")
Back to Journals » International Journal of Nanomedicine » Volume 11
Targeting melanoma with immunoliposomes coupled to anti-MAGE A1 TCR-like single-chain antibody
Authors Saeed M , van Brakel M, Zalba S, Schooten E, Rens J, Koning G , Debets R , ten Hagen TL
Received 10 September 2015
Accepted for publication 2 November 2015
Published 8 March 2016 Volume 2016:11 Pages 955—975
DOI https://doi.org/10.2147/IJN.S96123
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Thomas Webster
The supplementary video shows surface distribution of immunoliposomes.
Views: 757
Mesha Saeed,1 Mandy van Brakel,2 Sara Zalba,1 Erik Schooten,2 Joost AP Rens,1 Gerben A Koning,1,† Reno Debets,2 Timo LM ten Hagen1
1Laboratory of Experimental Surgical Oncology, Section Surgical Oncology, Department of Surgery, Erasmus MC, Rotterdam, the Netherlands; 2Laboratory of Tumor Immunology, Department of Medical Oncology, Erasmus MC Cancer Institute, Rotterdam, the Netherlands
†Dr Gerben A Koning passed away on December 29, 2015
Abstract: Therapy of melanoma using T-cells with genetically introduced T-cell receptors (TCRs) directed against a tumor-selective cancer testis antigen (CTA) NY-ESO1 demonstrated clear antitumor responses in patients without side effects. Here, we exploited the concept of TCR-mediated targeting through introduction of single-chain variable fragment (scFv) antibodies that mimic TCRs in binding major histocompatibility complex-restricted CTA. We produced scFv antibodies directed against Melanoma AntiGEn A1 (MAGE A1) presented by human leukocyte antigen A1 (HLA-A1), in short M1/A1, and coupled these TCR-like antibodies to liposomes to achieve specific melanoma targeting. Two anti-M1/A1 antibodies with different ligand-binding affinities were derived from a phage-display library and reformatted into scFvs with an added cysteine at their carboxyl termini. Protein production conditions, ie, bacterial strain, temperature, time, and compartments, were optimized, and following production, scFv proteins were purified by immobilized metal ion affinity chromatography. Batches of pure scFvs were validated for specific binding to M1/A1-positive B-cells by flow cytometry. Coupling of scFvs to liposomes was conducted by employing different conditions, and an optimized procedure was achieved. In vitro experiments with immunoliposomes demonstrated binding of M1/A1-positive B-cells as well as M1/A1-positive melanoma cells and internalization by these cells using flow cytometry and confocal microscopy. Notably, the scFv with nonenhanced affinity of M1/A1, but not the one with enhanced affinity, was exclusively bound to and internalized by melanoma tumor cells expressing M1/A1. Taken together, antigen-mediated targeting of tumor cells as well as promoting internalization of nanoparticles by these tumor cells is mediated by TCR-like scFv and can contribute to melanoma-specific targeting.
Keywords: cancer, immunotherapy, nanoparticles, cancer testis antigens, HLA-A1 targeting
Introduction
Melanoma patients with metastasized disease are in great need for new treatment modalities as survival rates are generally low. Recently, important progress has been made with the combination of so-called checkpoint inhibitors.1–4 The improved progression-free survival in previously untreated melanoma patients, when an antibody that blocks CTLA-4 combined with an antibody blocking PD-1 are administered, indicates the potential of immunotherapy in melanoma.1 Likewise, adoptive T-cell therapy, in which T-cells are administered after expansion outside the patient, shows promise. An improvement of that approach was achieved with the introduction of a genetically engineered T-cell receptor (TCR) in T-cells followed by expansion and administration. This method of TCR introduction results in T-cells which can be forced to target specific antigens expressed only by cancer cells.5–8 To mimic the TCR function, antibodies and single-chain variable fragments (scFv) were produced showing specific binding and cell kill. Previously, Willemsen et al5 demonstrated T-cell targeting to melanoma cells using the G8 Fab fragment. The use of antibodies has several advantages compared to TCRs. Antibodies are easier to produce, when introduced on T-cells do not recombine with wild-type TCRs, have a higher affinity, and can be used to target nanomaterials.9 Here, we used TCR-mimicking scFvs to target lipid-based nanoparticles, ie, liposomes, specifically to a melanoma peptide in the context of the major histocompatibility complex (MHC).
Among common nanosystems as classified by Torchilin,10 liposomes fall under the classification of nanoparticulate pharmaceutical drug delivery systems. Rigon et al11 discussed in their recent review the application of these nanotechnology-based drug delivery systems as therapy for melanoma. There have been many reported advantages including high drug stability, bioavailability, better tissue/organ distribution, blood retention, minimized toxic effects, and better blood circulation time. Targeted nanoparticle drug delivery as a concept and methods of enhancing the effect in melanoma has been discussed elsewhere.12 Development of multifunctional particles provide an opportunity to increased blood circulation time, better accumulation in tumors, and high efficacy, as shown in previous studies. Liposomes are used as carriers for chemotherapy, immunocytokines, and for instance, siRNA to enhance melanoma treatment efficacy.13 Chen et al13 further outline the use of nanoparticles in detection, diagnosis, and treatment of melanoma. In our study, we focus on liposomes for active targeting to melanoma.
Liposomes are lipid-based hollow nanoparticles which can be used to contain and deliver hydrophobic and hydrophilic compounds. Encapsulation of therapeutic compounds in liposomes dramatically changes their pharmacokinetics: it extends blood residence time and prevents degradation and clearance. For clinical use, and especially in the cancer field, these liposomes are around 100 nm in size and are coated with polyethylene glycol (PEG), which prevents opsonization and recognition by macrophages and further increases circulation time.14–16 Because of liposome size and their capacity to remain in the blood, passive accumulation in tumors is observed, which is believed to result from leaky and permeable vessels in tumors, and is also designated enhanced permeability and retention effect.17 Modified PEGylated liposomes can be further used to target antigens by decorating the liposome surface with antibodies,18–20 which results in increased accumulation in tumor cells as compared to healthy tissue.21 Targeting moieties on liposomes can facilitate prolonged retention of the nanocarrier in the tumor, but may also improve uptake by tumor cells.18,21 Through careful selection of the target and liposome composition, liposomal processing can be manipulated and tuned to need.
The aim of this study was the formulation of immunoliposomes targeting melanoma-specific peptides when expressed by MHC molecules on the surface of tumor cells. All nucleated cells express MHC molecules and present processed protein fragments to be recognized by T-cells through their TCR. This peptide–MHC has been used to generate TCR-like scFvs, which are coupled to the outside of liposomes. We focused on MHC class I to attract CD8+ T-cells, which can inflict direct damage to the cells they attack. Among the group of MHC class I presented peptides, we have selected peptides derived from cancer testis antigens (CTAs) due to the selective expression of certain CTAs by tumors and not healthy cells, except for MHC-negative germ line cells.22–24 CTAs are described to be restricted to human germ line cells, but are also present in certain malignant tumors.22 Melanoma AntiGEn A1, (MAGE A1), was the first CTA against which a cytotoxic T-lymphocyte reaction was discovered by van der Bruggen et al.25 MAGEs are however not restricted to melanoma but are expressed at the protein level by a range of tumors, such as esophageal carcinoma, lung adenocarcinoma, and head and neck carcinoma,22,26,27 making them an ideal target for nanocarrier-based therapy.
In an effort to obtain TCR-like binding molecules, we have selected M1/A1-specific Fab molecules via phage-display library (M1/A1 stands for MAGE A1 presented by HLA-A1 [human leukocyte antigen A1]).28,29 Fab fragments used for this study differ a log-scale in their affinity for M1/A1 (Fab G8 KD =250 nM and Fab Hyb3 KD =14 nM).29 The difference in ligand-binding affinity will allow us to compare the importance of this parameter for multivalent nanoparticles. These Fab fragments were reformatted into scFvs5,29 to facilitate coupling to liposomes. scFv are the smallest format of antibodies that retain a complete antigen binding site, but lack an Fc domain and a complement-activating region.19 Notably, scFv retain binding of antigen with high affinity, but with a potentially reduced immunogenicity.19 It is crucial to use relatively small targeting moieties, since previous studies demonstrated that using whole monoclonal antibodies on nanocarriers to target tumors resulted in rapid clearance from the bloodstream impairing the usefulness of these devices.19,30
Here, we describe the formulation of immunoliposomes that target uniquely expressed M1/A1 antigen on melanoma cells. Escherichia coli bacteria were used to prepare anti-M1/A1 scFvs. These fragments were assessed for their binding of M1/A1-positive cells by flow cytometry. When binding criteria were met, scFvs were coupled to liposomes and subjected to various characterization steps before use on cells. Characterization included quantitative and qualitative tests to determine the total lipid and protein levels, liposome characteristics, and assessment of binding to M1/A1-positive cells. Once validated, immunoliposome batches were released for further in vitro testing of their targeting ability toward melanoma cells. We demonstrate that immunoliposomes coupled to anti-M1/A1 scFvs represent a novel formulation based on TCR mimicry that allows successful targeting to tumor cells.
