Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 19
Targeting Mitochondria-Associated Endoplasmic Reticulum Membranes (MAMs): A Novel Therapeutic Strategy for Diabetic Complications
Authors Shi J, Sun X, You J ![]() , Ji X
, Ji X ![]() , Xie J, Li Y, Gu K, Wei X
, Xie J, Li Y, Gu K, Wei X
Received 4 February 2026
Accepted for publication 12 June 2026
Published 25 June 2026 Volume 2026:19 599670
DOI https://doi.org/10.2147/DMSO.S599670
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Rebecca Baqiyyah Conway
Jianmei Shi,1,2,* Xiaohan Sun,1,3,* Jiajun You,1,3 Xiaoxu Ji,1,3 Juanmei Xie,1 Yuxuan Li,1 Kailei Gu,1 Xiaojie Wei1,3
1College of Basic Medicine, Guangxi University of Chinese Medicine, Nanning, Guangxi, 530200, People’s Republic of China; 2Xiangxi Prefecture Institute of Ethnic Medicine, Xiangxi, Hunan, 416007, People’s Republic of China; 3Guangxi Key Laboratory of Translational Medicine for Treating Infectious Diseases with Integrative Medicine, Nanning, Guangxi, 530200, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xiaojie Wei, Email [email protected]
Abstract: The pathogenesis of diabetes mellitus and its complications involves multiple factors and complex mechanisms. Currently, clinical treatment options remain limited, posing an ongoing and severe challenge to global public health. Therefore, in-depth research and understanding are urgently needed to advance the development of therapeutic strategies. Extensive studies have indicated that the endoplasmic reticulum (ER) and mitochondria are key organelles driving the occurrence and progression of diabetes mellitus and its complications. Notably, the mitochondria-associated endoplasmic reticulum membranes (MAMs), serving as a physical and functional bridge connecting the ER and mitochondria, play a critical role in maintaining organelle functional homeostasis, which underscores the importance of inter-organelle communication. This review systematically summarizes the integrated structure and regulatory mechanisms of MAMs, as well as their physiological functions in mediating inter-organelle signal transduction and material exchange. Furthermore, it elaborates in detail on the diverse pathogenic roles of MAMs dysfunction in diabetes mellitus and its complications, with a focus on its involvement in calcium signaling, lipid synthesis and transport, mitochondrial dynamics, cellular stress, and cell death. In addition, this review specifically points out that multi-component and multi-target holistic regulatory strategies exhibit unique potential in restoring MAMs homeostasis, thereby providing novel perspectives and potential preclinical intervention approaches for the “holistic regulation” treatment of diabetes complications. In conclusion, clarifying the role of MAMs in diabetes mellitus and its complications is expected to lay an important theoretical foundation for the development of preclinical therapeutic strategies, although significant challenges remain for clinical translation, including tissue-specific targeting, safety, and the lack of validated biomarkers. The image illustrates the dysfunction of mitochondria-associated membranes (MAMs) due to high glucose levels. It begins with a depiction of high glucose, leading to MAMs dysfunction. The process is divided into several sections: Ca²⁺ dysregulation, mitochondrial fragmentation, lipid metabolism disorder, increased oxidative stress and endoplasmic reticulum stress (ERS). In Ca²⁺ dysregulation, the endoplasmic reticulum (ER) and mitochondrion are shown with IP3R, VDAC1 and MCU regulating calcium flow. Mitochondrial fragmentation involves MFN2 and Drp1 affecting the mitochondrion. Lipid metabolism disorder is depicted with PA and PS interacting with PSS1/2 between the ER and mitochondrion. Increased oxidative stress shows PERK, IRE1 and ATF6 leading to ROS production. ERS is linked to oxidative stress and involves p-eIF2α and ROS. The consequences of these dysfunctions are shown as apoptosis, inflammation and energy crisis, with arrows indicating the flow and impact of each process.High glucose causes MAMs dysfunction: Ca²⁺ issues, mitochondrial fragmentation, lipid disorder, stress, apoptosis.
Keywords: diabetes-related complications, multi-target therapy, holistic regulation, organelle homeostasis, endoplasmic reticulum stress
Introduction
Diabetes mellitus has become a major challenge that needs to be urgently addressed in the global public health field. Its global prevalence has been on a continuous rise, posing a severe threat to human health. According to the latest data from the International Diabetes Federation (IDF), the number of people with diabetes worldwide reached 537 million in 2021, and epidemiological models predict that this figure will surge to 1.31 billion by 2050, representing an increase of over 140%.1 As a metabolic disease characterized primarily by pancreatic β-cell dysfunction, the onset and progression of diabetes are closely associated with organelle dysfunctions such as mitochondrial abnormalities and endoplasmic reticulum stress (ERS). With the progression of the disease course, the pathological damage caused by metabolic disorders will gradually affect the systemic microvascular system and multiple parenchymal organs, ultimately leading to a series of disabling and life-threatening complications. Diabetic angiopathy mainly includes diabetic kidney disease (DKD), diabetic retinopathy (DR), diabetic peripheral neuropathy (DPN) and diabetic cardiovascular complications; while diabetic organ lesions cover diabetic encephalopathy, diabetes-related liver injury, diabetic foot and refractory wounds.2,3 Therefore, clarifying the core pathogenesis of diabetic complications is the premise and key to developing effective preclinical prevention and treatment strategies.
In recent years, the correlation between organelle homeostasis imbalance and human diseases has become a research hotspot in the field of life sciences. For living cells, it is crucial to maintain the structural integrity and functional coordination of various organelles. These organelles do not operate in isolation; instead, they form a dynamic interactive network through membrane contact sites (MCSs) and vesicular transport, enabling material exchange, signal transduction and functional regulation, thereby mediating the adaptive responses of cells to changes in the internal and external environment. In the pathological process of diabetes and its complications, mitochondrial and endoplasmic reticulum dysfunctions have been proven to be one of the core driving factors.4 As the most critical membrane contact structure between the two, mitochondria-associated endoplasmic reticulum membranes (MAMs) are not only the core hub for regulating basic biological processes such as intracellular Ca2+ homeostasis, lipid metabolism, mitochondrial dynamics, stress response and cell death, but also play a decisive role in the occurrence and development of diabetic complications through the abnormal activation or inhibition of the above-mentioned pathways.5
Based on this, this review systematically summarizes the structural characteristics and functional regulatory mechanisms of MAMs, focuses on elaborating their pathological roles in major diabetes-related complications, and highlights multi-target and holistic regulatory intervention strategies. It further explores in depth the potential molecular mechanisms underlying the regulation of MAMs dysfunction and related signaling networks. The purpose of this review is to provide a novel “targeted-holistic” synergistic strategy for the preclinical prevention and treatment of diabetes complications, as well as to lay a theoretical foundation for subsequent translational medicine research.
Accumulating evidence suggests that MAM alterations in diabetes are dynamic and context-dependent rather than unidirectional. Early stages may involve adaptive increases in ER–mitochondria coupling, whereas chronic stress leads to maladaptive hypercoupling or loss of MAM integrity. This stage-dependent perspective may help reconcile conflicting reports of increased and decreased MAM formation across different diabetic contexts.
Methods
This review aims to systematically summarize the mechanisms underlying the role of MAMs in diabetic complications and the research progress of their use as therapeutic targets. To ensure the systematicity and comprehensiveness of the literature search, we developed the following literature search strategy.
Databases Searched
We conducted literature searches using the following databases: PubMed (https://pubmed.ncbi.nlm.nih.gov), and Google Scholar (https://scholar.google.com). The search time range was set from January 2021 to March 2026. In addition, classic and highly influential articles published before 2021 were included to provide essential background information.
Search Keywords
The search strategy combined subject headings and free-text terms. The main keywords were as follows: (1) Core concept terms: mitochondria-associated endoplasmic reticulum membranes, MAMs, ER-mitochondria contacts, mitochondria-ER contact sites; (2) Disease-related terms: diabetic complications, diabetic nephropathy, diabetic retinopathy, diabetic cardiomyopathy, diabetic neuropathy, diabetic encephalopathy, diabetic liver injury, diabetic foot ulcers; (3) Mechanism-related terms: calcium signaling, lipid metabolism, mitochondrial dynamics, endoplasmic reticulum stress, autophagy, mitophagy, apoptosis; (4) Keywords were combined using the Boolean operators AND and OR.
Inclusion and Exclusion Criteria
Inclusion criteria: (1) Original research or review articles published in peer-reviewed journals; (2) Studies related to the role of MAMs in diabetes and its complications; (3) Studies involving the structure, function, regulatory mechanisms, or therapeutic intervention of MAMs; (4) Articles published in English. Exclusion criteria: (1) Non-English publications; (2) Incomplete studies such as conference abstracts, comments, and letters; (3) Studies not directly related to MAMs or diabetic complications; (4) Duplicate publications.
Literature Screening and Data Extraction
Literature screening and data extraction were performed independently by two researchers. Any discrepancies were resolved through discussion or consultation with a third researcher. The finally included articles were classified and integrated into corresponding thematic sections according to their research content.
Using the above search strategy, we included more than 90 relevant studies as the basic literature source for this review.
Structure, Function and Dysregulation of MAMs in Diabetes
Structure of MAMs
Mitochondria and the ER exhibit structural connections and functional interactions. Using electron microscopy combined with biochemical co-sedimentation assays, Mannella et al6 and Zhou et al7 observed that ER vesicles closely contact mitochondrial membranes, confirming the existence of specific physical links between these two organelles. This contact region was later identified as a vital functional hub, namely MAMs, which mediates critical life activities between the two organelles and is essential for maintaining cellular homeostasis.
Morphological studies have shown that this dynamic interface is not tightly apposed but is maintained at a precise distance of 10–50 nm by a series of tethering proteins. These include the inositol 1,4,5-trisphosphate receptor (IP3R)-glucose-regulated protein 75 (GRP75)-voltage-dependent anion channel 1 (VDAC1) complex, which plays a core tethering role, as well as regulatory proteins such as the Sigma-1 receptor (Sig-1R).8 The structure of this membrane contact site is not static; the two organelles are in close proximity but do not overlap. A contact site is considered functionally active only when the distance between MAMs is < 30 nm, whereas a distance > 30 nm may indicate MAMs dysfunction. Therefore, the molecular structure of MAMs depends on the tight spatial organization of the ER and mitochondria, and maintaining an appropriate distance (10–50 nm) is critical for efficient signal transduction and material exchange.9,10
In terms of composition, MAMs are a multi-layered platform enriched with various functional proteins, which can be roughly divided into three components: core tethering complexes, auxiliary stabilizing proteins, and specific signal regulators.8 This multi-layered architecture enables MAMs to act not only as a “channel” for inter-organelle material exchange but also as a highly integrated signaling hub, playing a key role in maintaining cellular homeostasis and responding to external perturbations.
Composition of MAMs
As a functional bridge between the ER and mitochondria, MAMs primarily rely on proteins to exert their biological functions. Based on localization characteristics and molecular functions, MAMs-associated proteins can be classified into three categories: first, exclusive resident proteins specifically localized to MAMs; second, proteins that are enriched in MAMs but also distributed in other cellular regions; third, proteins that are transiently recruited to MAMs in a condition-dependent manner. This localization diversity not only reflects the dynamic nature of MAMs structure but also lays the foundation for its functional diversity.11 At the functional level, these proteins mediate the tethering between mitochondrial and ER membranes, and directly determine the stability of physical connections between the two organelles by precisely regulating membrane contact distance, contact area, and the number of connection sites.12
The formation of MAMs is mediated by multiple membrane-anchored tethering protein complexes. To date, several key complexes have been identified and characterized in mammalian cells, including: (1) Vesicle-associated membrane protein-associated protein B (VAPB) and protein tyrosine phosphatase-interacting protein 51 (PTPIP51); (2) Oxysterol-binding protein-related proteins 5/8 (ORP5/8) and PTPIP51; (3) B-cell receptor-associated protein 31 (BAP31) and mitochondrial fission 1 protein (FIS1); (4) The calcium signaling core composed of IP3R, GRP75, voltage-dependent anion channel (VDAC), and mitochondrial calcium uniporter (MCU); (5) Mitofusin 2 (MFN2) and mitofusin 1 (MFN1), which mediate mitochondria-ER membrane tethering; (6) Ribosome-binding protein 1 (RRBP1) and synaptojanin 2-binding protein (SYNJ2BP).13
Proteomic studies have identified hundreds to thousands of MAMs-associated proteins across different species and tissues, with their core components being highly conserved.14–17 For example, in MAMs isolated from the retinas of diabetic rats, a total of 2664 unique proteins were quantified, among which 179 exhibited significant alterations under diabetic conditions. Functional annotation revealed that these differentially expressed proteins are involved in key biological processes such as cell survival, inflammatory response, and cellular homeostasis maintenance, indicating that diabetes can induce dysregulation of protein expression in retinal MAMs, thereby leading to functional abnormalities.16 In the hearts of db/db mice (a model of type 2 diabetes), MAMs proteomic analysis demonstrated a fundamental shift in molecular function: from calcium signaling processing and muscle contraction in the healthy state to energy metabolism disorders centered on mitochondrial dysfunction. This critical transition directly promotes the onset and progression of diabetic cardiomyopathy.18
In summary, MAMs possess a highly conserved protein composition and play a key role in maintaining cellular homeostasis. Their functional dysregulation has emerged as an important pathological basis for the development of various diabetic complications (Table 1).
