692–1703. doi:10.4049/jimmunol.1201767
103. Iwata S, Hajime Sumikawa M, Tanaka Y. B cell activation via immunometabolism in systemic lupus erythematosus. Front Immunol. 2023;14:1155421. doi:10.3389/fimmu.2023.1155421
104. Caro-Maldonado A, Wang R, Nichols AG, et al. Metabolic reprogramming is required for antibody production that is suppressed in anergic but exaggerated in chronically BAFF-exposed B cells. J Immunol. 2014;192(8):3626–3636. doi:10.4049/jimmunol.1302062
105. Wu T, Qin X, Kurepa Z, et al. Shared signaling networks active in B cells isolated from genetically distinct mouse models of lupus. J Clin Invest. 2007;117(8):2186–2196. doi:10.1172/JCI30398
106. Raybuck AL, Cho SH, Li J, et al. B Cell-Intrinsic mTORC1 Promotes Germinal Center-Defining Transcription Factor Gene Expression, Somatic Hypermutation, and Memory B Cell Generation in Humoral Immunity. J Immunol. 2018;200(8):2627–2639. doi:10.4049/jimmunol.1701321
107. Limon JJ, So L, Jellbauer S, et al. mTOR kinase inhibitors promote antibody class switching via mTORC2 inhibition. Proc Natl Acad Sci U S A. 2014;111(47):E5076–85. doi:10.1073/pnas.1407104111
108. Iwata S, Yamaoka K, Niiro H, et al. Amplification of Toll-like receptor-mediated signaling through spleen tyrosine kinase in human B-cell activation. J Allergy Clin Immunol. 2012;129(6):1594–601e2. doi:10.1016/j.jaci.2012.03.014
109. Iwata S, Yamaoka K, Niiro H, et al. Increased Syk phosphorylation leads to overexpression of TRAF6 in peripheral B cells of patients with systemic lupus erythematosus. Lupus. 2015;24(7):695–704. doi:10.1177/0961203314560424
110. Zhang M, Iwata S, Hajime M, et al. Methionine Commits Cells to Differentiate Into Plasmablasts Through Epigenetic Regulation of BTB and CNC Homolog 2 by the Methyltransferase EZH2. Arthritis Rheumatol. 2020;72(7):1143–1153. doi:10.1002/art.41208
111. Zhang X, Wang G, Bi Y, Jiang Z, Wang X. Inhibition of glutaminolysis ameliorates lupus by regulating T and B cell subsets and downregulating the mTOR/P70S6K/4EBP1 and NLRP3/caspase-1/IL-1beta pathways in MRL/lpr mice. Int Immunopharmacol. 2022;112:109133. doi:10.1016/j.intimp.2022.109133
112. Wu C, Fu Q, Guo Q, et al. Lupus-associated atypical memory B cells are mTORC1-hyperactivated and functionally dysregulated. Ann Rheum Dis. 2019;78(8):1090–1100. doi:10.1136/annrheumdis-2019-215039
113. Eriksson P, Wallin P, Sjowall C. Clinical Experience of Sirolimus Regarding Efficacy and Safety in Systemic Lupus Erythematosus. Front Pharmacol. 2019;10:82. doi:10.3389/fphar.2019.00082
114. Oaks Z, Winans T, Huang N, Banki K, Perl A. Activation of the Mechanistic Target of Rapamycin in SLE: explosion of Evidence in the Last Five Years. Curr Rheumatol Rep. 2016;18(12):73. doi:10.1007/s11926-016-0622-8
115. Wen HY, Wang J, Zhang SX, et al. Low-Dose Sirolimus Immunoregulation Therapy in Patients with Active Rheumatoid Arthritis: a 24-Week Follow-Up of the Randomized, Open-Label, Parallel-Controlled Trial. J Immunol Res. 2019;2019:7684352. doi:10.1155/2019/7684352
116. Peng L, Wu C, Hong R, et al. Clinical efficacy and safety of sirolimus in systemic lupus erythematosus: a real-world study and meta-analysis. Ther Adv Musculoskeletal Dis. 2020;12. doi:10.1177/1759720x20953336
117. Mao B, Zhang Q, Ma L, Zhao DS, Zhao P, Yan P. Overview of Research into mTOR Inhibitors. Molecules. 2022;27(16):5295. doi:10.3390/molecules27165295