Materials and methods
Restriction enzymes (NotI and SfiI), NEB buffer, BSA, and Nucleobond Xtra Midi were purchased from Bioke (Leiden, the Netherlands); pABC4 vector was a kind gift by Prof Kontermann (Stuttgart, Germany); DH5α E. coli was purchased from Invitrogen (Leek, the Netherlands); BL21 bacteria was from Cell Biology Department and HB2151 was from Tumor Immunology lab (Eramus MC, Rotterdam, the Netherlands); TG1 electrocompetent cells were purchased from Bio-connect (Huissen, the Netherlands); DNA Clean and Concentrator Kit and Zymoclean Gel DNA Recovery Kit was purchased from Baseclear (Leiden, the Netherlands); tryptone, yeast, NaCl, glucose, sucrose, β-mercaptanol, imidazole, EDTA, L-cysteine, HEPES, choloroform, and methanol from Sigma (Zwijndrecht, the Netherlands); tris glycine and bis-Tris precasted gels, tris-glycine buffer, and MOPS buffer from Novex (Amsterdam, the Netherlands); immobilized metal affinity chromatography (IMAC) Ni2+ columns and AKTA FPLC system from GE Healthcare (Hoevelaken, the Netherlands); phosphate-buffered saline (PBS) and Amicon filters MWCO 10,000 from Millipore (Amsterdam, the Netherlands); dialysis cassettes from Pierce scientific; and tris carboxyethyl phosphine (TCEP) beads and powder and Pierce spin cups cellulose acetate filters from Thermo Scientific (Bleiswijk, the Netherlands). Other chemicals used were of HPLC grade. Horseradish peroxidase (HRP) conjugated anti-HIS-tag antibody was purchased from Santa Cruz (Dallas, TX, USA), mouse anti-HIS-tag antibody was from Santa Cruz, Sanbio (Mountain View, CA, USA) and Abcam (Cambridge, MA, USA), goat anti-mouse PE from Southern Biotech (human ads-PE) (Uithoorn, the Netherlands), 7-AAD from Beckton and Dickinson (Breda, the Netherlands), and mouse anti-M13 antibody and HRP-tagged anti-M13 antibody from GE Healthcare (Leiderdorp, the Netherlands). The lipids, hydrogenated soy L-α-phosphatidylcholine (HSPC), cholesterol, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-PEG2000 (DSPE-PEG2000) were purchased from Lipoid (Ludwigshafen, Germany), and 1,2-distearoyl-sn- glycero-3-phosphoethanolamine-N-26 (ammonium salt) (DSPE-PEG2000Maleimide), 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N- (7-nitro-2-1,3 benzoxadiazol-4-yl) (NBD-PE), phosphatidylethanolamine dioleoyl- sulforhodamine B (Rho-PE) were purchased from Avanti Polar Lipid Inc. (Alabaster, AL, USA) and Nucleopore® Track-etched membranes were purchased from Whatman (Maidstone, UK). The study protocols were approved by the Erasmus MC Institutional Ethical Committee, the Netherlands.
Preparation, purification, and characterization of scFv
scFv G8 and Hyb3 were produced as Fab fragments by Willemsen et al5 by using phage display library, a technique that employs bacteriophages to study protein–protein interactions and connect the proteins to genetic information that encodes them. Fab fragments were reformatted into scFv for targeting T-cells to M1/A1 cells.5,28,29 scFv-DNAs were excised from pBullet vector via the restriction sites SfiI and NotI and cloned into pABC4 vector,31 derived from pAB1. The MYC tag was removed and an additional cysteine code was added behind the histidine tag.31 Upon protein production, the two tags are produced as part of the scFv at the C-terminus. The histidine tag was used to purify scFv from the periplasmic fraction (pf)32 of BL21 bacteria. The pf was subjected to IMAC33 over Ni2+ column. Briefly, all the proteins that hold a histidine tag have affinity for Ni2+ ions and hence get adsorbed in the agarose gel matrix.
Various optimization steps were carried out to obtain the protein, including 1) using three different strains of E coli bacteria including TG1, HB2151, and BL21, 2) growth at 37°C preinduction and utilization of different temperatures (30°C, 35°C and 37°C) postinduction, and 3) conditions such as duration of time after induction (4 hours or overnight). In addition to these parameters, cell compartments such as supernatant, pf, and cytoplasmic fractions (cfs) were analyzed to detect protein content. In previous studies, Messerschmidt et al34 and Baum et al35 used pABC4 vector in combination with TG1 bacteria to produce scFv by growing for 3 hours post induction at room temperature (23°C).36
Protein was produced as described previously by Ruger et al.36 Briefly, bacteria were grown in cultures at 37°C for 2.5 hours until an optical density of 0.6–0.8 was reached at 600 nm. Cultures were then induced with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) and grown at 37°C for an additional 4 hours. Cultures were pelleted at 4,500× g for 10 minutes at 4°C and frozen until further use. Pellets were thawed and resuspended in ice-cold periplasmic preparation buffer (30 mM tris-HCl, 20% sucrose, and 1 mM EDTA [pH 7]).36,37 The cells were lysed with 50 μg/mL lysozyme,37 and the spheroblasts were stabilized with 5 mM MgSO4. Samples were centrifuged at 10,000× g for 15 minutes at 4°C, and supernatants were collected, respun, and passed over 0.45 μm filters. Upon production of the protein, six histidine molecules and a cysteine molecule were present at the C-terminus. The histidine tag was used for purification of the molecule via IMAC, while cysteine was used for coupling to liposomes later. Proteins were purified by IMAC on AKTA system by using Ni+2 columns. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was done to determine the monomer of the produced protein and assess its purity. Concentration was determined either by Nanodrop® (Thermo Scientific) by measuring the absorbance at 280 nm or by using protein assays like Bicinchoninic acid Assay (BCA) and Bradford assay. Fractions with scFv protein were concentrated over Amicon filters (MWCO 10 kDa; Millipore) and kept at 4°C until further use.
Cell cultures
Four patient derived melanoma cell lines were used of which two cell lines, MZ2Mel43 and G43, are M1/A1positive and two cell lines, Mel78 and Mel2A, are M1negative but HLA-A1positive (Figure S1). Tumor cells were maintained in Dulbecco’s Modified Eagle’s Medium (Invitrogen) supplemented with 10% fetal bovine serum, nonessential amino acids, L-glutamine and penicillin–streptavidin solution. APD cells,6,8,38,39 which are an Epstein-Barr virus-transfected immortalized B-lymphoblast cell line, are HLA-A1positive. They were maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum, L-glutamine and penicillin–streptavidin solution. All cells were maintained at 37°C/5% CO2 in a humidified incubator. Cells were passaged upon reaching 80%–90% confluency. All the media had phenol red as a pH indicator.
In vitro binding of scFv
One of the steps in characterizing these scFvs was to evaluate their binding to APD cells and native tumor cells. APD cells were cultured in suspension at 500,000 cells per sample and pulsed with MAGE A1 peptide (10 μg/mL) for 30 minutes at 37°C. Cells were then subjected to a centrifugation at 450× g for 5 minutes, medium was aspirated, and various scFv concentrations ranging from 0.1 to 100 μg/mL in a volume of 200 μL were added to the cells for 1 hour at 4°C. For 500,000 tumor cells per sample, a concentration of 50 and 100 μg/mL of scFv was chosen to be incubated for 1 hour at 4°C. Cells were washed with 2 mL of ice-cold PBS once and incubated for 1 hour at 4°C with anti-HIS antibody (1:20) and then incubated with goat-anti-mouse PE antibody (1:500) for 1 hour at 4°C. Finally, cells were incubated for 10 minutes at room temperature with 10 μL of 7AAD per sample to exclude dead cells. Cells were washed with 2 mL ice-cold PBS and were either measured in 250 μL PBS or fixed in 250 μL of 1% PFA until measured. All samples were measured on BD FACS Canto™ (Becton, Dickinson Company, Franklin Lakes, NJ, USA) with BD FACS Diva software (San Jose, CA, USA) and analyzed using FCS Express (De Novo Software, Los Angeles, CA, USA). Phage-Hyb3 was used as a control and detected using anti-M13 antibody (1:500) and a secondary goat-anti-mouse PE.
Preparation and characterization of liposomes
A neutral liposome composition was chosen for this research; typically HSPC, cholesterol, DSPE-PEG2000, and maleimide-PEG2000 were used in a molar ratio 55:40:4:1.31,40 Liposomes were prepared by film hydration method as previously described.34,37,41,42 Briefly, all lipids were mixed in a round bottom flask and dissolved in chloroform:methanol (9:1 v/v). A thin and uniform lipid film was obtained by evaporating the solvents under pressure for 30 minutes at 42°C. This film was then flushed for 20 minutes under nitrogen and hydrated with 10 mM HEPES buffer at pH 6.7 for 20 minutes at 60°C. If necessary, the film was sonicated with silica beads for complete dissolution. The mixture was then extruded 17–20 times under pressure through a series of polycarbonate filters ranging from 200 to 50 nm to obtain a uniform suspension of particles. Size and polydispersity index (PDI)31,34,41,43 were determined by dynamic light scattering43 on a Zetasizer® Nano ZS (Malvern Instruments, Worcestershire, UK). Liposomes were subjected to Rouser phosphate assay44 where phospholipid was determined and a total lipid amount was calculated after cholesterol correction. For experiments, 0.3% Rhodamine-PE or 0.3% NBD-PE was used as fluorescent probe in either formulation.