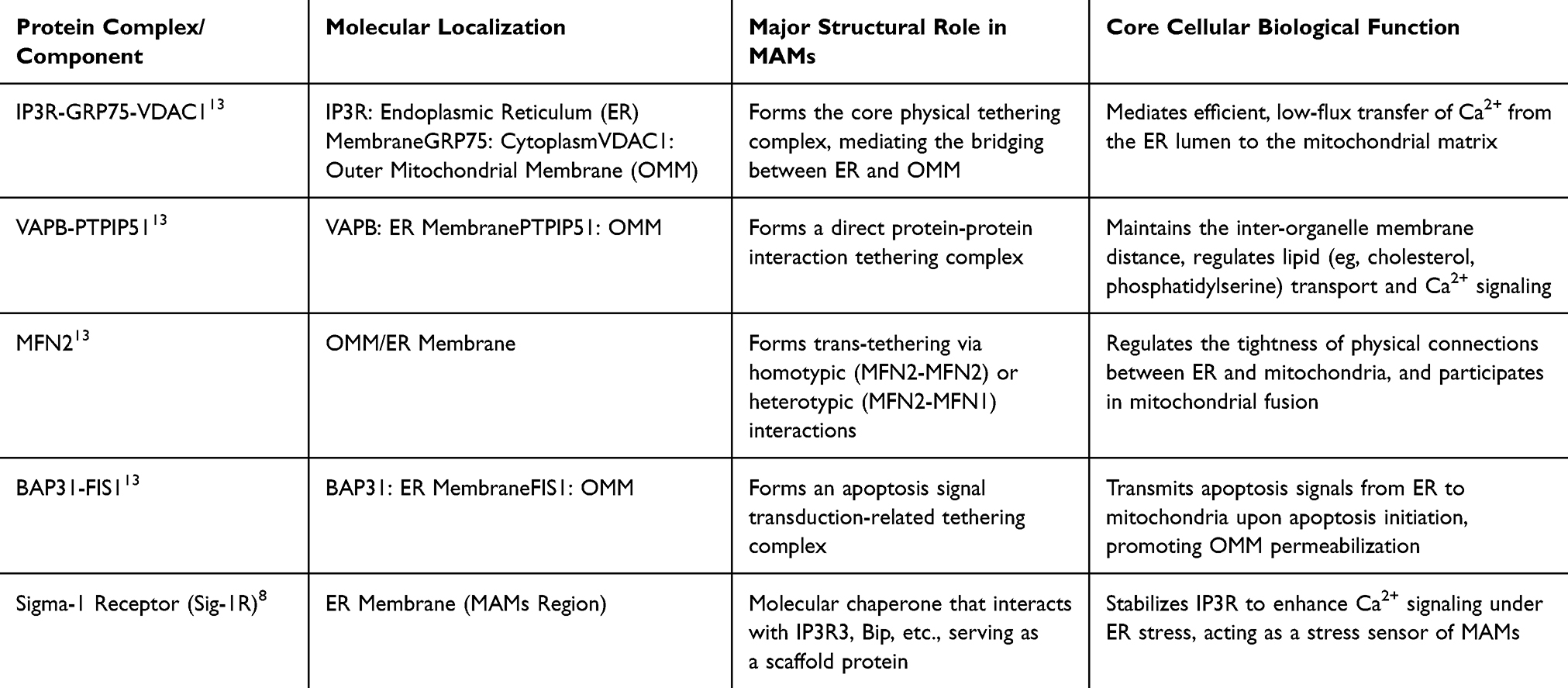 |
Table 1 Core Protein Composition of MAMs and Their Structural and Functional Roles |
Technical Challenges in MAMs Research
Despite remarkable progress in MAMs research in recent years, the field still faces a series of technical challenges. These challenges largely limit the comparability of results across different studies and may be a major cause of conflicting reports in the literature regarding the functional status of MAMs (increased vs decreased).
First, there is significant variability in morphological detection methods for MAMs. Transmission electron microscopy (TEM) serves as the gold standard for quantitative analysis of endoplasmic reticulum-mitochondria contact distances and contact lengths, as well as an essential tool for observing the ultrastructure of MAMs. However, its outcomes are highly dependent on sample processing protocols (eg, osmotic pressure of fixatives, dehydration steps), randomness in image acquisition, and the standardization of quantitative analysis methods.19 Studies have shown that limitations of TEM include low throughput, lengthy sample preparation time, limited field of view, and the requirement for sample dehydration and staining—all of which may compromise the in situ preservation of MAMs structure.19 Moreover, contact definition criteria (eg, distance thresholds set at 30 nm, 50 nm, or wider) vary substantially across laboratories, rendering direct comparison of quantitative results difficult.
Second, proximity ligation assay (PLA) and biochemical fractionation techniques each have limitations in specificity and resolution. PLA enables in situ detection of interactions between specific protein complexes in MAMs regions, but its signal intensity is influenced by factors such as antibody quality, fixation conditions, and threshold settings, and it cannot provide precise subcellular localization information.20 Biochemical fractionation methods (eg, density gradient centrifugation) are commonly used to obtain MAMs-enriched fractions at present. Nevertheless, lysis conditions, gradient media (eg, Percoll, Nycodenz, OptiPrep), and centrifugation parameters differ widely among studies, resulting in variable purity of isolated MAMs fractions. This may account for the substantial discrepancies in MAMs-associated proteins identified across different investigations.21 Notably, it is widely agreed among researchers that combining multiple techniques (eg, TEM coupled with proteomics, PLA integrated with functional assays) is necessary to draw more reliable conclusions.19
Third, the selection of cell models and disease stages exerts a decisive influence on MAMs phenotypes. In in vitro experiments, variations in high-glucose stimulation concentration (5.5 mM vs 25–30 mM), exposure duration (acute vs chronic), and cell type (primary cells vs immortalized cell lines) can all trigger distinct adaptive changes in MAMs. For instance, short-term high-glucose stimulation may induce compensatory enhancement of MAMs, whereas prolonged stimulation may lead to structural disruption.20 In in vivo studies, different diabetic animal models (eg, STZ-induced type 1 diabetes vs db/db or ob/ob type 2 diabetes models) and different disease stages (early vs advanced) within the same model may exhibit completely opposite alterations in MAMs.20,21 A recent study found that in diet-induced obese and type 2 diabetic mouse models, basal MAMs contacts in colonic L-cells were paradoxically enhanced but lost their normal responsiveness to glucose stimulation, indicating functional remodeling of MAMs at different disease stages.20
Fourth, MAMs research is also plagued by the lack of standardized measurement techniques. Currently, parameters adopted for MAMs quantitative analysis across laboratories (eg, contact length, contact area, number of contact sites, distance thresholds) lack uniform standards, making cross-study comparison of research results challenging.19 Establishing standardized workflows for MAMs quantification, developing more specific live-cell dynamic imaging technologies (eg, contact probes based on FRET or FLIM), and performing cross-validation using multi-omics techniques will be key directions to advance the field in the future.
Therefore, when interpreting and comparing MAMs-related studies, full consideration must be given to the aforementioned technical and methodological differences. Understanding these technical challenges is crucial for promoting the development of the MAMs field and facilitating the transition from mechanistic research to clinical translation.21
Core Cytological Functions of MAMs
As a key connection hub between mitochondria and the endoplasmic reticulum, the core physiological functions of MAMs involve the regulation of cellular physiological processes such as calcium signaling, lipid synthesis and transport, mitochondrial dynamics, cellular stress, and cell death.
MAMs Participate in the Regulation of Calcium Signaling
Calcium ions (Ca2+), as ubiquitous second messengers, are crucial for cell survival and function.22 By forming a highly efficient microdomain enriched with key calcium-handling proteins, MAMs serve as a central hub for the precise regulation of intracellular Ca2+ signaling.8,23 Under physiological conditions, MAMs-mediated calcium signaling is a highly coordinated multi-step process: extracellular signal stimulation triggers the activation of IP3R or ryanodine receptors (RyR) on the endoplasmic reticulum membrane, mediating the release of Ca2+ from the ER lumen into the MAMs region. Subsequently, the released Ca2+ is captured by GRP75 in the cytoplasm. Acting as a molecular bridge, GRP75 binds IP3R at one end and VDAC1 on the outer mitochondrial membrane at the other end, thereby “delivering” Ca2+ to the mitochondrial surface. Ultimately, upon sensing the high-Ca2+ microenvironment in the mitochondrial intermembrane space, the MCU complex located on the inner mitochondrial membrane opens, driving Ca2+ influx into the mitochondrial matrix.12
The precise regulation of this process relies on multiple MAMs-resident proteins: tethering proteins including MFN2 and VAPB-PTPIP51 structurally ensure the efficiency and fidelity of calcium transfer by maintaining an optimal membrane distance of 10–30 nm. Meanwhile, the Sig-1R, acting as a molecular chaperone, stabilizes IP3R under ER stress to enhance calcium flux, whereas Bcl-2 family proteins can interact with IP3R to inhibit its excessive opening, serving as a functional “brake”.
MAMs-mediated mitochondrial calcium uptake is of critical physiological significance. On the one hand, a moderate increase in Ca2+ within the mitochondrial matrix acts as a potent signal to activate key dehydrogenases in the tricarboxylic acid cycle (eg, pyruvate dehydrogenase, isocitrate dehydrogenase) as well as F1F0-ATP synthase, thereby directly promoting ATP synthesis to meet cellular energy demands. On the other hand, acting as a “calcium buffer”, mitochondria rapidly uptake Ca2+ from the MAMs region, which helps terminate ER calcium release and prevent cytoplasmic calcium overload, thus shaping the frequency and amplitude of calcium oscillations and ensuring the proper decoding of downstream signaling pathways.11
Of note, calcium signaling homeostasis mediated by MAMs is closely associated with insulin signal transduction. MAMs play a dual role in insulin resistance. On the one hand, proper structure and function of MAMs are essential for maintaining insulin sensitivity. MAMs provide the necessary ATP support for insulin signaling pathways (such as the PI3K/Akt pathway) by regulating mitochondrial calcium uptake and energy metabolism. Meanwhile, MAMs also serve as an important component of the insulin receptor signaling complex and participate in the regulation of glucose uptake in response to insulin stimulation.11 On the other hand, under pathological conditions, dysfunctional MAMs can directly induce the development of insulin resistance. Studies have shown that high-fat diet or high-glucose stimulation can alter the expression or localization of key proteins in MAMs (such as PTP1B and MFN2), triggering disordered endoplasmic reticulum-mitochondrial calcium transport and mitochondrial dysfunction. This in turn inhibits the normal transmission of insulin signaling pathways by activating stress kinases including JNK and PKCθ.5,24 Notably, the transition between “protective MAMs” and “pathological MAMs” depends on the nature, intensity, and duration of the stimulus. Recent studies have found that short-term high-glucose stimulation induces compensatory enhancement of MAMs, whereas prolonged stimulation leads to structural disruption of MAMs and impaired insulin signaling.11 In addition, downregulation of MAMs-associated proteins (such as PACS-2 and MFN2) in hepatocytes of obese and type 2 diabetic mice is positively correlated with the severity of insulin resistance.25,26 Therefore, MAMs are the intersection of cellular energy metabolism and calcium signal integration. In the hyperglycemic stress environment of diabetes, disruption of MAMs structure or functional abnormalities of regulatory proteins can easily shift calcium signaling from “precise regulation” to “uncontrolled transfer”, triggering mitochondrial calcium overload, ROS burst and energy crisis, and ultimately initiating the apoptotic program. These are the common cellular events underlying various diabetic complications.