118. Shi G, Li D, Li X, et al. mTOR inhibitor INK128 attenuates systemic lupus erythematosus by regulating inflammation-induced CD11b+Gr1+ cells. Biochim Biophys Acta - Molecular Basis Disease. 2019;1865(1):1–13. doi:10.1016/j.bbadis.2018.10.007
119. Ji L, Xie W, Zhang Z. Efficacy and safety of sirolimus in patients with systemic lupus erythematosus: a systematic review and meta-analysis. Semin Arthritis Rheum. 2020;50(5):1073–1080. doi:10.1016/j.semarthrit.2020.07.006
120. Yap DYH, Tang C, Chan GCW, et al. Longterm Data on Sirolimus Treatment in Patients with Lupus Nephritis. J Rheumatol. 2018;45(12):1663–1670. doi:10.3899/jrheum.180507
121. Fernandez DR, Telarico T, Bonilla E, et al. Activation of mammalian target of rapamycin controls the loss of TCRzeta in lupus T cells through HRES-1/Rab4-regulated lysosomal degradation. J Immunol. 2009;182(4):2063–2073. doi:10.4049/jimmunol.0803600
122. Gergely Jr P, Grossman C, Niland B, et al. Mitochondrial hyperpolarization and ATP depletion in patients with systemic lupus erythematosus. Arthritis Rheum. 2002;46(1):175–190. doi:10.1002/1529-0131(200201)46:1<175::AID-ART10015>3.0.CO;2-H
123. Mokhtari V, Afsharian P, Shahhoseini M, Kalantar SM, Moini A. A Review on Various Uses of N-Acetyl Cysteine. Cell J. 2017;19(1):11–17. doi:10.22074/cellj.2016.4872
124. Perl A, Hanczko R, Lai ZW, et al. Comprehensive metabolome analyses reveal N-acetylcysteine-responsive accumulation of kynurenine in systemic lupus erythematosus: implications for activation of the mechanistic target of rapamycin. Metabolomics. 2015;11(5):1157–1174. doi:10.1007/s11306-015-0772-0
125. Wang H, Wang G, Liang Y, et al. Redox regulation of hepatic NLRP3 inflammasome activation and immune dysregulation in trichloroethene-mediated autoimmunity. Free Radic Biol Med. 2019;143:223–231. doi:10.1016/j.freeradbiomed.2019.08.014
126. Suwannaroj S, Lagoo A, Keisler D, McMurray RW. Antioxidants suppress mortality in the female NZB x NZW F1 mouse model of systemic lupus erythematosus (SLE). Lupus. 2001;10(4):258–265. doi:10.1191/096120301680416940
127. Shi D, Li X, Chen H, et al. High level of reactive oxygen species impaired mesenchymal stem cell migration via overpolymerization of F-actin cytoskeleton in systemic lupus erythematosus. Pathol Biol. 2014;62(6):382–390. doi:10.1016/j.patbio.2014.07.009
128. Doherty E, Oaks Z, Perl A. Increased mitochondrial electron transport chain activity at complex I is regulated by N-acetylcysteine in lymphocytes of patients with systemic lupus erythematosus. Antioxid Redox Signal. 2014;21(1):56–65. doi:10.1089/ars.2013.5702
129. Garcia RJ, Francis L, Dawood M, Lai ZW, Faraone SV, Perl A. Attention deficit and hyperactivity disorder scores are elevated and respond to N-acetylcysteine treatment in patients with systemic lupus erythematosus. Arthritis Rheum. 2013;65(5):1313–1318. doi:10.1002/art.37893
130. Abbasifard M, Khorramdelazad H, Rostamian A, et al. Effects of N-acetylcysteine on systemic lupus erythematosus disease activity and its associated complications: a randomized double-blind clinical trial study. Trials. 2023;24(1):129. doi:10.1186/s13063-023-07083-9
131. Inoki K, Kim J, Guan KL. AMPK and mTOR in cellular energy homeostasis and drug targets. Annu Rev Pharmacol Toxicol. 2012;52:381–400. doi:10.1146/annurev-pharmtox-010611-134537
132. Lieberman LA, Tsokos GC. The IL-2 defect in systemic lupus erythematosus disease has an expansive effect on host immunity. J Biomed Biotechnol. 2010;2010:740619. doi:10.1155/2010/740619