Immunoliposome formulation and characterization
Immunoliposomes were produced by postattachment method of coupling,18,45 also known as conventional coupling, to maleimide-PEG liposomes.36,37 Fresh, preformed liposomes were incubated with prereduced scFv. ScFvs were reduced using TCEP (10 mM at a final concentration)34,37 under N2 at room temperature for 1 hour prior to the coupling. We found that reduction of scFv was a necessary step, as we observed greatly reduced binding without reduction, although the majority of scFv was present as monomer (data not shown). TCEP was removed from the mixture by washing five times with deoxygenated coupling buffer (10 mM Na2HPO4/NaH2PO4 buffer, 0.2 mM EDTA, 30 mM NaCl [pH 6.7]) and concentrating at the same time over Amicon filter (MWCO 10 kDa, Millipore) for 10 minutes, after each wash step, at 4,000× g and 4°C. Final concentration was determined using Nanodrop®. An amount of 340 μg of scFv was mixed with 10 μmol of freshly made liposomes for a total of 16 hours. The mixture was overlaid by N2 and left on a shaker incubator for 2 hours at room temperature and then overnight at 4°C without shaking. After 16 hours of total coupling, free maleimide groups were quenched with 1 mM L-cysteine31,34,36,37,42 for 10 minutes at room temperature, and uncoupled scFv was removed by gel filtration over Sepharose CL4B (retailer) column (GE Healthcare),34,36,37,42 pretreated with 10 mM HEPES buffer pH 7.4. The immunoliposomes obtained were subjected to phospholipid determination once more and the total protein coupled was determined by Lowry–Peterson assay.46 The number of scFv molecules attached was calculated using the protein and lipid determinations. Size and charge was determined on the Zetasizer®.
ELISA
A concentration range of 1–100 μg/mL of free scFv and 0.05 to 1 μM of total lipids was tested on ELISA (enzyme-linked immunosorbent assay). Briefly, Nunc 96 (Sanbio, Mountain View, CA, USA) flat well plates with streptavidin coating were washed three times with 0.1% Tween-PBS and twice with PBS and incubated with biotinylated M1/A1 complex (1 μM) for 30 minutes at room temperature or overnight at 4°C. All further steps were carried out at room temperature. Wells were blocked with 2% milk–PBS for 30 minutes. Dilutions of free scFv and immunoliposomes were made in 4% milk-PBS in 100 μL volume and incubated for 1.5 hours in each well after which wells were washed. Anti his-HRP antibody was diluted in 2% milk-PBS, added to the wells, and incubated for 1 hour. Wells were subsequently washed with PBS and 0.1% Tween-PBS and supplemented with ELISA substrate. The reaction was stopped with 1 M H2SO4. As control, w6/32 antibody was used, which only showed a positive signal if the M1/A1 complex bound to the plate successfully. HRP conjugated anti-His tag antibody was used to recognize the His tag on scFv, and substrate development was read with a TECAN ELISA reader (BioTek, Winooski, VT, USA).
In vitro binding of immunoliposomes
APD cells were cultured in suspension at 500,000 cells per mL and pulsed with MAGE A1 peptide (1 μM) for 30 minutes at 37°C. Cells were then subjected to centrifugation at 450× g for 5 minutes, medium was aspirated, and liposomes were added to the cells. A range of concentrations from 0.05 μM to 1 μM total lipid in a volume of 200 μL was added for 1 and 2 hours at 4°C and 37°C. For tumor cells, a concentration of 0.5 μM and 1 μM (total lipid) was chosen to be incubated for 1 and 2 hours at 4°C and 37°C. These cells were washed with ice-cold PBS and incubated for 10 minutes at room temperature with 7AAD to exclude dead cells. Cells were again washed with ice-cold PBS and were either measured in PBS or fixed in 1% PFA. All samples were measured on BD FACS Canto with BD FACS Diva software and analyzed by FCS express software.
Confocal microscopy
For confocal microscopy, liposomes were formulated with Rhodamine PE (λex 560 nm, λem 583 nm). Glass cover slips (25 mm diameter) were coated with either 500 μL of 0.1% collagen37 or 0.1% gelatin for 30 minutes at 37°C and washed with sterile PBS. Tumor cells were harvested and grown on the coated cover slips at a concentration of 0.5×106 in 6-well plates overnight at 37°C in 5% CO2. Next day, cells were incubated with immunoliposomes at a concentration of 1 μM (total lipid) for 2 hours at 37°C, carefully washed with ice-cold PBS, and overlaid with medium at 37°C until they were imaged. Cells were imaged with confocal microscope (Zeiss LSM 510 META, Zeiss, Jena, Germany) with a 40× oil objective lens (NA 1.3) using a 543 nm helium–neon laser and 560–615 band pass filter. Images were analyzed with LSM Image browser (Zeiss).
Statistical analysis
ELISA readings were analyzed using Graphpad Prism software (La Jolla, CA, USA). All data was analyzed using Kruskal–Wallis and Mann–Whitney U tests. Data was processed using GraphPad Prism and SPSS software (version 21) (IBM Corporation, Armonk, NY, USA).
Results
Optimized production of cys-containing and M1:A1-binding scFv
The first step toward obtaining specific immunoliposomes was to produce TCR-mimicking scFv. An additional cysteine group was required at the 3′ end of scFv to facilitate site-directed coupling to liposomes.31,34 As reported by Messerschmidt et al,31 pABC4 vector, derived from pAB1 vector, adds a 3′ cysteine group to scFv inserts, followed by a His tag to enable protein purification.31 Various optimization steps were carried out to successfully produce scFv molecules. The parameters optimized for scFv production included: 1) bacterial strains, 2) duration of bacterial growth, 3) temperature after protein induction, and 4) type of cellular compartments. Bacterial cultures were induced with IPTG and grown either at 30°C, 35°C or 37°C for 4 hours or overnight. We observed an increase in total protein production when bacteria were grown at 37°C for 4 hours. Bacterial strains included TG1, HB2151, and BL21, and pfs of 4 hours cultures of BL21 at 37°C proved to be most optimal with respect to yield of scFv (Figure 1A). The two compartments checked for protein were the pf and cf, where the pf contained the highest protein yield, which was also reported in previous studies.34 pf and cf were run on gel and subsequently assessed by immune staining. A comparison of Hyb3-containing fractions from three bacterial strains grown under similar conditions, pre- and postinduction at 37°C for 4 hours show a dense band depicting higher yield (Figure 1A [i]) from the pf of BL21 bacterial culture. Protein content reached up to 1 mg per liter of BL21 bacterial culture in comparison with a ~250 μg protein yield from HB2151, while there was negligible amount of protein in TG1 cultures (~120 μg). The yield of scFv G8 in the pf of BL21 bacteria grown at 37°C post induction for 4 hours was comparable to Hyb3 production as shown in Figure 1A (ii) on 14% bis-Tris SDS gel.