MAMs Participate in Lipid Synthesis and Transport
Lipid metabolic homeostasis is essential for maintaining membrane integrity, energy storage and signal transduction in cells. MAMs serve as a core intracellular platform for lipid synthesis and distribution, and are particularly responsible for regulating the metabolism of phospholipids, cholesterol and sphingolipids, as well as their directional transport to mitochondria. Structurally, MAMs are enriched with a variety of key enzymes and carrier proteins involved in lipid synthesis and transport. For instance, after phosphatidylserine (PS) is synthesized in the endoplasmic reticulum, it can be further modified by phosphatidylserine synthases that are locally enriched in MAMs. Subsequently, lipid transfer proteins (eg, the ORP5/8 and VAPB-PTPIP51 complexes) located on MAMs mediate its transport to the outer mitochondrial membrane. Within mitochondria, PS is rapidly decarboxylated to form phosphatidylethanolamine (PE), which can be transported back to the endoplasmic reticulum for the synthesis of other phospholipids.27
Under conditions of hyperglycemia and insulin resistance in diabetes, the lipid metabolic function of MAMs undergoes significant dysregulation. On the one hand, hyperglycemia can induce the abnormal activation of the sterol regulatory element-binding protein (SREBP) pathway in the MAMs region, leading to the over-synthesis of cholesterol and triglycerides.28 On the other hand, impaired lipid transport function of MAMs—such as abnormal expression or function of the VAPB-PTPIP51 complex—reduces the efficiency of phospholipid (eg, cardiolipin) delivery to mitochondria.13 Insufficient cardiolipin content in mitochondria directly impairs the stability and activity of electron transport chain complexes, exacerbating oxidative phosphorylation disorders and ROS production.29 Meanwhile, the ectopic accumulation of lipids in non-adipose tissues (eg, liver, kidney, myocardium) is a crucial initiating factor for diabetes-related steatosis, lipotoxicity and organ damage.30–32 Therefore, MAMs-mediated lipid metabolic imbalance constitutes a key link connecting metabolic disorders in diabetes with organelle dysfunction.
MAMs Participate in the Regulation of Mitochondrial Dynamics
Mitochondria are in a dynamic balance of continuous fusion and fission, a process known as mitochondrial dynamics. MAMs act as a strategic command center for regulating mitochondrial dynamics, which is essential for maintaining mitochondrial network integrity, function and quality control.33
Mitochondrial fusion is mainly mediated by mitofusin 1 and 2 (MFN1/2) as well as optic atrophy 1 (OPA1).34 Among these, MFN2 has a unique dual localization: it is not only located on the outer mitochondrial membrane to facilitate mitochondria-mitochondria fusion, but also resides on the endoplasmic reticulum membrane. By forming MFN2-MFN1/2 trans-complexes, MFN2 directly tethers the endoplasmic reticulum to mitochondria, thereby shaping the MAMs structure.35,36 Therefore, MFN2 is a core molecule linking mitochondrial dynamics and MAMs integrity. Mitochondrial fission is dominated by dynamin-related protein 1 (Drp1), which is recruited to the fission sites on the outer mitochondrial membrane.37,38 Emerging evidence indicates that the endoplasmic reticulum pre-wraps and constricts mitochondria to mark future fission sites, a process that is highly dependent on MAMs.38 The INF2 protein on the endoplasmic reticulum membrane can promote actin polymerization, and cooperates with the Spire1C protein in the MAMs region to jointly provide a structural scaffold for the recruitment and oligomerization of Drp1.39
In diabetic complications, the hyperglycemic environment generally shifts mitochondrial dynamics toward excessive fission (fragmentation).40–44 The underlying mechanisms involve: 1) Downregulated expression or functional inhibition of MFN2, which impairs MAMs tethering and mitochondrial fusion capacity;40 2) Enhanced activation of Drp1, with increased phosphorylation levels (eg, via PKCδ and CaMKII), leading to greater recruitment to mitochondria-ER contact sites with augmented MAMs formation.43,44 The resulting mitochondrial fragmentation is characterized by low respiratory efficiency, increased ROS production, and susceptibility to autophagic clearance. When the autophagic capacity of cells is overwhelmed, dysfunctional mitochondria accumulate and trigger apoptosis.42 This phenomenon has been confirmed in diabetic cardiomyocytes, renal tissues and podocytes in diabetic kidney disease, as well as retinal ganglion cells in diabetic retinopathy.
MAMs Participate in the Regulation of Autophagy and Mitophagy
In addition to participating in the regulation of endoplasmic reticulum stress and oxidative stress, MAMs also play a key role in the initiation and regulation of autophagy and mitophagy. Autophagy is a highly conserved cellular degradative process that maintains cellular homeostasis by forming double-membraned autophagosomes to encapsulate and eliminate damaged organelles, misfolded proteins, and pathogens. Based on cargo specificity, autophagy can be classified into non-selective macroautophagy and selective types such as mitophagy.45
Studies have demonstrated that MAMs serve as a critical platform for autophagosome membrane formation. Under autophagy-inducing conditions, the endoplasmic reticulum membrane generates pre-autophagosomal structures at MAMs, which then interact with other organelles including mitochondria to promote autophagosome elongation and maturation. Multiple proteins located at MAMs are involved in this process: for instance, the VAPB-PTPIP51 complex not only mediates ER-mitochondria tethering but also participates in the recruitment of autophagy-related proteins such as LC3 and ATG14;46,47 meanwhile, the BAP31-FIS1 complex plays an important role in autophagic signal transduction.48
MAMs also act as a core regulator in mitophagy. The PINK1/Parkin pathway is the best-characterized mechanism for selective mitophagy. In healthy mitochondria, PINK1 is rapidly degraded; however, upon loss of mitochondrial membrane potential, PINK1 stably accumulates on the outer mitochondrial membrane and recruits Parkin to damaged mitochondria, thereby triggering mitophagy. Both PINK1 and Parkin localize to MAMs, and the structural integrity of MAMs is essential for PINK1/Parkin-mediated mitophagy.49 Furthermore, other MAMs-associated proteins, including MFN2 and FUNDC1, regulate mitophagy: MFN2 acts as a receptor for Parkin to facilitate the removal of damaged mitochondria, whereas FUNDC1 mediates PINK1/Parkin-independent mitophagy under hypoxic conditions.50
Dysregulation of autophagy and mitophagy is one of the major pathogenic mechanisms in diabetic complications. A high-glucose environment impairs MAMs structure and suppresses PINK1/Parkin-mediated mitophagy, leading to the accumulation of damaged mitochondria and further exacerbating oxidative stress and apoptosis.51 Therefore, restoring MAMs-mediated autophagy and mitophagy represents a promising preclinical therapeutic strategy for diabetic complications, although its clinical applicability requires further validation. For example, black raspberry anthocyanins protect the retina by activating mitophagy via inhibiting the PTP1B-ERS axis;52 ginsenoside Ro alleviates endothelial injury in diabetic retinopathy by promoting mitophagy through the Epac1/AMPK pathway.53
MAMs Participate in Cellular Stress
Cellular stress, particularly ERS and oxidative stress, plays a central role in the pathogenesis of diabetic complications. MAMs serve as a key hub for integrating and coordinating these two stress responses.
When ERS occurs, the three major sensors of the unfolded protein response (UPR)—PERK, IRE1α and ATF6—are activated. Among them, PERK has been confirmed to be partially localized at MAMs.54 Activated PERK not only globally regulates protein synthesis by phosphorylating eIF2α, but also directly phosphorylates key proteins on MAMs (eg, altering the conformation of MFN2), thereby modulating the tightness and function of MAMs.54,55 Meanwhile, ERS leads to increased release of calcium from the calcium store in the MAMs region, exacerbating mitochondrial calcium influx through the IP3R-GRP75-VDAC1 channel.56 On the other hand, excessive ROS generated by mitochondrial dysfunction can diffuse back to the MAMs region, oxidatively modifying proteins on the endoplasmic reticulum membrane (eg, IP3R, SERCA), disrupting calcium homeostasis and further aggravating ERS, thus forming an ERS-ROS vicious cycle.57 The Sig-1R on MAMs acts as a stress sensor and molecular chaperone; it is activated in the early stage of ERS, maintains moderate calcium signaling by stabilizing IP3R3, and helps preserve the structural integrity of MAMs, which constitutes an important protective mechanism for cells against stress.58 However, under the chronic stress of sustained hyperglycemia in diabetes, this protective mechanism may be exhausted, transforming MAMs from a stress coordination hub into an amplifier of apoptotic signals.
MAMs Participate in Cell Death
MAMs are a core platform for regulating cell fate, especially mediating mitochondria-dependent apoptosis. They integrate multiple apoptotic signals from the endoplasmic reticulum and cytoplasm, and decisively control mitochondrial outer membrane permeabilization (MOMP)—the irreversible point of apoptosis.59,60 The activation and mitochondrial localization of the pro-apoptotic proteins Bax/Bak are critical steps in the execution of apoptosis.60 Studies have found that the early activation of a subset of Bax occurs in the MAMs region.61 BAP31, which resides on the endoplasmic reticulum, forms a pro-apoptotic complex with FIS1 on the outer mitochondrial membrane at MAMs. Upon receiving death signals, this complex can be cleaved to generate p20 BAP31, which promotes the recruitment and activation of Caspase-8 and facilitates the transmission of apoptotic signals to mitochondria.62,63 More importantly, MAMs-mediated calcium signaling is a potent trigger of apoptosis.64 As described in section 2.3.1, the pathological, excessive calcium flux from the endoplasmic reticulum into mitochondria via MAMs is the direct cause of mitochondrial membrane potential collapse, persistent opening of the mitochondrial permeability transition pore (mPTP), and cytochrome C release. In addition, Bcl-2 family proteins on MAMs (eg, the anti-apoptotic Bcl-2 and Bcl-xL) directly interact with IP3R to modulate its calcium channel activity, thereby inhibiting excessive pro-apoptotic calcium transfer under physiological conditions.65
In diabetic complications, hyperglycemia creates a pro-apoptotic microenvironment by disrupting MAMs structure (eg, downregulating MFN2 and PACS-2) and function (eg, enhancing IP3R-VDAC1 coupling and inhibiting protective autophagy).46,56,66,67 This exacerbates calcium-mediated mitochondrial damage, while weakening the protective “brake” effect of proteins such as Bcl-2, leading to a marked increase in cellular sensitivity to apoptotic stimuli.68 Therefore, the functional integrity of MAMs is a critical switch that determines cell survival in the hyperglycemic environment of diabetes (Figure 1).
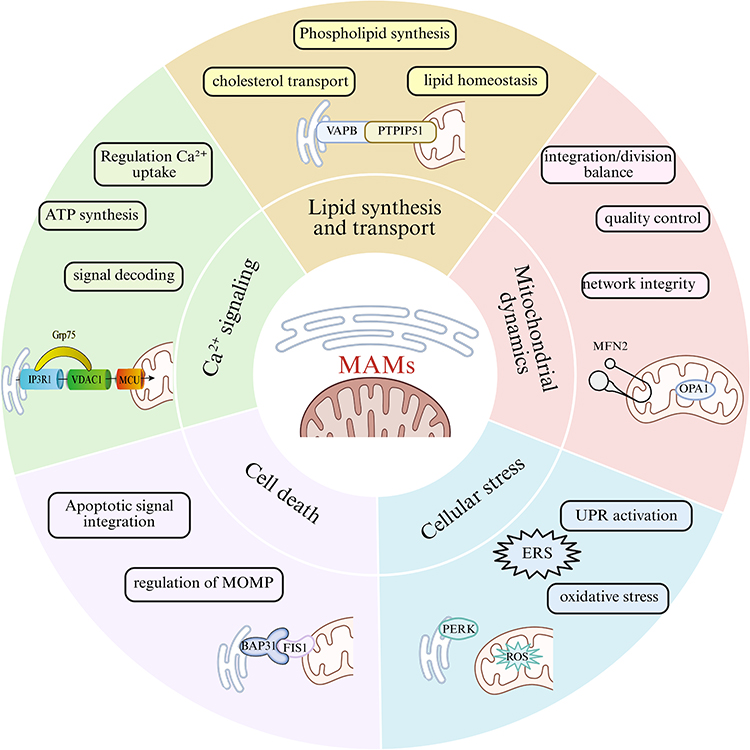 |
Figure 1 The core cytological functional network of MAMs. Figures were created using BioRender (https://www.biorender.com/). No artificial intelligence tools were used in the generation of any figures or images. |
MAMs Dysfunction: A Common Pathway Driving Diabetic Complications
The pathological changes of MAMs in diabetic complications do not follow a uniform pattern, but instead exhibit a seemingly paradoxical phenomenon: the number and extent of ER-mitochondria contacts are reduced in some tissues or disease stages, while excessive formation occurs in others. This discrepancy is not merely due to experimental error, but likely reflects the complex regulatory functions of MAMs under different pathophysiological conditions. Specifically, the “decrease” or “increase” in MAMs may correspond to two distinct pathological states. On the one hand, impaired structural integrity of MAMs (eg, increased contact distance, downregulated expression of key tethering proteins) reduces the efficiency of material exchange and signal transduction between the ER and mitochondria. This “hypofunctional” state is frequently observed in the early stage of disease or in certain tissues under chronic stress, such as renal tissue in diabetic nephropathy69 and hippocampal neurons in diabetic encephalopathy.70 The consequences include disrupted calcium transport, disordered energy metabolism, and increased cellular susceptibility to apoptotic stimuli. On the other hand, “excessive formation” or “hyper-coupling” of MAMs is commonly seen in the progressive stage of disease or in specific cell types, such as cardiomyocytes in diabetic cardiomyopathy71 and retinal vascular endothelial cells in diabetic retinopathy.21 This “hyper functional” state triggers abnormal massive Ca2+ influx from the ER into mitochondria, leading to mitochondrial Ca2+ overload, explosive ROS production, and irreversible apoptosis.