133. Iwata S, Tanaka Y. Therapeutic perspectives on the metabolism of lymphocytes in patients with rheumatoid arthritis and systemic lupus erythematosus. Expert Rev Clin Immunol. 2021;17(10):1121–1130. doi:10.1080/1744666X.2021.1964957
134. Lee SY, Moon SJ, Kim EK, et al. Metformin Suppresses Systemic Autoimmunity in Roquin(san/san) Mice through Inhibiting B Cell Differentiation into Plasma Cells via Regulation of AMPK/mTOR/STAT3. J Immunol. 2017;198(7):2661–2670. doi:10.4049/jimmunol.1403088
135. Wang H, Li T, Chen S, Gu Y, Ye S. Neutrophil Extracellular Trap Mitochondrial DNA and Its Autoantibody in Systemic Lupus Erythematosus and a Proof-of-Concept Trial of Metformin. Arthritis Rheumatol. 2015;67(12):3190–3200. doi:10.1002/art.39296
136. Gonzalez Moret YA, Lo KB, Tan IJ. Metformin in Systemic Lupus Erythematosus: investigating Cardiovascular Impact and Nephroprotective Effects in Lupus Nephritis. ACR Open Rheumatol. 2024;6(8):497–503. doi:10.1002/acr2.11698
137. Niculescu F, Nguyen P, Niculescu T, Rus H, Rus V, Via CS. Pathogenic T cells in murine lupus exhibit spontaneous signaling activity through phosphatidylinositol 3-kinase and mitogen-activated protein kinase pathways. Arthritis Rheum. 2003;48(4):1071–1079. doi:10.1002/art.10900
138. Suarez-Fueyo A, Rojas JM, Cariaga AE, et al. Inhibition of PI3Kdelta reduces kidney infiltration by macrophages and ameliorates systemic lupus in the mouse. J Immunol. 2014;193(2):544–554. doi:10.4049/jimmunol.1400350
139. Barber DF, Bartolome A, Hernandez C, et al. PI3Kgamma inhibition blocks glomerulonephritis and extends lifespan in a mouse model of systemic lupus. Nat Med. 2005;11(9):933–935. doi:10.1038/nm1291
140. Chen H, Shi B, Feng X, et al. Leptin and Neutrophil-Activating Peptide 2 Promote Mesenchymal Stem Cell Senescence Through Activation of the Phosphatidylinositol 3-Kinase/Akt Pathway in Patients With Systemic Lupus Erythematosus. Arthritis Rheumatol. 2015;67(9):2383–2393. doi:10.1002/art.39196
141. Taniguchi M, Kitatani K, Kondo T, et al. Regulation of autophagy and its associated cell death by “sphingolipid rheostat”: reciprocal role of ceramide and sphingosine 1-phosphate in the mammalian target of rapamycin pathway. J Biol Chem. 2012;287(47):39898–39910. doi:10.1074/jbc.M112.416552
142. Wenderfer SE, Stepkowski SM, Braun MC. Increased survival and reduced renal injury in MRL/lpr mice treated with a novel sphingosine-1-phosphate receptor agonist. Kidney Int. 2008;74(10):1319–1326. doi:10.1038/ki.2008.396
143. Okazaki H, Hirata D, Kamimura T, et al. Effects of FTY720 in MRL-lpr/lpr mice: therapeutic potential in systemic lupus erythematosus. J Rheumatol. 2002;29(4):707–716.
144. Hedrich CM, Tsokos GC. Epigenetic mechanisms in systemic lupus erythematosus and other autoimmune diseases. Trends Mol Med. 2011;17(12):714–724. doi:10.1016/j.molmed.2011.07.005
145. Koga T, Mizui M, Yoshida N, et al. KN-93, an inhibitor of calcium/calmodulin-dependent protein kinase IV, promotes generation and function of Foxp3(+) regulatory T cells in MRL/lpr mice. Autoimmunity. 2014;47(7):445–450. doi:10.3109/08916934.2014.915954
146. Ichinose K, Juang YT, Crispin JC, Kis-Toth K, Tsokos GC. Suppression of autoimmunity and organ pathology in lupus-prone mice upon inhibition of calcium/calmodulin-dependent protein kinase type IV. Arthritis Rheum. 2011;63(2):523–529. doi:10.1002/art.30085
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.