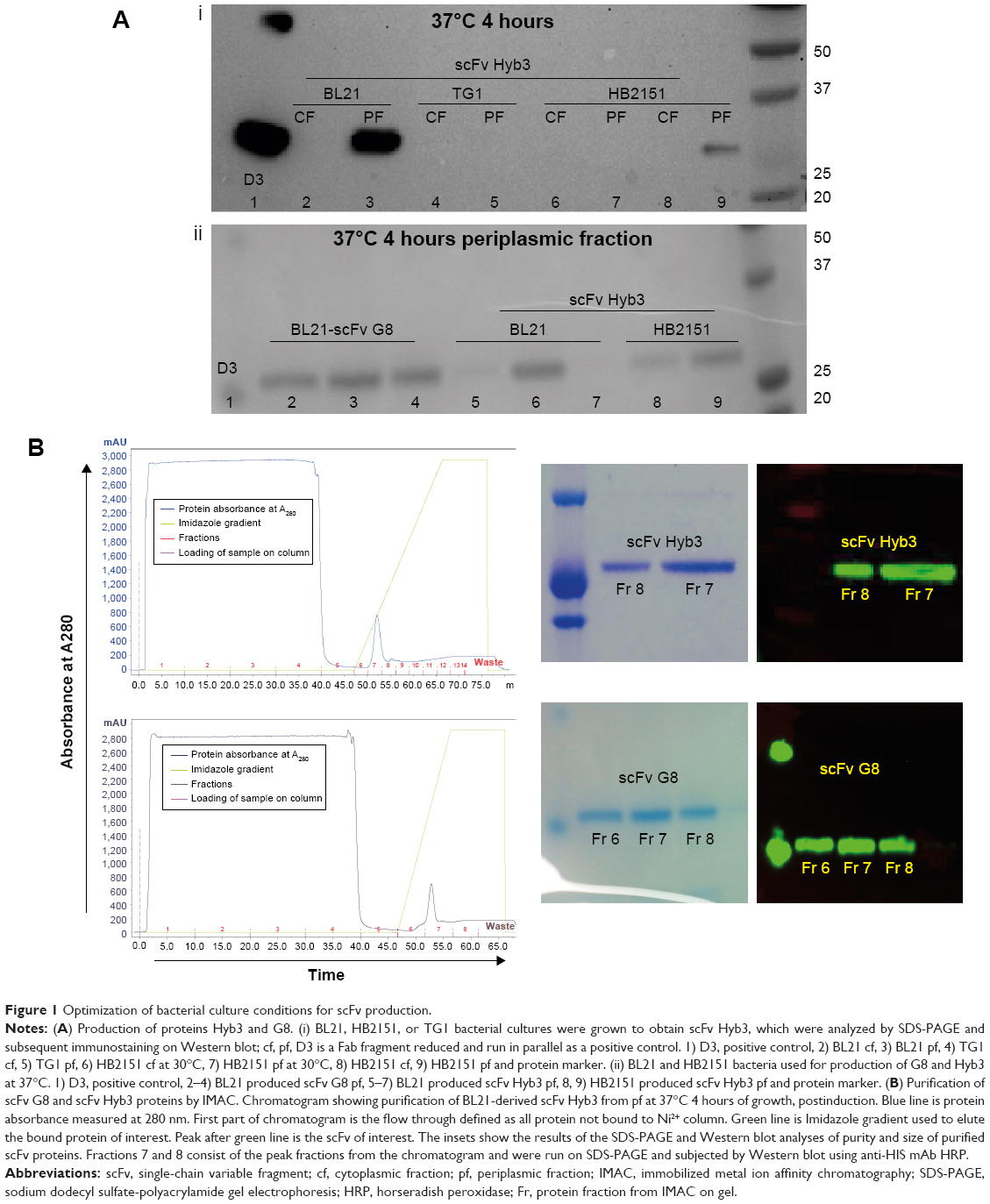 | Figure 1 Optimization of bacterial culture conditions for scFv production. |
Chromatography resulted in good separation, and a single band was detected on SDS-PAGE in the form of a monomer at a molecular mass of 27 kDa (Figure 1B). Collected fractions were pooled, concentrated, and tested for their binding to EBV-transformed B-cells (APD) before proceeding with their coupling to liposomes. B-cells have a high expression of HLA-A1 surface molecules,47 and when pulsed with MAGE A1 peptide (EADPTGHSY), they are recognized by Fab-Hyb3 and G8 as positive targets.28,29 Hyb3 and G8, following their reformatting into scFv and used at a concentration of 10 μg/mL, bound readily to APD cells pulsed with peptide MAGE A1 (Figure 2A). As a positive control, Phage Hyb3 was used. All cells were gated on their respective control, and APD cells without peptide were used as a negative control showing almost no binding of scFvs. A complete shift in the histogram toward the right indicates that the complete population of APD cells interacted with scFvs. At the different concentrations tested, scFv G8 and Hyb3 demonstrated comparable binding to APD cells pulsed with peptide. However, Hyb3 showed a trend toward better binding and specificity toward these cells (Figure 2B). Binding of scFv Hyb3 at lower concentrations is attributed to its affinity maturation and hence higher affinity and sensitivity to recognize the peptide:MHC (pMHC).28 Once scFvs passed this validation, they were subjected to reduction with TCEP, to guarantee efficient availability of the cysteine sulfhydryl, prior to coupling to liposomes.
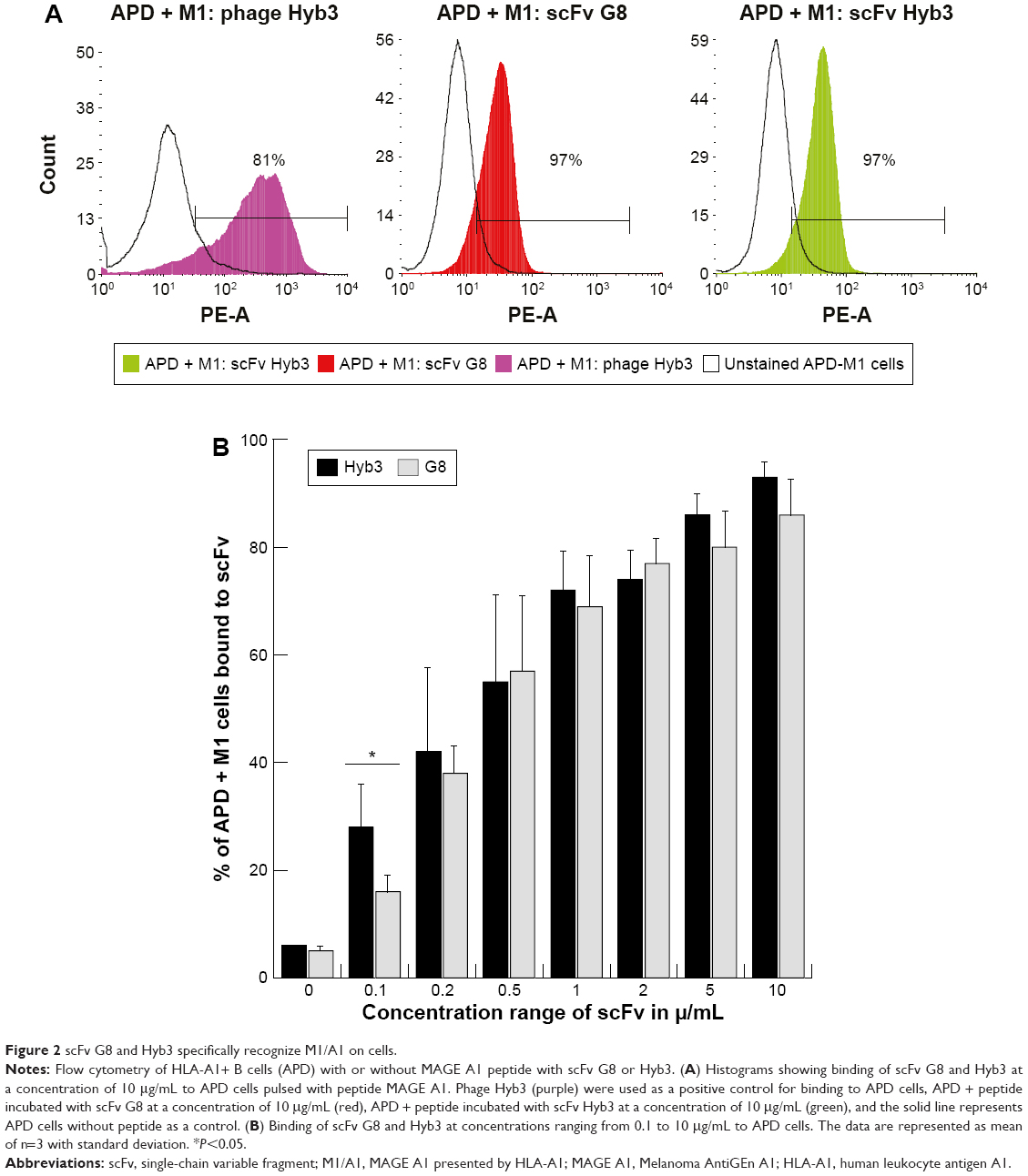 | Figure 2 scFv G8 and Hyb3 specifically recognize M1/A1 on cells. |
Optimized generation of scFv-coupled immunoliposomes and their binding to M1:A1
Liposomes were composed of neutral lipids31,34,35,42 as shown in Table 1, and all formulations were made by the lipid film hydration method.48 Liposomal parameters such as size, PDI, and surface charge were determined. When within-set criteria was size between 80±5 nm before coupling, PDI below 0.1, and surface charge between −12 and −16 mV, liposomes were used for coupling. Initial immunoliposome validations with flow cytometry were done using NBD-PE (0.3%) anchored in the lipid bilayer to determine fluorescence of liposomes after binding to cells. Size of particles was also measured post coupling, and immunoliposomes were only used for subsequent experiments when each batch met the set criteria previously mentioned, but with a size range between 80 and 95 nm. Table 1 summarizes three independent batches of immunoliposomes as an example before and after the coupling out of 16 batches that met the criteria.