Therefore, changes in the structure and function of MAMs may follow a biphasic regulatory mode: at the initial stage of disease, cells may enhance MAMs formation to compensatory maintain ER-mitochondrial communication in response to metabolic stress. However, when pathological stimuli such as high glucose and high lipid persist and exceed the cellular compensatory capacity, MAMs may shift toward structural disruption or hyperfunction, eventually acting as a “catalyst” that amplifies damaging signals. Understanding this dynamic evolutionary process is critical for precisely targeting MAMs function and avoiding potential risks caused by inappropriate therapeutic timing.
Dysfunction of MAMs has been increasingly recognized as a key and shared pathological mechanism underlying the development and progression of various diabetic complications. The hyperglycemic environment can directly disrupt the structural integrity and functional coordination of MAMs, thereby impairing calcium ion transfer, lipid exchange, and energy metabolism coupling between the ER and mitochondria, and ultimately triggering extensive organelle dysfunction and tissue damage.
In the central nervous system, structural integrity of MAMs in hippocampal neurons is impaired in diabetic encephalopathy models, characterized by increased distance and reduced contact area between the ER and mitochondria. This leads to downregulated expression of the IP3R-GRP75-VDAC1-MCU calcium channel complex, which in turn causes impaired mitochondrial calcium uptake, excessive ROS production, and decreased ATP synthesis, ultimately compromising synaptic plasticity and cognitive function.70 In pancreatic β-cells, prolonged hyperglycemic exposure induces ER stress, excessive mitochondrial fission, and energy depletion through MAMs-mediated dysregulated Ca2+ transfer, accelerating β-cell dysfunction and apoptosis, and promoting the onset and progression of type 2 diabetes. Meanwhile, impaired MAMs structural integrity in peripheral tissues such as the liver, muscle, and adipose tissue also interferes with insulin signaling transduction and exacerbates systemic insulin resistance.72
At the target organ level, MAMs abnormalities are widely involved in multiple diabetic complications including DKD, diabetic cardiomyopathy (DCM), diabetic retinopathy, impaired skin wound healing, and diabetic neuropathy. Using in situ proximity ligation assay, Yang et al found that the number of MAMs in renal biopsy tissues of DKD patients decreased significantly with the progression of pathological grades (from IIa to class III), which was closely associated with the downregulated expression of key regulatory proteins such as DsbA-L, PACS-2, and MFN-2.73 Cao et al further confirmed that under high-glucose stimulation, downregulated MFN2 expression in podocytes attenuates its interaction with PERK, leading to MAMs structural damage, mitochondrial dysfunction, and cell apoptosis.54 Liu et al demonstrated in renal tubular epithelial cells that high glucose upregulates MAPK1 and inhibits PACS-2 expression; MAPK1 can directly bind to PACS-2 to promote its degradation, disrupt MAMs integrity, and induce mitochondrial fragmentation. Notably, treatment with the MAPK1 inhibitor VX-11e effectively restores PACS-2 levels, reconstructs MAMs structure, and significantly alleviates proteinuria and renal injury.74
In diabetic cardiomyopathy, Wei et al discovered that the hyperglycemic and hyperlipidemic environment promotes excessive MAMs formation by upregulating acid sphingomyelinase (ASMase), resulting in disordered and massive Ca2+ influx from the ER into mitochondria and subsequent calcium overload. This process is regulated by MICU1 and ultimately triggers oxidative stress and cardiomyocyte apoptosis.75 In addition, Zhang et al reported that the circadian rhythm gene Bmal1 is downregulated under high glucose conditions, which inhibits Bcl2 transcription, attenuates the inhibitory effect of Bcl2 on IP3R, promotes excessive Ca2+ release to mitochondria via MAMs, and induces calcium overload and cell death.76 Other studies have indicated that hyperglycemia enhances MAMs connectivity by inhibiting AMPKα2 and upregulating FUNDC1, which also leads to cardiac energy metabolism disorders.77 Meanwhile, the PACS2/IP3R2/FUNDC1/VDAC1 signaling axis has been confirmed to mediate excessive MAMs assembly, inhibit oxidative phosphorylation, and activate the mitochondria-dependent apoptotic pathway.78
In diabetic skin wound models, Xian et al found that oxidative stress-induced endothelial cell senescence is associated with excessive MAMs formation, specifically characterized by enhanced interaction of the IP3R1/GRP75/VDAC1 complex, which causes mitochondrial Ca2+ overload and functional impairment.75
In terms of diabetic retinopathy, Wang et al revealed through proteomic analysis that prolonged hyperglycemia significantly alters the retinal MAMs-associated protein profile, affecting pathways related to inflammation, calcium homeostasis, and energy metabolism.16 Meanwhile, Ca2+/calmodulin-dependent protein kinase II (CaMKII) is abnormally activated to promote cell apoptosis. Moreover, decreased MFN2 expression and upregulated Drp1 drive mitochondrial fragmentation, which together facilitate Bax translocation to mitochondria and cytochrome C release, thereby activating the canonical apoptotic program.79
In diabetic bladder dysfunction, a complication related to peripheral neuropathy, damage to Schwann cells is particularly critical. Hyperglycemia impairs MAMs structure and function in Schwann cells, inducing PERK pathway activation (increased ER stress), IP3R-mediated abnormal calcium signaling, and excessive mitochondrial fission caused by Drp1/MFN2 imbalance, ultimately leading to Schwann cell damage. Since Schwann cells are essential for maintaining the myelin sheath and function of peripheral nerves, their damage can further impair the nerves innervating the bladder, inducing or exacerbating bladder dysfunction.77
In summary, although the specific manifestations of MAMs in different complications may present as “decreased quantity” or “excessive formation”, their core pathological consequences are highly consistent: enhanced ER stress, impaired calcium homeostasis, abnormal mitochondrial dynamics, energy metabolism disorders, and activated cell apoptosis. Therefore, MAMs not only serve as a key hub linking diabetic metabolic disorders to multi-organ damage, but also provide promising preclinical intervention targets and strategies for the development of novel therapies, such as targeting PACS-2, MFN2, FUNDC1, MAPK1, or regulating calcium channel complexes. However, significant challenges remain before these strategies can be translated into clinical practice (Table 2 and Figure 2).
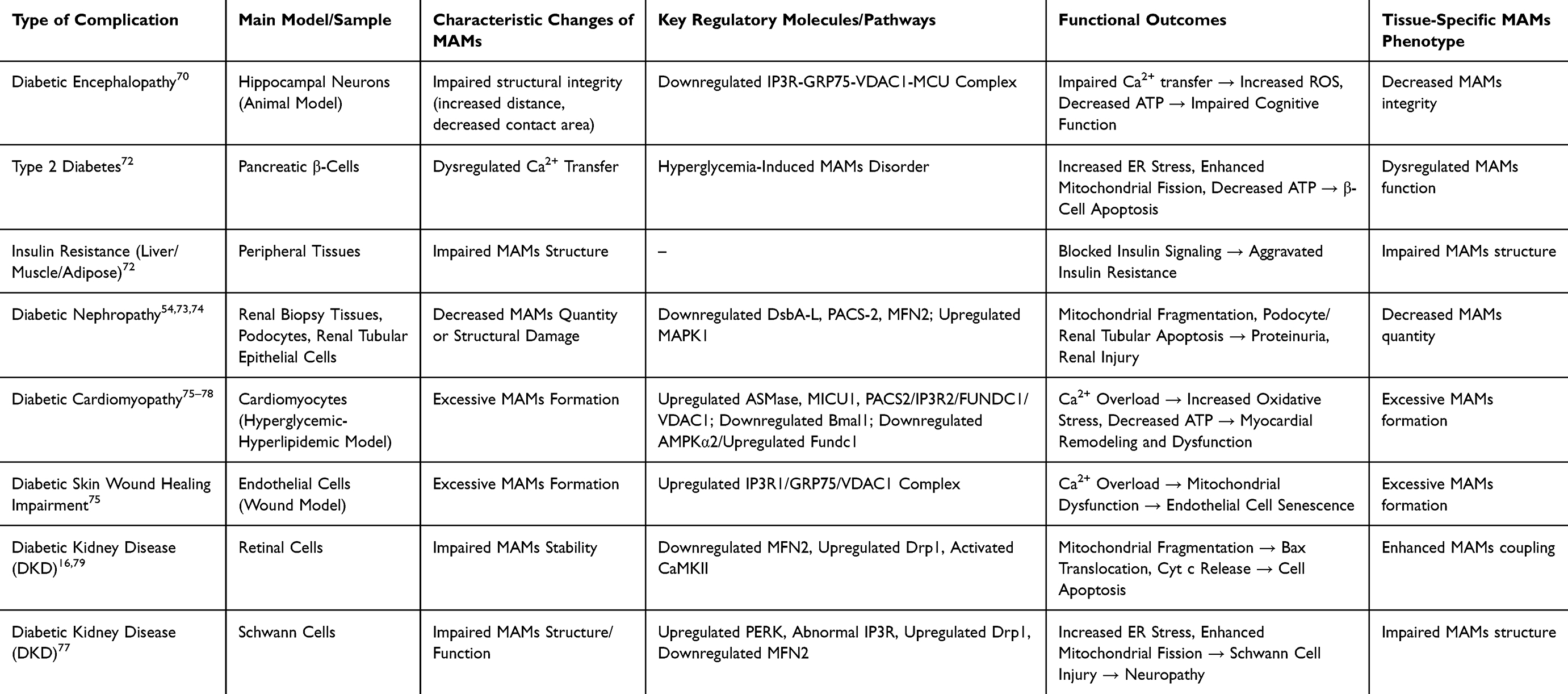 |
Table 2 MAMs Dysfunction as a Common Pathogenic Pathway in Diabetic Complications |
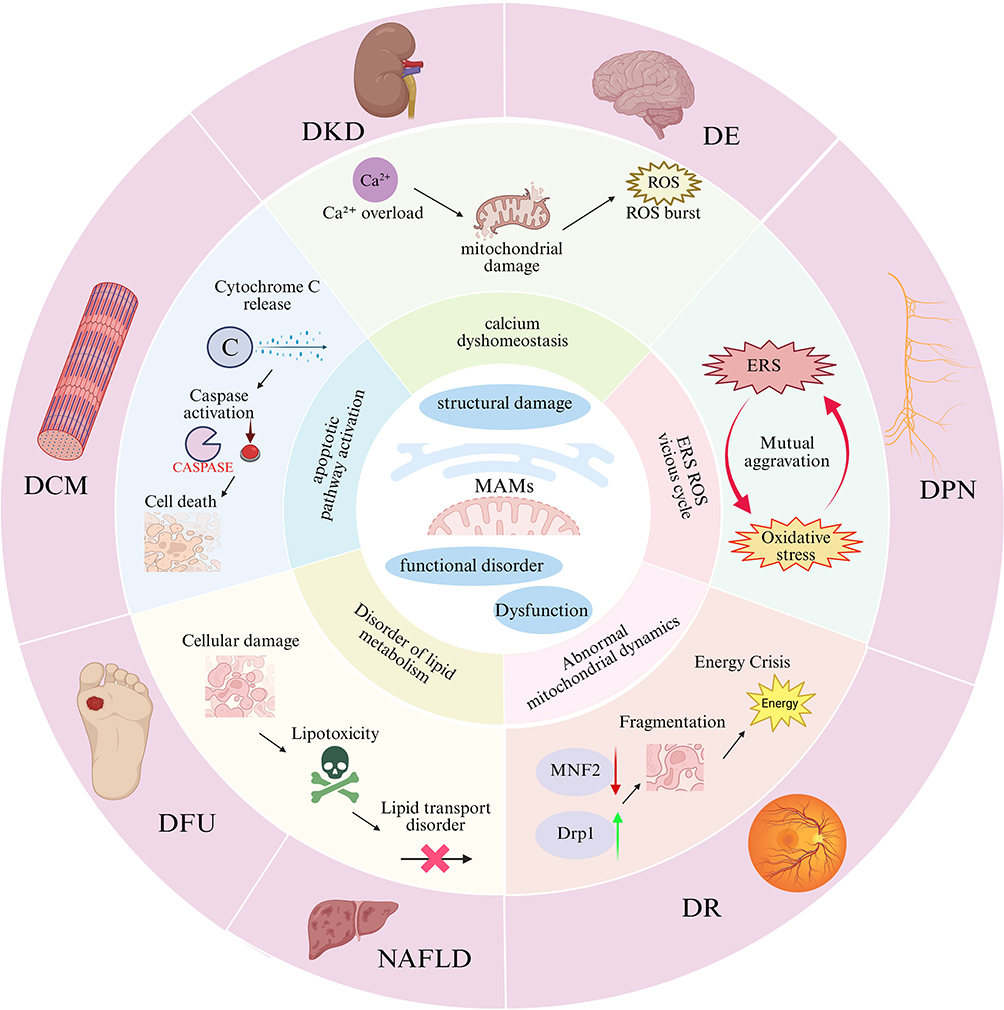 |
Figure 2 MAMs dysfunction drives the common pathways of diabetic complications. Figures were created using BioRender (https://www.biorender.com/). No artificial intelligence tools were used in the generation of any figures or images. |
Clinical Evidence for Targeting MAMs in the Prevention and Treatment of Diabetic Complications
Diabetic Kidney Disease
DKD is one of the most common and devastating microvascular complications of diabetes, and also a leading cause of End-Stage Renal Disease (ESRD) worldwide. Recent studies have demonstrated that structural and functional disorders of MAMs play a central role in the pathogenesis and progression of DKD. In a hyperglycemic microenvironment, the stability of MAMs is disrupted, inducing mitochondrial dysfunction, ERS, and cell death, which ultimately lead to renal fibrosis and progressive loss of renal function. A study by Li et al69 found that stabilizing the VDAC1/GRP75/IP3R complex can restore intracellular calcium homeostasis, thereby effectively alleviating the degree of renal fibrosis. Research by Dai et al46 confirmed that down-regulating the overexpression of VAPB-PTPIP51 can ameliorate the abnormal tight junction of pathological MAMs and podocyte injury. In addition, overexpression of DsbA-L can up-regulate the expression level of MFN2, exerting a protective effect on renal tubular cells.80 As a structural scaffold protein of MAMs, the deletion of PACS-2 expression can exacerbate mitochondrial dysfunction, whereas PACS-2 overexpression can promote mitophagy;66 A study by Liu et al74 revealed that MAPK1 impairs the structural integrity of MAMs by degrading PACS-2, and its specific inhibitor VX-11e exerts a significant renoprotective effect. Up-regulating the expression of SERCA2 can restore the function of endoplasmic reticulum calcium pumps, thereby reducing podocyte apoptosis.66 In diabetic nephropathy, MAMs predominantly exhibit reduced number and structural disruption, which is in sharp contrast to the excessive formation of MAMs observed in diabetic cardiomyopathy, reflecting the distinct response patterns of different organs to high glucose stress.73
Diabetic Retinopathy
DR is a common microvascular complication of diabetes, characterized clinically by retinal vascular leakage, tissue ischemia, neovascularization, and neurodegeneration, which can lead to irreversible vision loss in severe cases. In contrast to the reduced number of MAMs characteristic of diabetic nephropathy, MAMs in diabetic retinopathy exhibit a state of hyper-coupling, characterized by enhanced ER-mitochondria contact and aberrant activation of the IP3R1-GRP75-VDAC1 axis.81 This tissue-specific difference may be related to the high sensitivity of retinal vascular endothelial cells to calcium signaling. As a dynamic contact region between the endoplasmic reticulum and mitochondria, MAMs are involved in regulating key physiological processes such as intracellular calcium homeostasis, oxidative stress response, mitochondrial quality control, and cell apoptosis, and their dysfunction is closely associated with the pathological progression of DR. Studies have shown that under DR pathological conditions, MAMs exhibit the characteristic of “excessive coupling”, driven by the activation of the IP3R1-GRP75-VDAC1 axis, which ultimately results in mitochondrial Ca2+ overload, ROS burst, opening of the mPTP, and Caspase-3-dependent cell apoptosis; the calcium chelator BAPTA-AM can effectively reverse this pathological process, confirming the central role of calcium overload in the pathogenesis of DR.82 A vicious cycle exists between ERS and MAMs dysfunction: the ERS inducer tunicamycin can mimic the pathological effects of hyperglycemia and significantly enhance the degree of MAMs coupling.82 PTP1B, as a key regulatory factor of the ERS pathway, is highly expressed in the retinal tissues of DR patients; small vesicles derived from mesenchymal stem cells (MSCs) can target and inhibit PTP1B expression by delivering miR-125a-5p, activate the PINK1/Parkin-mediated mitophagy pathway, and thus alleviate retinal injury.83 Black raspberry anthocyanins can also exert a retinal protective effect by inhibiting the PTP1B-ERS axis.52 In addition, ginsenoside Ro can activate the Epac1/AMPK pathway to promote mitophagy; whereas the hyperglycemic environment can activate the PKCδ/Drp1 signaling pathway, which on the one hand inhibits PINK1/Parkin-mediated mitophagy, and on the other hand promotes the dissociation of hexokinase II (HK-II) from mitochondria, exerting a dual effect that hinders the clearance of damaged mitochondria.53 Meanwhile, hyperglycemia can also induce excessive mitochondrial fission by activating the PKCδ/Drp1 signaling pathway, further exacerbating mitochondrial homeostasis imbalance; in this process, Drp1 can promote the dissociation of HK-II from mitochondria, further weakening the HK-II-PINK1-mediated mitophagy pathway.84 The above-mentioned mitochondrial homeostasis imbalance process exacerbates retinal vascular endothelial cell apoptosis and vascular lesions, suggesting that MAMs dysfunction plays a key regulatory role in the development of DR.
Diabetic Peripheral Neuropathy
DPN is currently the most prevalent and severe complication of diabetes, and its pathogenesis is closely associated with Schwann cell injury and axonal degeneration.85 In diabetic peripheral neuropathy, MAMs in Schwann cells mainly show impaired structural integrity, which is distinct from the excessive formation of MAMs observed in diabetic cardiomyopathy. Downregulation of MFN2 results in disrupted ER-mitochondrial tethering, leading to enhanced endoplasmic reticulum stress and excessive mitochondrial fission.86 Studies have shown that in a hyperglycemic environment, abnormal expression of MFN2 impairs the structural stability of MAMs, leading to enhanced PERK-mediated endoplasmic reticulum stress, dysregulated IP3R-controlled calcium signaling, and Drp1-mediated excessive mitochondrial fission. These perturbations further trigger oxidative stress responses, energy metabolism disorders, and apoptosis in Schwann cells, ultimately exacerbating nerve damage and demyelinating lesions.77 Tang Bi Formula (TBF) has been shown to stabilize the balance between mitochondrial fission and fusion, inhibit Schwann cell apoptosis and oxidative stress responses by activating the AMPK-PGC-1α-MFN2 signaling pathway, thereby improving mitochondrial function and promoting the repair of nerve injury.86 In addition, inhibition of Drp1-mediated excessive mitochondrial fission can alleviate calcium overload and oxidative stress in the MAMs region, exerting a protective effect on peripheral nerve function.87 Paeoniflorin (PF), the main active component of Paeonia lactiflora, can enhance mitochondrial protein processing capacity, direct antioxidant activity, and the regulatory function of the mitochondrial interactome by upregulating the expression of mitochondrial thioredoxin 2 (Trx2) in Schwann cells. This consequently improves mitochondrial function under hyperglycemic conditions, reduces ROS production, and enhances mitochondrial membrane potential as well as mtDNA stability.88 The total glucosides of paeony (TGP) capsule has also been shown to upregulate Trx2 system protein expression, alleviate demyelinating lesions, and improve pain threshold and nerve conduction velocity in DPN model rats.88 On the other hand, Compound Qiying Granules (CQYG) exerts a neuroprotective effect by inhibiting endoplasmic reticulum stress and apoptotic pathways. Results of proteomic analysis demonstrated that CQYG can significantly regulate the abnormally expressed proteins in the sciatic nerves of DPN rats, inhibit abnormal endoplasmic reticulum protein processing, excessive activation of the PERK pathway, and activation of apoptosis-related signaling pathways. This further improves the ultrastructure of mitochondria and endoplasmic reticulum, reduces the inflammatory response and apoptosis of Schwann cells, and ultimately promotes the repair and functional recovery of the myelin sheath structure of nerve fibers.89
Diabetic Cardiomyopathy
Diabetic Cardiomyopathy (DCM) is primarily characterized by cardiomyocyte metabolic disorders and progressive cardiac dysfunction. In diabetic cardiomyopathy, MAMs predominantly exhibit excessive formation, which is in sharp contrast to the tissue-specific feature of reduced MAMs number observed in diabetic nephropathy. A high-glucose and high-fat environment promotes excessive MAMs formation by upregulating acid sphingomyelinase (ASMase), leading to mitochondrial calcium overload and cardiomyocyte apoptosis.75 In the pathological progression of DCM, MAMs can exhibit two abnormal states: excessive formation or functional impairment, both of which lead to mitochondrial calcium overload and cardiomyocyte apoptosis. Ferulic acid (FeA), a monophenolic active component derived from various plants, can regulate the PACS2/IP3R2/FUNDC1/VDAC1 signaling pathway of MAMs, inhibit hyperglycemia-induced MAMs structural abnormalities, restore the balance of mitochondrial biogenesis and dynamics, and improve oxidative phosphorylation function and ATP synthesis efficiency. Consequently, it significantly suppresses mitochondria-mediated cardiomyocyte apoptosis and alleviates cardiomyocyte injury.90 Brain and muscle Arnt-like protein 1 (Bmal1), as a core circadian clock transcription factor, can directly bind to and promote the transcription of the Bcl2 gene, thereby enhancing the inhibitory effect of Bcl2 on IP3R. This action reduces MAMs-mediated Ca2+ release, inhibits mitochondrial calcium overload, further improves mitochondrial function, reduces ROS accumulation and cell apoptosis, and ultimately alleviates cardiac hypertrophy, fibrosis and diastolic dysfunction in diabetic cardiomyopathy.76 Protein kinase RNA-like endoplasmic reticulum kinase (PERK) is a key sensor of endoplasmic reticulum stress that is localized on MAMs. Through its dominated signaling pathway (PERK-ATF4-CHOP), PERK plays a central role in ROS-mediated ER stress-induced cardiomyocyte apoptosis, thus significantly accelerating the development of cardiac systolic and diastolic dysfunction in DCM.91
Diabetic Encephalopathy
Diabetic Encephalopathy (DE) is mainly clinically manifested as cognitive decline, and the vulnerability of hippocampal neurons is a key link in its pathogenesis. Calcium signal transduction disorders and mitochondrial dysfunction caused by MAMs structural damage are important molecular bases for the occurrence of DE. In diabetic encephalopathy, MAMs in hippocampal neurons mainly present impaired structural integrity (increased contact distance and decreased contact area), which is distinct from the hyper-coupling of MAMs observed in diabetic retinopathy.70 Furthermore, Lipin1 can enhance IP3R-GRP75-VDAC1-MCU-mediated Ca2+ transfer by maintaining the integrity of MAMs, improve mitochondrial calcium homeostasis and redox balance, thereby alleviating hyperglycemia-induced MAMs structural abnormalities, mitochondrial dysfunction, synaptic plasticity damage, and cognitive deficits in DE.70
Diabetic Liver Injury
Diabetes is often accompanied by the occurrence of Non-Alcoholic Fatty Liver Disease (NAFLD). In liver tissue, MAMs are key sites for coordinating lipid synthesis, transport, and oxidative metabolism. In diabetes-associated liver injury, MAMs dysfunction is mainly manifested as impaired lipid transport, which differs from the pathological mechanism dominated by calcium overload in cardiac and retinal tissues. The efficiency of MAMs-mediated phosphatidylserine transport from the endoplasmic reticulum to mitochondria decreases, leading to insufficient mitochondrial cardiolipin synthesis and subsequent oxidative phosphorylation dysfunction.92 Under the hyperglycemic state of diabetes, dysfunction of hepatic MAMs leads to decreased efficiency of lipid transport from the endoplasmic reticulum to mitochondria, thereby exacerbating intracellular lipid accumulation and oxidative stress response in hepatocytes. Studies suggest that SIRT1 can maintain the structural stability of MAMs by inhibiting MDM2 expression and disrupting its interaction with MAMs resident proteins, reduce IP3R1-GRP75-VDAC1-mediated excessive Ca2+ transfer, improve mitochondrial calcium homeostasis and redox balance, thereby alleviating high-calorie intake-induced MAMs abnormal enrichment, mitochondrial dysfunction, hepatic steatosis, and metabolic disorders in NAFLD.93 Melatonin can maintain MAMs structural integrity by binding to IP3R and downregulating its expression, reduce IP3R-GRP75-VDAC1-mediated excessive Ca2+ transfer, and then promote autophagy by activating the AMPK/mTOR pathway, thereby alleviating lipid load-induced MAMs abnormal enrichment, hepatic lipid accumulation, and metabolic disorders in NAFLD.94 Dimethyl fumarate (DMF), a methyl ester derivative with anti-inflammatory and antioxidant properties, can inhibit high-energy stimulation-induced excessive MAMs enrichment and abnormal expression of IP3R1/GRP75/VDAC1/MCU1 proteins by activating the SIRT1 signaling pathway, reduce mitochondrial calcium overload and oxidative stress, improve mitochondrial respiratory chain function and membrane potential, thereby significantly inhibiting lipid accumulation and alleviating non-alcoholic fatty liver disease.95
Diabetic Foot Ulcers
In diabetic foot ulcers (DFU), MAMs are predominantly characterized by excessive formation, a feature similar to that in diabetic cardiomyopathy, but distinct from the pattern of reduced MAMs quantity in diabetic nephropathy. Excessive MAMs formation mediated by IP3R1/GRP75/VDAC1 leads to endothelial cell senescence and impaired angiogenesis.78
DFU are often accompanied by an abnormal increase in extracellular mitochondria-derived vesicles (MDVs). In DFU tissues, MDVs are key mediators affecting mitochondrial function and oxidative stress response. Under the hyperglycemic state of diabetes, MDV secretion increases under the regulation of SNX9, which promotes fibroblast apoptosis, exacerbates intracellular oxidative stress, disrupts mitochondrial structure, and inhibits aerobic metabolism. Studies suggest that SNX9 inhibitors can block MDV generation and its mediated mitochondrial dysfunction, restore mitochondrial dynamic balance and redox homeostasis, thereby improving hyperglycemia-induced abnormal MDV increase, mitochondrial damage, oxidative stress, and delayed wound healing in diabetic mouse models.96 Histatin 1 (Hst1) is a 38-amino acid histidine-rich short peptide in human saliva; it can inhibit IP3R1/GRP75/VDAC1-mediated excessive formation of MAMs by activating ERK-mediated Nrf2 nuclear translocation, reduce mitochondrial calcium overload and oxidative stress, improve mitochondrial function and membrane potential, thereby significantly inhibiting endothelial cell senescence, promoting angiogenesis, and accelerating diabetic wound healing.78 Future research needs to further explore whether traditional wound-healing medicinal materials directly regulate MAMs function in wound cells to promote wound healing (Table 3).