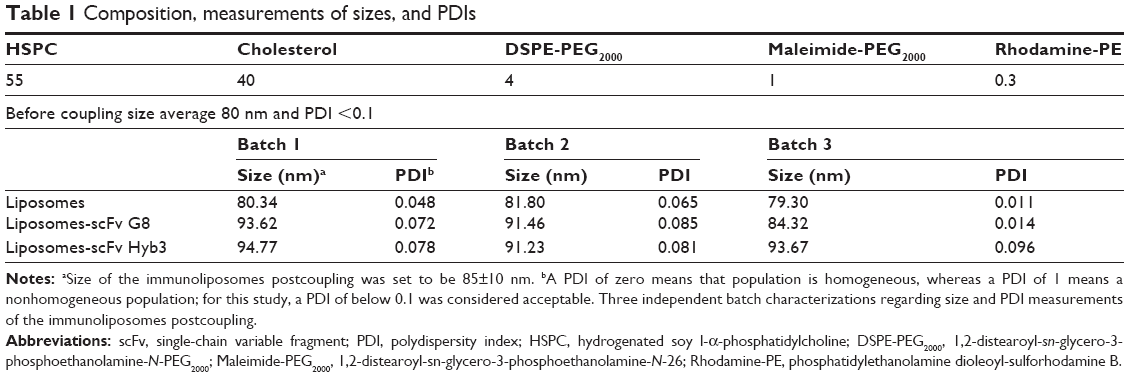 | Table 1 Composition, measurements of sizes, and PDIs |
The generation of immunoliposomes was based on site-directed coupling of scFvs via their C-terminal cysteines to maleimide on the outer end of the PEG molecule. Immunoliposomes were reduced with β-mercaptoethanol and gel-analyzed to determine size change31 before and after coupling36 (Figure 3A). The actual molecular weight of scFv was around 27 kDa; however, after coupling we observed the expected shift to 30 kDa due to attachment of the 3 kDa PEG molecule.31,49 Size change of the scFv after coupling was confirmed by dynamic light scattering, which showed an acceptable increase in the liposome size (around 10 nm) and PDI (Table 1). Quantification of total lipid and protein levels of immunoliposomes resulted in a calculated assessment of the number of scFv (G8 and Hyb3) molecules to be between 13 and 23 per liposomes. For APD binding and confocal experiments, liposomes-scFv G8 (L-scFv G8) had 18, 19 and 21 molecules per liposome and liposomes-scFv Hyb3 (L-scFv Hyb3) had 22, 23 and 20 molecules, whereas for tumor cells binding and confocal experiments L-scFv G8 had 13 and 18 molecules per liposomes and L-scFv Hyb3 had 15 and 22. ELISA was performed for all batches. Batches which had a similar coupling efficiency and number of post coupling molecules of scFv per carrier, were selected for further experiments. After coupling, immunoliposomes were stored in 10 mM HEPES pH 7.4 buffer, under N2 (vacuum), at 4°C in the dark. Postcoupling liposome sizes and PDI did not change in 2 weeks (Table S1).
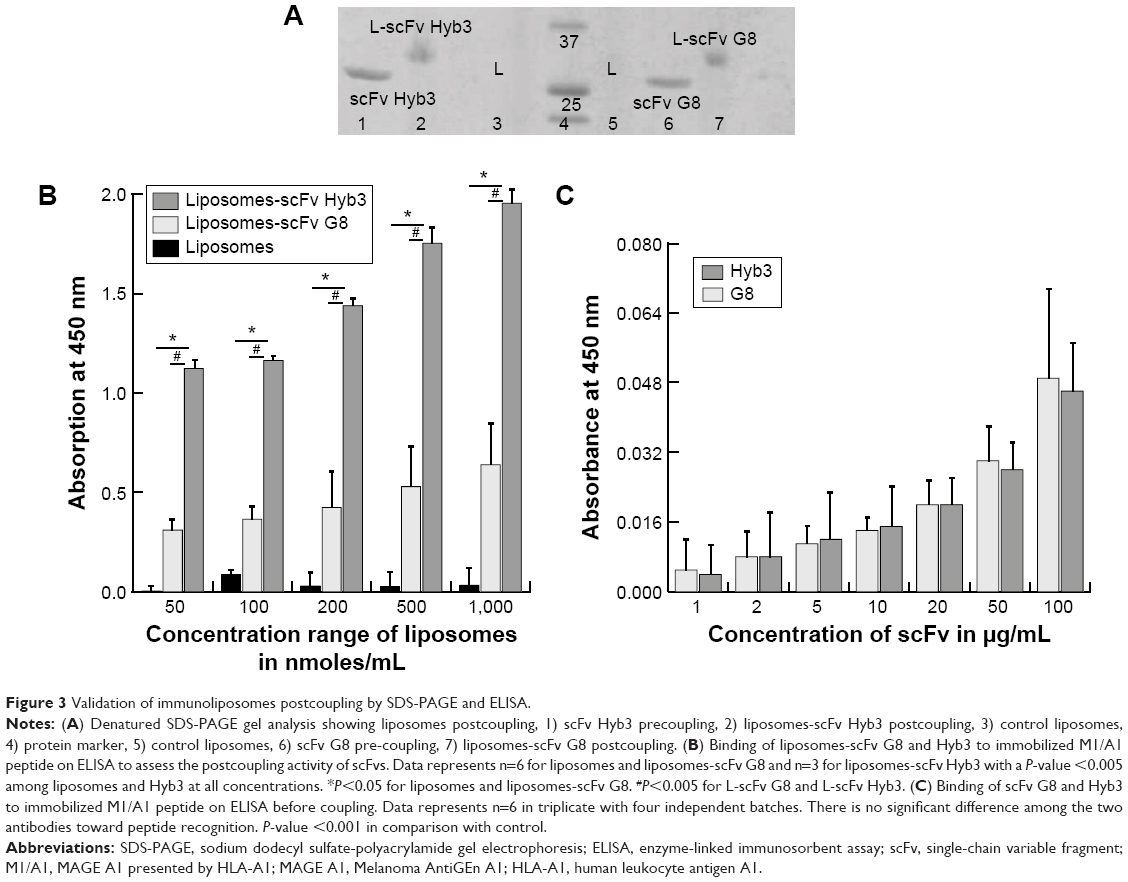 | Figure 3 Validation of immunoliposomes postcoupling by SDS-PAGE and ELISA. |
Immunoliposomes were further subjected to ELISA (Figure 3B). The main aim of testing liposomes-scFv on ELISA was to show whether scFvs remains functional after coupling. It has been observed that certain nanobodies lost functionality after they were coupled with liposomes. Also, it has been reported that PEG molecules can form a mushroom like structure which may cover the antigen binding domain and affect the biological function of scFv molecules. With the ELISA, we prove that scFv remain functional postcoupling to liposomes and bind to immobilized pMHC. A concentration from 0.05 μM to 1 μM based on total lipids shows an increasing signal on ELISA, which was then used to determine a suitable concentration to be used in later experiments. We assessed immunoliposomes binding to immobilized M1/A1 complexes covalently bound to ELISA plates. Nontargeted conventional liposomes show almost no binding, whereas immunoliposomes showed significant and liposome concentration-dependent binding (Figure 3B). Notably, L-scFv Hyb3 showed improved binding compared to L-scFv G8, (P<0.001) indicating a significant difference between the two targeted formulations (Figure 3B). Free scFvs were also tested for pMHC recognition on ELISA. A concentration-related binding was observed, indicating specific and increased binding to pMHC. However, when compared with liposomal scFv, it was negligible, thus showing that liposomal scFv is a more sensitive system in recognizing pMHC complexes. No significant differences were found among free scFvs (Figure 3C).
Following these validations, immunoliposomes were tested for their in vitro binding to cells. To this end, they were incubated with APD cells pulsed with MAGE A1 peptide. Flow cytometry revealed minimal binding of nontargeted liposomes (6%), whereas targeted immunoliposomes (L-scFv Hyb3 70%, L-scFv G8 84%) bound to a greater extent at 4°C after 1 and 2 hours (Figure 4A and B).