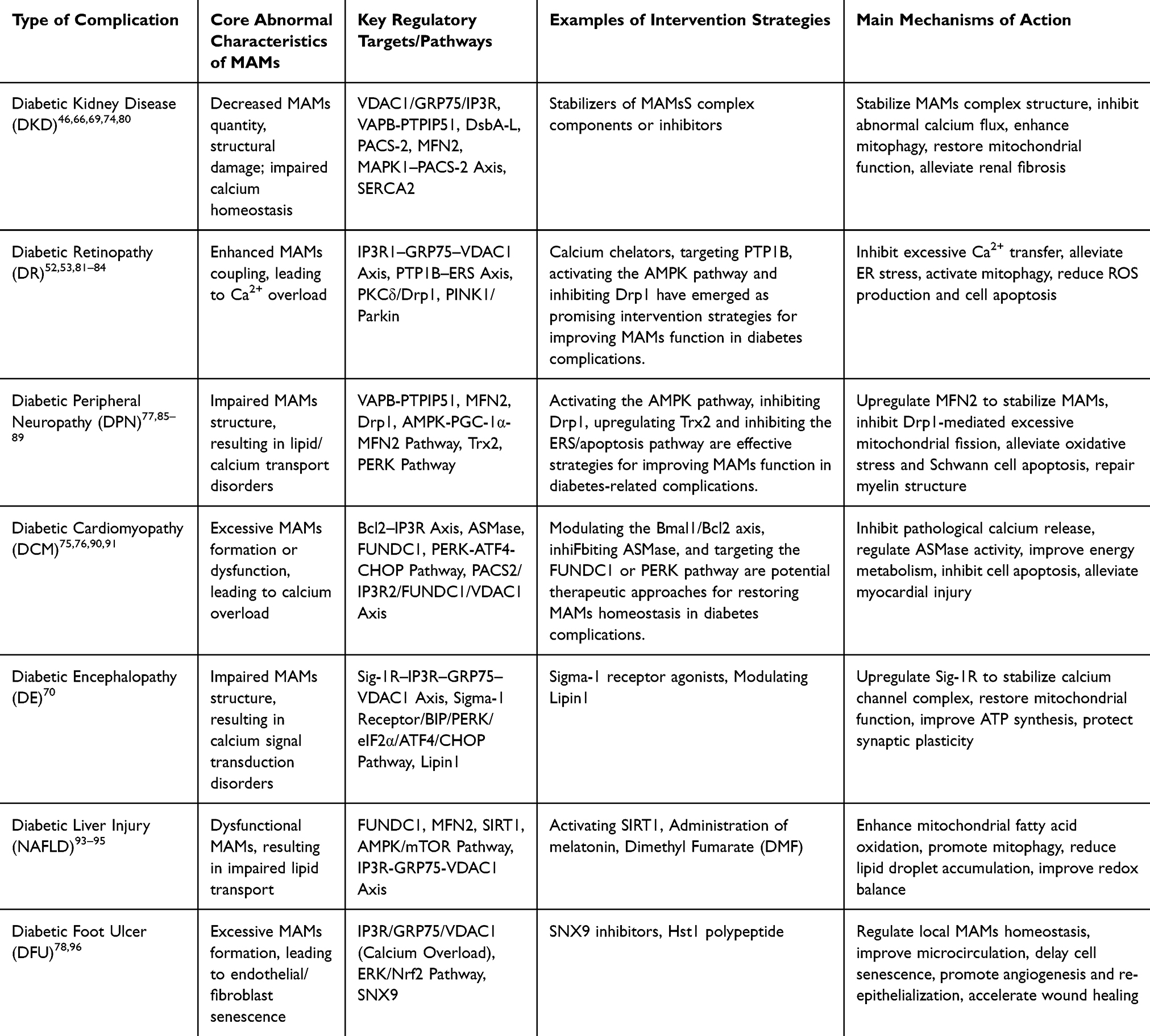 |
Table 3 Experimental Evidence of Targeting MAMs in Diabetic Complications |
Conclusion
Although current studies have fully established the central role of MAMs in diabetic complications, this field still faces a series of critical unresolved issues, especially the dynamic evolutionary patterns of MAMs during disease progression and the translational feasibility of precise regulatory strategies.
First, MAMs undergo temporal dynamic changes during disease progression, making the timing of intervention crucial. Studies have shown that the structure and function of MAMs are not static, but are dynamically remodeled according to the duration and intensity of pathological stimuli. In the early stage of disease, cells may enhance MAMs formation to compensatory maintain endoplasmic reticulum-mitochondrial communication, which represents a protective adaptive response. However, when pathological stimuli such as high glucose and high lipids persist and exceed cellular compensatory capacity, MAMs may shift toward structural disruption or functional hyperactivity, ultimately acting as a “catalyst” amplifying injury signals.97 Recent studies further confirm that in early diabetic cardiomyopathy, increased MAMs contacts may represent a compensatory response, whereas sustained calcium overload eventually leads to cardiomyocyte apoptosis as the disease progresses.75 Therefore, understanding the temporal dynamic evolutionary patterns of MAMs is clinically significant for identifying the optimal intervention window.
Second, nonselective modulation of MAMs carries potential risks, and precise regulatory strategies urgently need to be established. As a hub integrating multiple signaling pathways, MAMs function is bidirectional: appropriate MAMs activity is essential for maintaining cellular homeostasis, whereas either excessive enhancement or inhibition of MAMs function can trigger severe consequences. On the one hand, excessive MAMs formation induces mitochondrial calcium overload, reactive oxygen species burst, and cell apoptosis;98 on the other hand, complete ablation of MAMs structure may disrupt material exchange and signal transduction between the endoplasmic reticulum and mitochondria, leading to energy metabolic disorders and insulin resistance.99 Thus, an ideal therapeutic strategy should be “precise regulation” rather than “complete activation or inhibition”—that is, targeting specific protein complexes or signaling pathways of MAMs to restore functional homeostasis rather than simply enhancing or blocking them.
Third, the clinical translation of MAMs research faces multiple challenges. At present, MAMs-targeted intervention strategies remain largely at the preclinical stage, with considerable progress needed before clinical application. Specific challenges include: (1) Target specificity and delivery efficiency: Designing small molecules or nanoformulations that precisely target MAMs while avoiding nonspecific effects on other membrane structures represents a key technical hurdle for clinical translation; (2) Systematic elucidation of mechanisms: Multi-omics technologies such as proteomics, lipidomics, and spatial transcriptomics are required to comprehensively map the regulatory landscape of drug effects on MAMs protein interaction networks and local lipid microenvironments, and to identify core therapeutic targets;71 (3) Efficacy evaluation and biomarkers: There is an urgent need to establish a biomarker system based on MAMs functional status to objectively assess intervention effects, and to validate safety and efficacy in diabetic patients through well-designed clinical trials;21 (4) Precise matching with disease stages: Given that MAMs may exhibit opposite patterns at different disease stages, future therapeutic strategies require personalized design based on individual disease stages to avoid a “one-size-fits-all” intervention model.
In summary, as a critical hub connecting the endoplasmic reticulum and mitochondria, MAMs play a central role in the development and progression of diabetic complications. MAMs-targeted interventions have opened new avenues for the preclinical treatment of diabetic complications. Nevertheless, it must be emphasized that these strategies remain at an experimental stage and are not yet ready for clinical application. However, translating mechanistic research into clinical application still requires in-depth elucidation of the dynamic evolutionary patterns of MAMs during disease progression, establishment of precise regulatory strategies, and overcoming technical challenges including targeted delivery and efficacy evaluation. Integrating the concept of “holistic regulation” with modern cell biology research is expected to drive the development of more comprehensive and effective preclinical prevention and treatment strategies, providing novel experimental options for the large global population with diabetes, although further validation is required before clinical application.
Data Sharing Statement
Data sharing is not applicable to this article as no data were created or analysed in this study.
Author Contributions
Jianmei Shi: Conceptualization, Investigation, Formal analysis, Writing–original draft. Xiaohan Sun: Conceptualization; Investigation; Data curation; Writing-original draft. Jiajun You: Validation; Data curation; Writing-review & editing. Xiaoxu Ji: Investigation; Resources; Writing-review & editing. Juanmei Xie: Resources; Validation; Writing-review & editing. Yuxuan Li: Visualization; Writing-review & editing. Kailei Gu: Writing-review & editing; Investigation. Xiaojie Wei: Conceptualization; Supervision; Project administration; Funding acquisition; Writing – review & editing. All authors gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Natural Science Foundation of China (No. 82560873), the Platform Building “Action Plan” Guangxi Science and Technology Innovation Platform Program (Guangxi Science LT2600640032), the third batch of “Qihuang Project” High-level Talent Team Cultivation Project Funded by Guangxi University of Chinese Medicine (202402) and the innovation training program for college students (202510600027, X202510600055, X202510600011).).
Disclosure
The author(s) report no conflicts of interest in this work. The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Ong KL, Stafford LK, Mclaughlin SA, et al. Global, regional, and national burden of diabetes from 1990 to 2021, with projections of prevalence to 2050: a systematic analysis for the Global Burden of Disease Study 2021. Lancet. 2023;402(10397):203–22.
2. Jia G, Bai H, Mather B, et al. Diabetic vasculopathy: molecular mechanisms and clinical insights. Int J Mol Sci. 2024;25(2):804. doi:10.3390/ijms25020804
3. Li L, Wu Y-Q, Yang J-E. Stress-related LncRNAs and their roles in diabetes and diabetic complications. Int J Mol Sci. 2025;26(5):2194.
4. Mao H, Chen W, Chen L, et al. Potential role of mitochondria-associated endoplasmic reticulum membrane proteins in diseases. Biochem Pharmacol. 2022;199:115011. doi:10.1016/j.bcp.2022.115011
5. Yang S, Zhou R, Zhang C, et al. Mitochondria-associated endoplasmic reticulum membranes in the pathogenesis of type 2 diabetes mellitus. Front Cell Dev Biol. 2020;8:571554. doi:10.3389/fcell.2020.571554
6. Mannella CA, Buttle K, Rath BK, Marko M. Electron microscopic tomography of rat-liver mitochondria and their interaction with the endoplasmic reticulum. Biofactors. 1998;8:225–228. doi:10.1002/biof.5520080309
7. Zhou H, Wang S, Hu S, et al. ER–mitochondria microdomains in cardiac ischemia–reperfusion injury: a fresh perspective. Front Physiol. 2018;9:755. doi:10.3389/fphys.2018.00755
8. Bui V, Santerre M, Shcherbik N, et al. Mitochondria-associated membranes (MAMs): molecular organization, cellular functions, and their role in health and disease. FEBS Open Bio. 2025;16:11–24. doi:10.1002/2211-5463.70121
9. Liu Y, Mao ZH, Huang J, et al. Mitochondria-associated endoplasmic reticulum membranes in human health and diseases. MedComm. 2025;6(7):e70259.
10. Chen C, Dai G, Fan M, et al. Mitochondria-associated endoplasmic reticulum membranes and myocardial ischemia: from molecular mechanisms to therapeutic strategies. J Transl Med. 2025;23(1):277.
11. Cheng H, Gang X, He G, et al. The molecular mechanisms underlying mitochondria-associated endoplasmic reticulum membrane-induced insulin resistance. Front Endocrinol. 2020;11:592129. doi:10.3389/fendo.2020.592129
12. Guo M, Liu R, Zhang F, et al. A new perspective on liver diseases: focusing on the mitochondria-associated endoplasmic reticulum membranes. Pharmacol Res. 2024;208:107409.
13. Yeo HK, Park TH, Kim HY, et al. Phospholipid transfer function of PTPIP51 at mitochondria-associated ER membranes. EMBO Rep. 2021;22(6):EMBR202051323. doi:10.15252/embr.202051323
14. Liu J, Liu Y, Gao C, et al. The ultrastructural and proteomic analysis of mitochondria-associated endoplasmic reticulum membrane in the midbrain of a Parkinson’s disease mouse model. Aging Cell. 2024;24(4):e14436.
15. Lu X, Gong Y, Hu W, et al. Ultrastructural and proteomic profiling of mitochondria-associated endoplasmic reticulum membranes reveal aging signatures in striated muscle. Cell Death Dis. 2022;13(4):296. doi:10.1038/s41419-022-04746-4
16. Wang JJ, Park KS, Dhimal N, et al. Proteomic analysis of retinal mitochondria-associated ER membranes identified novel proteins of retinal degeneration in long-term diabetes. Cells. 2022;11(18):2819. doi:10.3390/cells11182819
17. Dia M, Givre L, Leon C, et al. Comparative proteomic analysis of cardiac mitochondria-associated reticulum membranes in type 2 diabetes. Cardiovasc Res. 2022;118:cvac066–223. doi:10.1093/cvr/cvac066.223
18. Gomes KP, Jadli AS, De Almeida LGN, et al. Proteomic analysis suggests altered mitochondrial metabolic profile associated with diabetic cardiomyopathy. Front Cardiovasc Med. 2022;9:791700. doi:10.3389/fcvm.2022.791700
19. Zhang S, Shao Y, Su M, et al. The application of different biotechnologies in detecting the changes in MAMs and their classic discoveries. Anal Biochem. 2025;698:115744. doi:10.1016/j.ab.2024.115744
20. Humbert A, Nawrot M, Bendridi N, et al. Endoplasmic reticulum–mitochondria contact sites are signalling hubs connecting nutrient sensing and GLP-1 secretion in L cells of the mouse gut: from physiology to obesity and type 2 diabetes. Diabetologia. 2026;69:2044–2060. doi:10.1007/s00125-026-06693-7
21. Wang Y, Zhang Y, Jin Q, et al. The role of mitochondrial-associated endoplasmic reticulum membranes (MAMs) in diabetic microvascular complications: a review. Cell Death Dis. 2025;17(1):81. doi:10.1038/s41419-025-08236-1
22. Boyman L, Karbowski M, Lederer WJ. Regulation of mitochondrial ATP production: Ca2+ signaling and quality control. Trends Mol Med. 2020;26(1):21–39. doi:10.1016/j.molmed.2019.10.007
23. De Ridder I, Kerkhofs M, Lemos FO, et al. The ER-mitochondria interface, where Ca2+ and cell death meet. Cell Calcium. 2023;112:102743. doi:10.1016/j.ceca.2023.102743
24. Yang M, Li C, Sun L. Mitochondria-Associated Membranes (MAMs): a novel therapeutic target for treating metabolic syndrome. Curr Med Chem. 2021;28(7):1347–1362. doi:10.2174/0929867327666200212100644
25. Li C, Li L, Yang M, et al. PACS-2: a key regulator of mitochondria-associated membranes (MAMs). Pharmacol Res. 2020;160:105080. doi:10.1016/j.phrs.2020.105080
26. Arruda AP, Pers BM, Parlakgül G, et al. Chronic enrichment of hepatic endoplasmic reticulum–mitochondria contact leads to mitochondrial dysfunction in obesity. Nat Med. 2014;20(12):1427–1435. doi:10.1038/nm.3735
27. Mavuduru VA, Vadupu L, Ghosh KK, et al. Mitochondrial phospholipid transport: role of contact sites and lipid transport proteins. Prog Lipid Res. 2024;94:101268. doi:10.1016/j.plipres.2024.101268
28. Thakur M, Tupe RS. Lipoxin and glycation in SREBP signaling: insight into diabetic cardiomyopathy and associated lipotoxicity. Prostaglandins Other Lipid Mediat. 2023;164:106698. doi:10.1016/j.prostaglandins.2022.106698
29. Reyna-Bolaños I, Solís-García EP, Vargas-Vargas MA, et al. Polydatin prevents electron transport chain dysfunction and ROS overproduction paralleled by an improvement in lipid peroxidation and cardiolipin levels in iron-overloaded rat liver mitochondria. Int J Mol Sci. 2024;25(20):11104. doi:10.3390/ijms252011104
30. Cao X, Wang N, Yang M, et al. Lipid accumulation and insulin resistance: bridging metabolic dysfunction-associated fatty liver disease and chronic kidney disease. Int J Mol Sci. 2025;26(14):6962. doi:10.3390/ijms26146962
31. Thongnak L, Pongchaidecha A, Lungkaphin A. Renal Lipid Metabolism and Lipotoxicity in Diabetes. Am J Med Sci. 2020;359(2):84–99. doi:10.1016/j.amjms.2019.11.004
32. Opazo-Ríos L, Mas S, Marín-Royo G, et al. Lipotoxicity and diabetic nephropathy: novel mechanistic insights and therapeutic opportunities. Int J Mol Sci. 2020;21(7):2632. doi:10.3390/ijms21072632
33. Latino D, Venditti M, Falvo S, et al. Steroidogenesis upregulation through mitochondria-associated endoplasmic reticulum membranes and mitochondrial dynamics in rat testes: the role of D-Aspartate. Cells. 2024;13(6):523. doi:10.3390/cells13060523
34. Gordaliza-Alaguero I, Sànchez-Fernàndez-De-Landa P, Radivojevikj D, et al. Endogenous interactomes of MFN1 and MFN2 provide novel insights into interorganelle communication and autophagy. Autophagy. 2024;21(5):957–978. doi:10.1080/15548627.2024.2440843
35. Sloat SR, Whitley BN, Engelhart EA, et al. Identification of a mitofusin specificity region that confers unique activities to Mfn1 and MFN2. Mol Biol Cell. 2019;30(17):2309–2319. doi:10.1091/mbc.E19-05-0291
36. Hoppins S, Edlich F, Cleland MM, et al. The soluble form of bax regulates mitochondrial fusion via MFN2 homotypic complexes. Mol Cell. 2011;41(2):150–160. doi:10.1016/j.molcel.2010.11.030
37. Graef M. A dividing matter: drp1/Dnm1-independent mitophagy. J Cell Biol. 2016;215(5):599–601. doi:10.1083/jcb.201611079
38. Ilamathi HS, Benhammouda S, Lounas A, et al. Contact sites between endoplasmic reticulum sheets and mitochondria regulate mitochondrial DNA replication and segregation. iScience. 2023;26(7):107180. doi:10.1016/j.isci.2023.107180
39. Coscia SM, Thompson CP, Tang Q, et al. Myo19 tethers mitochondria to endoplasmic reticulum-associated actin to promote mitochondrial fission. J Cell Sci. 2023;136(5):jcs260612. doi:10.1242/jcs.260612
40. Yu H, Hong X, Liu L, et al. Cordycepin decreases ischemia/reperfusion injury in diabetic hearts via upregulating AMPK/MFN2-dependent mitochondrial fusion. Front Pharmacol. 2021;12:754005. doi:10.3389/fphar.2021.754005
41. Niu W, Liu X, Deng B, et al. Piezo1 deletion mitigates diabetic cardiomyopathy by maintaining mitochondrial dynamics via ERK/Drp1 pathway. Cardiovasc Diabetol. 2025;24(1):127. doi:10.1186/s12933-025-02625-8
42. Yue R, Yan Z, Zha H, et al. Inhibition of Drp1-mediated mitochondrial fission by P110 ameliorates renal injury in diabetic nephropathy. Int Immunopharmacol. 2025;152:114342. doi:10.1016/j.intimp.2025.114342
43. Zhang M-Y, Zhu L, Zheng X, et al. TGR5 activation ameliorates mitochondrial homeostasis via regulating the PKCδ/Drp1-HK2 signaling in diabetic retinopathy. Front Cell Dev Biol. 2022;9:759421. doi:10.3389/fcell.2021.759421
44. Tang S, Huang M, Wang R, et al. Drp1-dependent mitochondrial fragmentation mediates photoreceptor abnormalities in type 1 diabetic retina. Exp Eye Res. 2024;242:109860. doi:10.1016/j.exer.2024.109860
45. Alim Al-Bari A, Ito Y, Thomes PG, et al. Emerging mechanistic insights of selective autophagy in hepatic diseases. Front Pharmacol. 2023;14:1149809. doi:10.3389/fphar.2023.1149809
46. Dai L, Wang M, Wang B, et al. Yiqi Huoxue recipe ameliorates diabetic nephropathy by mediating VAPB–PTPIP51 complex to activate autophagy and regulate MAMs contact. Front Nutr. 2025;12:1634555. doi:10.3389/fnut.2025.1634555
47. Kulkarni PG, Mohire VM, Waghmare PP, et al. Interplay of mitochondria-associated membrane proteins and autophagy: implications in neurodegeneration. Mitochondrion. 2024;76:101874. doi:10.1016/j.mito.2024.101874
48. Liu J, Wang L, Ge L, et al. Lanthanum decreased VAPB-PTPP51, BAP31-FIS1, and MFN2-MFN1 expression of mitochondria-associated membranes and induced abnormal autophagy in rat hippocampus. Food Chem Toxicol. 2022;161:112831. doi:10.1016/j.fct.2022.112831
49. Thayer JA, Petersen JD, Huang X, et al. A unified mechanism for mitochondrial damage sensing in PINK1-Parkin–mediated mitophagy. EMBO J. 2025;45(1):64–105. doi:10.1038/s44318-025-00604-z
50. He H, Zhang X, Xiong L, et al. A review of FUN14 domain-containing 1 involvement in mitochondrial biological processes and mechanisms across various systems. Cell Biochem Funct. 2025;43(10):e70125. doi:10.1002/cbf.70125
51. Zhu J-Y, Van De Leemput J, Han Z. Promoting mitochondrial dynamics by inhibiting the PINK1–PRKN pathway to relieve diabetic nephropathy. Dis Models Mech. 2024;17(4):dmm050471. doi:10.1242/dmm.050471
52. Xiao T, Zhi Y, Tian F, et al. Ameliorative effect of black raspberry anthocyanins on diabetes retinopathy by inhibiting axis protein tyrosine phosphatase 1B-endoplasmic reticulum stress. J Funct Foods. 2023;107:105696. doi:10.1016/j.jff.2023.105696
53. Liu J, Zhang Y, Xu X, et al. Ginsenoside Ro prevents endothelial injury via promoting Epac1/AMPK- mediated mitochondria protection in early diabetic retinopathy. Pharmacol Res. 2025;211:107562. doi:10.1016/j.phrs.2024.107562
54. Cao Y, Chen Z, Hu J, et al. MFN2 regulates high glucose-induced MAMs dysfunction and apoptosis in podocytes via PERK pathway. Front Cell Dev Biol. 2021;9:769213. doi:10.3389/fcell.2021.769213
55. Ju S-M, Jo Y-S, Jeon Y-M, et al. Phosphorylation of eIF2α suppresses cisplatin-induced p53 activation and apoptosis by attenuating oxidative stress via ATF4-mediated HO-1 expression in human renal proximal tubular cells. IntJ Mol Med. 2017. doi:10.3892/ijmm.2017.3181
56. Yuan M, Gong M, He J, et al. IP3R1/GRP75/VDAC1 complex mediates endoplasmic reticulum stress-mitochondrial oxidative stress in diabetic atrial remodeling. Redox Biol. 2022;52:102289. doi:10.1016/j.redox.2022.102289
57. Yan M, Zhang Y, Niu W, et al. Reactive oxygen species-mediated endoplasmic reticulum stress contributes to osteocyte death induced by orthodontic compressive force. Microsc Res Tech. 2023;86(11):1529–1541. doi:10.1002/jemt.24382
58. Zhang Q, Chen W, Zhang B, et al. Lonp1 and Sig-1R contribute to the counteraction of ursolic acid against ochratoxin A-induced mitochondrial apoptosis. Food Chem Toxicol. 2023;172:113592. doi:10.1016/j.fct.2022.113592
59. Means RE, Katz SG. Balancing life and death: BCL-2 family members at diverse ER–mitochondrial contact sites. FEBS J. 2021;289(22):7075–7112. doi:10.1111/febs.16241
60. Yapryntseva MA, Zhivotovsky B, Gogvadze V. Permeabilization of the outer mitochondrial membrane: mechanisms and consequences. Biochim Biophys Acta Mol Basis Dis. 2024;1870(7):167317. doi:10.1016/j.bbadis.2024.167317
61. Légiot A, Céré C, Dupoiron T, et al. Mitochondria-Associated Membranes (MAMs) are involved in Bax mitochondrial localization and cytochrome c release. Microb Cell. 2019;6(5):257–266. doi:10.15698/mic2019.05.678
62. Iwasawa R, Mahul-Mellier AL, Datler C, et al. Fis1 and Bap31 bridge the mitochondria–ER interface to establish a platform for apoptosis induction. EMBO J. 2010;30(3):556–568. doi:10.1038/emboj.2010.346
63. Breckenridge DG, Stojanovic M, Marcellus RC, et al. Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J Cell Biol. 2003;160(7):1115–1127. doi:10.1083/jcb.200212059
64. Wang C-H, Wei Y-H. Role of mitochondrial dysfunction and dysregulation of Ca2+ homeostasis in the pathophysiology of insulin resistance and type 2 diabetes. J Biomed Sci. 2017;24(1):70. doi:10.1186/s12929-017-0375-3
65. Jabbour L, Nguyen T, Gadet R, et al. The endoplasmic reticulum pool of Bcl-xL dampens the unfolded protein response through IP3R-dependent calcium release. bioRxiv Cell Biol. 2021.