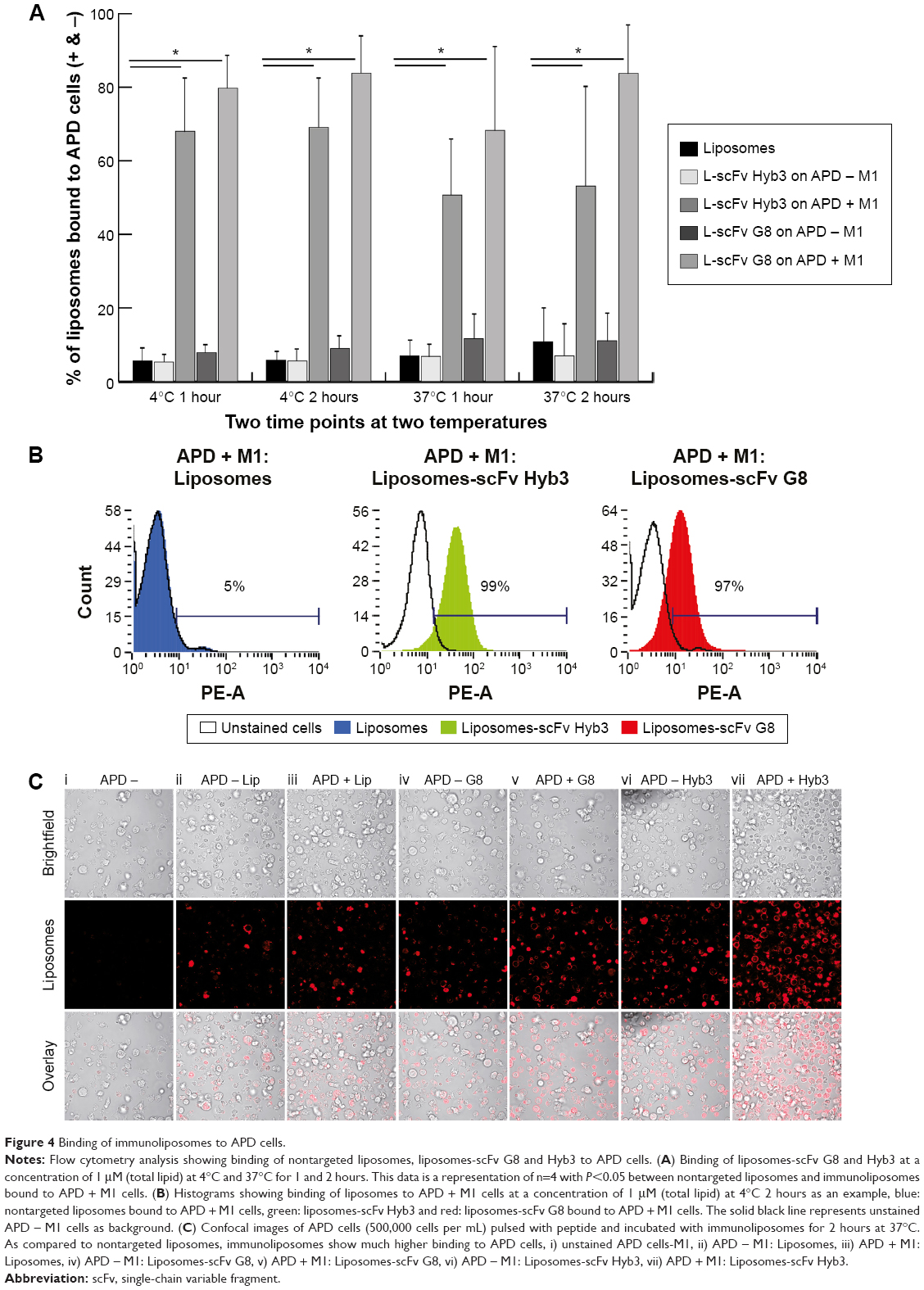 | Figure 4 Binding of immunoliposomes to APD cells. |
When liposome-scFv Hyb3 was used, binding to APD control cells (APD cells without the M1 peptide) was observed at 4°C and 37°C after 1 or 2 hours, which we designated off-target binding (Figure S2). This binding (56%) was not as profound as when the peptide M1 was added (100%), but was higher than binding of control liposomes or cell only (5%) (Figure 4B, Figure S2). When liposome-scFv G8 was added to APD cells without peptide M1, these APD cells remained negative (9%) when compared to experiments using nontargeted liposomes or cells only (5%). Addition of the M1 peptide to APD cells resulted in specific binding of the liposome-scFv G8 (97%). It is hypothesized that this off-target binding of L-scFv Hyb3 to antigen-positive B-cells may be attributed to affinity maturation of scFv Hyb3.
We subsequently investigated cellular internalization of fluorescently-labeled immunoliposomes by confocal microscopy. These experiments demonstrated that immunoliposomes are distributed on the surface of antigen-positive B-cells (Figure 4C), and some surface distribution was also seen on antigen-negative cells, which is in line with our flow cytometric data (Figure 4A). Minimal binding to APD cells was also observed with nontargeted liposomes. The 3D projection of these cells confirms surface distribution of immunoliposomes (Video S1 and S2). Binding of immunoliposomes to APD cells was observed but internalization was not evident in agreement with reports by others. Mommaas et al,47 showed that B-cells present antigen but there is no antigen mediated uptake by these cells which suggest B-cells do not internalize through MHC class I molecule.50 Also, the present study suggests that there is no MHC-mediated endocytosis in B-cells when incubated with scFv-coupled liposomes. As discussed earlier, L-scFv Hyb3 show some off-target binding with APD cells without peptide (Figure 4C); however, positive cells show higher signal in accordance with our flow cytometry data (Figure 4A). From ELISA and flow cytometry results, we concluded that activity of scFv after coupling to liposomes was preserved up to 1 week of storage (Figure S3).
scFv-coupled immunoliposomes specifically target and are internalized by M1/A1-positive melanoma cells
Following experiments with antigen-high APD cells, we studied in vitro binding of immunoliposomes to antigen-low tumor cells, which included two M1/A1-positive and two negative cell lines (assessed for MAGE A1 and HLA-A1 expression by polymerase chain reaction (Figure S1). The flow cytometric binding experiments were conducted at 4°C and 37°C for 1 and 2 hours. Minimal binding was seen with nontargeted liposomes to all cell lines, however L-scFv Hyb3 bound 8 times greater to melanoma cells MZ2Mel43 and G43 (Figure 5A and B), while binding was much lower to negative melanoma cells Mel78 and Mel2A. The results also demonstrate that L-scFv Hyb3 showed (in contrast to findings with APD cells) improved binding compared to L-scFv G8, most likely due to the more physiological low surface expression of antigen on melanoma cells. Again, some off-target recognition was observed using antigen-negative melanoma with L-scFv Hyb3. Strikingly, binding at 37°C was greatly reduced in antigen-positive melanoma cell lines for both immunoliposomes (~6 times lower), which we attribute to the internalization and processing of these immunoliposomes as shown in Figure 5C.
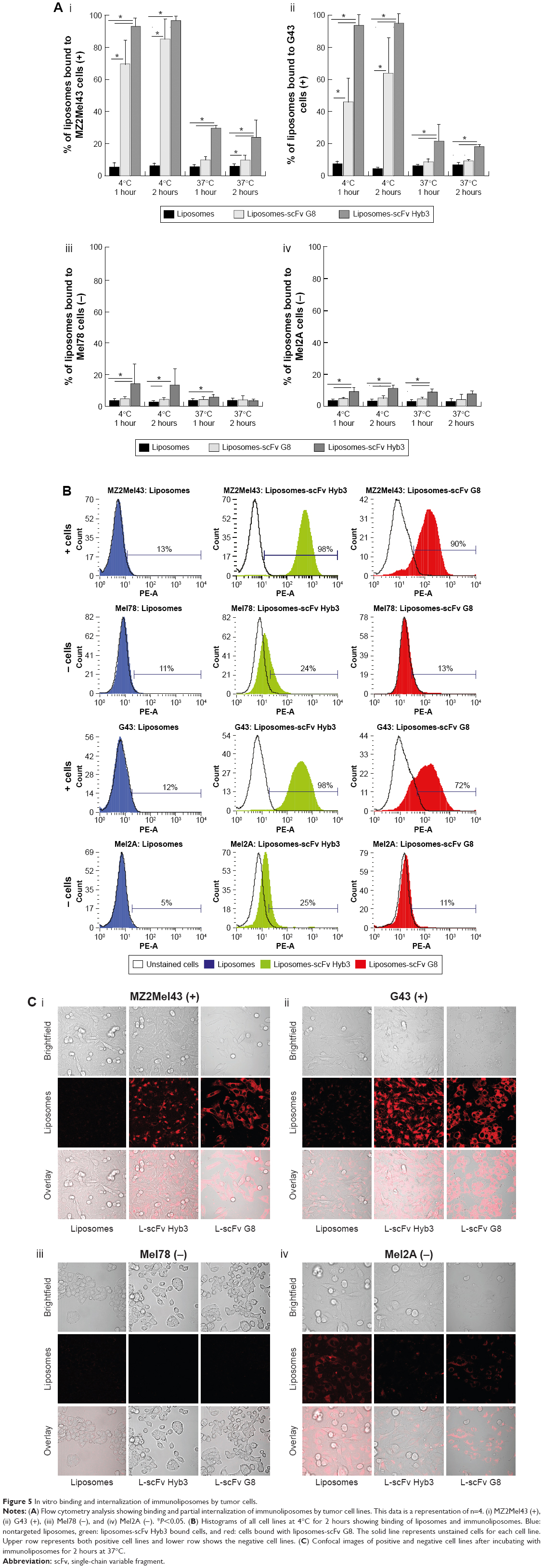 | Figure 5 In vitro binding and internalization of immunoliposomes by tumor cells. |
To complete our analysis, immunoliposomes were further tested on melanoma cells and imaged with confocal microscopy to visualize binding and internalization at 37°C for 2 hours (Figure 5C). M1/A1-positive melanoma cells internalized L-scFv Hyb3 and L-scFv G8 rapidly, while internalization by M1/A1-negative melanoma cells was negligible (Figure 5C), corresponding with the flow cytometry results. Moreover, M1/A1-negative melanoma cells hardly bound nor internalized both nontargeted liposomes as well as L-scFv Hyb3 and L-scFv G8.
Discussion
Several antibody-mediated immunotherapies are in development or already approved. In spite of the relative success, most patients with metastasized melanoma do not respond to therapy or will progress under treatment. Therefore, it is essential that new therapies are developed, which need to be specific, can be dosed repeatedly, and have as little side effect as possible. A highly specific process is TCR-mediated targeting of cancer cells. Previously, we showed specific targeting with TCR-engineered T-cells using G8 Fab fragment to M1/A1-positive melanoma cells, which induces antigen-specific killing of melanoma cells.5 These T-cells were capable of inducing cytokine production, such as TNFα, IL-2, and INFγ, in targeted cells. A study by Willemsen et al5 showed the successful production of MHC Class I specific T-cells grafted with TCR αβ after retroviral transduction. Although forced preferred pairing of the TCR, to prevent unwanted mispairing, provides a strategy to increase efficacy and safety of retargeted T-cells,6 using a mimicking antibody solves TCR mismatch and potential self-reactivity. Using the TCR as a model, we proposed to decorate liposomes with TCR-mimicking scFv to target melanoma cells. This introduces the possibility of establishing a clean and tunable system for therapy.
In this study, we show that novel TCR-like scFvs, and the high- and low-affinity Hyb3 and G8 specific for M1/A1, were successfully produced, characterized, and coupled to nanoparticles, ie, liposomes, to produce targetable devices, also called immunoliposomes, that allow in vitro targeting of antigen-low melanoma cells. To acquire scFv-based immunoliposomes which are of consistent quality and display specific binding, the production of scFv had to be optimized by testing several bacterial strains, growth time, temperature postinduction, and bacterial compartments to yield a better quality and quantity protein. Finally, the pf of BL21 bacteria was used for protein production at 37°C for 4 hours to obtain both proteins. Previously, production of scFv by growing bacterial culture at room temperature (23°C) for 3 hours postinduction has been described, while others used the pABC4 vector, but the same conditions to produce scFv.35 Importantly, our optimization results in a better yield of the antibodies using pABC4 vector in combination with BL21 bacteria and growing cultures at 37°C for 4 hours.
As reported in previous studies, scFv molecules retain complete antigen binding while the Fc domain is missing. Removal of the Fc domain renders these fragments less immunogenic, and no Fc receptor-mediated signaling and complement activation occurs. Previous studies demonstrated that interaction of macrophages with whole mAb coupled to liposomes is Fc domain mediated.51 Likewise, Kupffer cells in vitro interact with antibody decorated liposomes through the Fc domain. Uptake of these liposomes is shown to be less when mAb are coupled via the Fc domain to liposomes, which shields recognition somewhat, indicating its involvement in uptake.52 Fc receptor coupling to liposomes favors binding to tumor cells but does not exclude the recognition by macrophages. Cheng and Allen53 compared liposomes coupled with whole antibody or with scFv against B-cell antigen CD-19. They show that mAb-liposomes were cleared rapidly from the bloodstream due to Fc receptor-mediated uptake, while circulation time of scFv-liposomes was prolonged.
Since scFvs are less stable than whole antibodies19 or Fab fragments, freshly prepared scFvs were used to form immunoliposomes. The scFv produced were tested for their activity and then coupled to preformed liposomes, and coupling was considered successful when they conformed to strict validation criteria. Immunoliposomes made by conventional methods have limitations.36,42,45 In particular, antibodies or scFv molecules may randomly bind with liposomal maleimide, and the true number of scFv per carrier cannot be accurately determined. We set strict validation criteria in terms of size, PDI, quantity of lipids, protein, and activity of scFv using easy readouts to minimize errors in calculating the number of scFv molecules per liposome and thereby minimizing batch-to-batch variation. Total lipid and protein concentration was determined, and the number of scFv molecules coupled to the liposomes was calculated. As a final validation step, immunoliposomes were tested for binding of M1/A1 complexes by ELISA. Only those batches that met the above stated criteria were used for subsequent experimentation, taking into account further therapeutic use. A schematic presentation of our conjugation, production, and the criteria used for analysis is shown in Figure 6. After coupling, the scFvs still remained functional, which was proven by ELISA and flow cytometry. It was important to determine the functionality of these scFv after coupling, before proceeding to further advanced experiments. It has been reported in literature that coupling of scFv to maleimide groups can result in a shielding effect where scFv cannot access antigen due to PEG molecules forming a mushroom-like barrier54 and hindering interaction with antigens.30 Here, we show via a simple ELISA assay that it can be determined whether or not scFvs are functional after coupling.
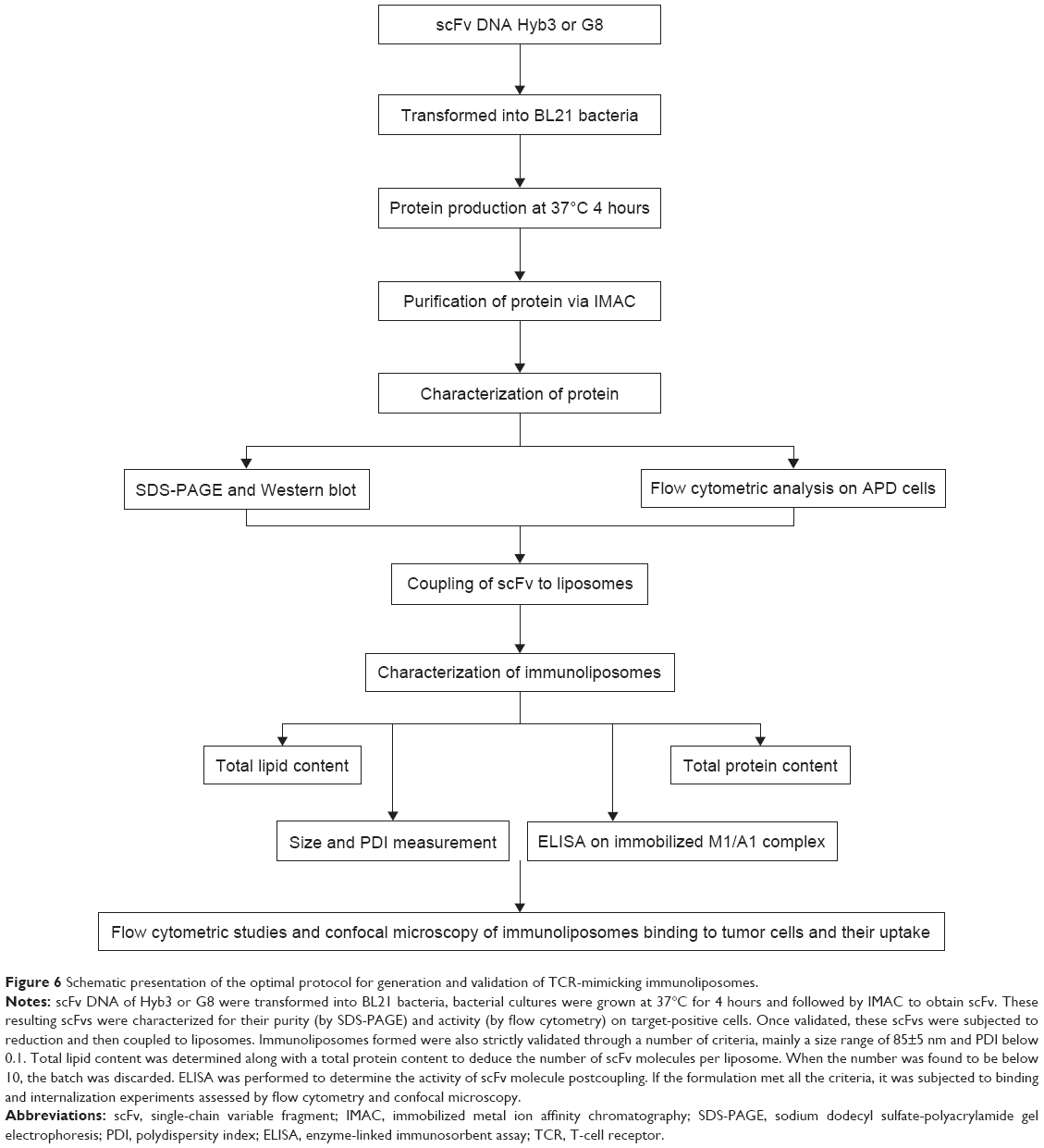 | Figure 6 Schematic presentation of the optimal protocol for generation and validation of TCR-mimicking immunoliposomes. |
Importantly, we observed that immunoliposomes showed specific and significant binding to peptide-pulsed APD cells, whereas negligible binding occurred to peptide-negative cells. There was no internalization observed in APD cells, which corresponds with a study by Mommaas et al47 where they incubated APD cells and (antigen stimulated) T-cells with endorphin gold and observed only binding in APD cells and uptake in T-cells, indicating that APD cells do not internalize MHC class I molecules. We also showed immunoliposome binding to antigen-positive melanoma cells in comparison with negligible binding to antigen-negative melanoma cells at 4°C, whereas internalization occurred at 37°C as determined by flow cytometry and confocal microscopy. Binding to tumor cells depends on many factors, such as the ligand-binding affinity of the scFv used, density of scFv on the carriers, and quantity of pMHC on tumor cells.30 Binding and internalization of liposomes is expected to be more efficient as multiple binding sites are available, increasing the avidity, as compared to single scFv. Uptake is dependent on targeted moieties on the cell membrane, while intracellular processing of liposomes is also dependent on the physicochemical properties of the liposome. In spite of low expression of pMHC on tumor cells when compared to peptide-pulsed B-cells, we see pronounced and significant binding of immunoliposomes by tumor cells, which was followed by internalization. Here, we have generated two recombinant scFvs, which we have used to couple to liposomes: G8 and Hyb3. Fab-Hyb3 represents an affinity-matured variant of G8 Fab, shows preserved fine peptide specificity, and mediates an increased T-cell function when constructed into a chimeric antigen receptor and transduced into T-cells.29 In the same study, antibody genes were displayed on phages and phage-Fab-G8 demonstrated binding toward cells pulsed with MAGE A1 peptide, which was about two logs less compared to phage-Fab-Hyb3. In the present study, we produced scFv-G8 and Hyb3 proteins and showed that both scFvs, used at a concentration ~10 μg/mL, bind to peptide-pulsed B-cells, but only the high-affinity scFv-Hyb3 binds cells at lower concentrations. Expectedly, liposomes-scFv Hyb3 always showed enhanced binding to antigen-positive cells compared to liposomes-scFv G8. Of note, however, liposome-scFv Hyb3 but not G8 demonstrated off-target binding to antigen-negative cells, in particular when using antigen-high B-cells, but this was also noticeable