66. Xue M, Fang T, Sun H, et al. PACS-2 attenuates diabetic kidney disease via the enhancement of mitochondria-associated endoplasmic reticulum membrane formation. Cell Death Dis. 2021;12(12):1107. doi:10.1038/s41419-021-04408-x
67. Pei X, Liu J, Wei Y, et al. PINK1 affects mitochondrial oxidative phosphorylation by regulating MFN2 to alleviate diabetic kidney disease. Int Immunopharmacol. 2025;157:114786. doi:10.1016/j.intimp.2025.114786
68. Cheng W, Cai C, Xu Y, et al. The TRIM21-FOXD1-BCL-2 axis underlies hyperglycaemic cell death and diabetic tissue damage. Cell Death Dis. 2023;14(12):825. doi:10.1038/s41419-023-06355-1
69. Li H, Dong A, Liu C, et al. Schisandra chinensis mixture attenuates diabetic kidney disease via VDAC1/Grp75/IP3R-mediated MAMs stabilization and apoptosis-autophagy regulation. Phytomedicine. 2025;147:157164. doi:10.1016/j.phymed.2025.157164
70. Zhuang Z, Huang S, Zhang X, et al. Lipin1 ameliorates cognitive ability of diabetic encephalopathy via regulating Ca2+ transfer through mitochondria-associated endoplasmic reticulum membranes. Int Immunopharmacol. 2025;150:114266. doi:10.1016/j.intimp.2025.114266
71. Guedouari H, Dia M, Geoffray J, et al. Structural and functional characterization of the cardiac mitochondria-associated reticular membranes in the ob/ob mouse model. J Mol Cell Cardiol Plus. 2025;12:100453. doi:10.1016/j.jmccpl.2025.100453
72. Madec AM, Perrier J, Panthu B, Dingreville F. Role of mitochondria-associated endoplasmic reticulum membrane (MAMs) interactions and calcium exchange in the development of type 2 diabetes, inter-organellar Ca2+ signaling in health and disease - Part B. Int Rev Cell Mol Biol. 2021;363:169–202.
73. Yang M, Han Y, Luo S, et al. MAMs protect against ectopic fat deposition and lipid-related kidney damage in DKD patients. Front Endocrinol. 2021;12:609580.
74. Liu S, Han S, Wang C, et al. MAPK1 mediates MAMs disruption and mitochondrial dysfunction in diabetic kidney disease via the PACS-2-dependent mechanism. Int J Bio Sci. 2024;20(2):569–584. doi:10.7150/ijbs.89291
75. Wei Y, Ji Y, Meng J, et al. Acid sphingomyelinase promotes diabetic cardiomyopathy via disruption of mitochondrial calcium homeostasis. Cardiovasc Diabetol. 2025;24(1):272. doi:10.1186/s12933-025-02801-w
76. Zhang N, Yu H, Liu T, et al. Bmal1 downregulation leads to diabetic cardiomyopathy by promoting Bcl2/IP3R-mediated mitochondrial Ca2+ overload. Redox Biol. 2023;64:102788. doi:10.1016/j.redox.2023.102788
77. Fu H, Ou Z, Wang F, et al. Mitochondrial fusion protein 2-modified bone marrow mesenchymal stem cells improved hyperglycemia-induced schwann cell injury via regulating mitochondria-associated endoplasmic reticulum membranes. Neurourol Urodyn. 2025;44(5):1211–1218. doi:10.1002/nau.70067
78. Xian T, Liu Y, Ye Y, et al. Human salivary histatin 1 regulating IP3R1/GRP75/VDAC1 mediated mitochondrial-associated endoplasmic reticulum membranes (MAMs) inhibits cell senescence for diabetic wound repair. Free Radic Biol Med. 2024;225:164–180. doi:10.1016/j.freeradbiomed.2024.09.046
79. Kowluru RA, Mishra M. Therapeutic targets for altering mitochondrial dysfunction associated with diabetic retinopathy. Expert Opin Ther Targets. 2018;22(3):233–245. doi:10.1080/14728222.2018.1439921
80. Yang M, Zhao L, Gao P, et al. DsbA-L ameliorates high glucose induced tubular damage through maintaining MAMs integrity. EBioMedicine. 2019;43:607–619. doi:10.1016/j.ebiom.2019.04.044
81. Liang T, Hang W, Chen J, et al. ApoE4 (Δ272–299) induces mitochondrial-associated membrane formation and mitochondrial impairment by enhancing GRP75-modulated mitochondrial calcium overload in neuron. Cell Biosci. 2021;11(1):50. doi:10.1186/s13578-021-00563-y
82. Li Y, Li H-Y, Shao J, et al. GRP75 modulates endoplasmic reticulum–mitochondria coupling and accelerates Ca2+-dependent endothelial cell apoptosis in diabetic retinopathy. Biomolecules. 2022;12(12):1778. doi:10.3390/biom12121778
83. Liu C, Xiang J, Chen Y, et al. MiR-125a-5p in MSC-derived small extracellular vesicles alleviates Müller cells injury in diabetic retinopathy by modulating mitophagy via PTP1B pathway. Cell Death Discov. 2025;11(1):226. doi:10.1038/s41420-025-02439-3
84. Zhang M-Y, Zhu L, Bao X, et al. Inhibition of Drp1 ameliorates diabetic retinopathy by regulating mitochondrial homeostasis. Exp Eye Res. 2022;220:109095. doi:10.1016/j.exer.2022.109095
85. Abd Razak NH, Idris J, Hassan NH, et al. Unveiling the role of schwann cell plasticity in the pathogenesis of diabetic peripheral neuropathy. Int J Mol Sci. 2024;25(19):10785. doi:10.3390/ijms251910785
86. Yang C, Yu H, Zhou L, et al. Tang Bi formula alleviates diabetic sciatic neuropathy via AMPK/PGC-1α/MFN2 pathway activation. Sci Rep. 2025;15(1):25069.
87. Ben Y, Hao J, Zhang Z, et al. Astragaloside IV inhibits mitochondrial-dependent apoptosis of the dorsal root ganglion in diabetic peripheral neuropathy rats through modulation of the SIRT1/p53 signaling pathway. Diabetes Metab Syndr Obes. 2021;14:1647–1661. doi:10.2147/DMSO.S301068
88. Yang X, Li X, Zhu Y, et al. Paeoniflorin upregulates mitochondrial thioredoxin of schwann cells to improve diabetic peripheral neuropathy indicated by 4D label-free quantitative proteomics. Oxid Med Cell Longev. 2022;2022(1):4775645.
89. Hu Y, Chen C, Liang Z, et al. Compound Qiying Granules alleviates diabetic peripheral neuropathy by inhibiting endoplasmic reticulum stress and apoptosis. Mol Med. 2023;29(1):98. doi:10.1186/s10020-023-00698-3
90. Salin Raj P, Nair A, Preetha Rani MR, et al. Ferulic acid attenuates high glucose-induced MAMs alterations via PACS2/IP3R2/FUNDC1/VDAC1 pathway activating proapoptotic proteins and ameliorates cardiomyopathy in diabetic rats. Int J Cardiol. 2023;372:101–109. doi:10.1016/j.ijcard.2022.12.003
91. Liu ZW, Zhu HT, Chen KL, et al. Protein kinase RNA-like endoplasmic reticulum kinase (PERK) signaling pathway plays a major role in reactive oxygen species (ROS)-mediated endoplasmic reticulum stress-induced apoptosis in diabetic cardiomyopathy. Cardiovasc Diabetol. 2013;12:158. doi:10.1186/1475-2840-12-158
92. Rowe IA, D’amico G. Taking a risk-based approach to testing for liver disease in primary care, a step in the right direction. J Hepatol. 2022;77(2):293–295. doi:10.1016/j.jhep.2022.03.038
93. Zhang R, Zhang Q, Cui Z, et al. Regulatory role of sirtuin-1 by targeting MDM2 to mitochondria-associated membranes formation in the treatment of NAFLD. Cell Commun Signal. 2025;23(1):514. doi:10.1186/s12964-025-02251-7
94. Jin M-Y, Yu H, Wei Y-F, et al. Melatonin regulates accumulation of lipids in the liver via the IP3R on the mitochondria-associated membranes (MAMs). Cell Signal. 2025;136:112109. doi:10.1016/j.cellsig.2025.112109
95. Zhang R, Zhang Q, Cui Z, et al. Dimethyl fumarate improves non-alcoholic fatty liver disease by regulating SIRT1 signal to inhibit MAMs over-enrichment. Eur J Pharmacol. 2025;1000:177693. doi:10.1016/j.ejphar.2025.177693
96. Zhang H, Yan Z, Zhu J, et al. Extracellular mitochondrial-derived vesicles affect the progression of diabetic foot ulcer by regulating oxidative stress and mitochondrial dysfunction. Adv Sci. 2025;12(10):2407574.
97. Luan Y, Guo G, Luan Y, et al. Single-cell transcriptional profiling of hearts during cardiac hypertrophy reveals the role of MAMs in cardiomyocyte subtype switching. Sci Rep. 2023;13(1):8339. doi:10.1038/s41598-023-35464-2
98. Wang J, Yin Z, Wang M, et al. Tunable reconstruction of metal-organic frameworks for advanced electrocatalytic degradation of antibiotics: key role of structural defects and pollutant properties. J Hazard Mater. 2025;494:138456.
99. Wang C-H, Wang C-H, Hung P-J, et al. Disruption of mitochondria-associated ER membranes impairs insulin sensitivity and thermogenic function of adipocytes. Front Cell Dev Biol. 2022;10:965523. doi:10.3389/fcell.2022.965523
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.