when using antigen-low melanoma cells. The unwanted off-target binding mediated by scFv-Hyb3 is most likely caused by its high affinity. Interestingly, a study by Mareeva et al55 showed that TCR-like antibodies bind to the pMHC recognizing mainly its canonical structure based on the interaction of the MHC molecule with the peptide. Unlike most TCR-like antibodies however, Fab Hyb3 binds via a noncanonical orientation to the pMHC recognizing mainly MHC.56 Complementary to these studies, we here demonstrate that liposome-scFv Hyb3 not only binds to cells that are M1positive, HLA-A1positive, (MZ2Mel43 and G43) but also, however to a low degree, to cells that are M1negative, HLA-A1positive (Mel78 and Mel2A). In contrast, liposomes-scFv G8 show no off-target binding at all and only bound antigen-positive cells; a binding that is less when compared to Hyb3, but in all cases specific for antigen. Our findings with liposomes-scFv G8 are in extension to previous studies by our laboratory, which demonstrate that Fab G8 protein as well as T-cells transduced with Fab G8 bind to M1/A1positive-cells and not HLA-A1positive-cells pulsed with peptides that differ from M1 only by a few amino acids.5,28 Notably, when gene-engineered T-cells with scFv Hyb3 were coupled to costimulatory domains, such peptide fine-specificity was lost.38
Another line of evidence pointing to the risk of affinity maturation comes from a study by Linette et al,57 which demonstrated off-target toxicity in two patients treated with T-cells gene-engineered with affinity-enhanced TCRs directed against MAGE-A3:HLA-A1. Both patients died due to cross-reactivity of T-cells recognizing a striated muscle protein called titin, which is highly similar to the targeted MAGE-A3 peptide. Other studies have reported that affinity enhancement may also result in self-reactivity in T-cells. A recent study by Oren et al58 reported that they used a high-affinity TCR-like scFv and found that this antibody, once transduced into T-cells, resulted in loss of antigen specificity, whereas the low-affinity antibody maintained T-cell cytotoxic potential and specificity.
However, we used two scFv with different affinities to promote interaction of immunoliposomes with tumor cells instead of interaction with T-cells. It is reported that binding-site barrier phenomenon is common when proteins with higher affinities are used, resulting in higher affinity molecules binding to the first molecules in line and not penetrating deeper in the tissue.19 Fujimori et al59 reported by a mathematical model showing that high-affinity monoclonal antibodies and their interactions with tumor antigens may affect their localization in tumors eventually. Adams et al60 demonstrated the binding-site barrier phenomenon by constructing high-affinity variants of antibody fragments (scFv) and testing them in vivo. They showed that higher affinity of scFv toward the tumor antigens limits its localization and diffusion into tumors. High affinity of scFv is another factor affecting the scFv movement within tumor in addition to its size, avidity, and tumor hydrostatic pressure.60
Apart from the potential this novel formulation shows, production of optimal conjugated immunoliposomes is a laborious process. Therefore, an optimized protocol with robust, reliable, and easy to conduct screening as we provide here is imperative. We show that using validated batches of immunoliposomes results in consistent in vitro targeting performance. In future, these immunoliposomes can be exploited for delivering compounds to the tumor microenvironment, to tumor cells, or to the intracellular environment. When encapsulating contrast agents, these liposomes can be used for imaging and aid in detection of micrometastasis and sentinel nodes during surgery. Upon coentrapment of therapeutic and imaging molecules, these nanocarriers can be used for image-guided drug delivery or immunomodulation. Immunoliposomes provide a versatile platform for combination therapy or transport of immune molecules that can inhibit cell growth, proliferation, and survival. After delivery of chemotherapeutics, liposomes can be used to interfere directly (or through the delivery of compounds that interfere with protumorigenic pathways) with, for instance, block expression of AkT and PD-L1 molecules, which have been linked with tumor progression.61,62 Sun et al63 showed successful siRNA and doxorubicin co-delivery to cells in vitro. With these niosomes, siRNA delivery resulted in sensitization of tumor cells for doxorubicin. The co-delivery system showed increased therapeutic efficacy in vivo.
Conclusion
In this study, we have successfully produced and coupled TCR-like antibodies, and scFv Hyb3 and G8, to liposomes. Antibody fragments maintained their functionality postcoupling, as proven by ELISA. Further experiments show that these novel immunoliposomes bind to M1/A1-positive ADP cells, but also bind to and are internalized by positive melanoma cells, hence providing promising data for further use of these immunoliposomes. Melanoma-specific immunoliposomes can be further used in drug delivery by entrapment of therapeutic molecules in the liposome or as imaging agents by entrapping contrast agents. These liposomes can also act as carriers for viral particles or molecules that are prone to degradation. Here, we show that targeted liposomal therapy to melanoma using scFv is promising and that scFv-targeted immunoliposomes are a novel formulation that can be further exploited for the benefit of science.
Acknowledgment
The authors thank Dr Ann Seynhaeve at Experimental Surgery Laboratory for her help and advice. The study was financially supported by MRACE grants (Erasmus MC, Rotterdam, the Netherlands).
Disclosure
The authors report no conflicts of interest in this work.
References
Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23–34. | ||
Mullard A. New checkpoint inhibitors ride the immunotherapy tsunami. Nat Rev Drug Discov. 2013;12:489–492. | ||
Fife BT, Pauken KE, Eagar TN, et al. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat Immunol. 2009;10:1185–1192. | ||
Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995;182:459–465. | ||
Willemsen RA, Debets R, Hart E, Hoogenboom HR, Bolhuis RL, Chames P. A phage display selected fab fragment with MHC class I-restricted specificity for MAGE-A1 allows for retargeting of primary human T lymphocytes. Gene Ther. 2001;8:1601–1608. | ||
Sebestyen Z, Schooten E, Sals T, et al. Human TCR that incorporate CD3zeta induce highly preferred pairing between TCRα and β chains following gene transfer. J Immunol. 2008;180:7736–7746. | ||
Govers C, Sebestyén Z, Coccoris M, Willemsen RA, Debets R. T cell receptor gene therapy: strategies for optimizing transgenic TCR pairing. Trends Mol Med. 2010;16:77–87. | ||
Roszik J, Sebestyen Z, Govers C, et al. T-cell synapse formation depends on antigen recognition but not CD3 interaction: studies with TCR:zeta, a candidate transgene for TCR gene therapy. Eur J Immunol. 2011;41:1288–1297. | ||
Denkberg G, Reiter Y. Recombinant antibodies with T-cell receptor-like specificity: novel tools to study MHC class I presentation. Autoimmun Rev. 2006;5:252–257. | ||
Torchilin VP. Multifunctional, stimuli-sensitive nanoparticulate systems for drug delivery. Nat Rev Drug Discov. 2014;13:813–827. | ||
Rigon RB, Oyafuso MH, Fujimura AT, et al. Nanotechnology-based drug delivery systems for melanoma antitumoral therapy: a review. Biomed Res Int. 2015:841817. | ||
Li J, Wang Y, Liang R, et al. Recent advances in targeted nanoparticles drug delivery to melanoma. Nanomedicine. 2015;1:769–794. | ||
Chen J, Shao R, Zhang XD, Chen C. Applications of nanotechnology for melanoma treatment, diagnosis, and theranostics. Int J Nanomedicine. 2013;8:2677–2688. | ||
Maruyama K, Okuizumia S, Ishidaa O, Yamauchib H, Kikuchib H, Iwatsurua M. Phosphatidyl polyglycerols prolong liposome circulation in vivo. Int J Pharm. 1994;111:103–107. | ||
Torchilin VP, Shtilman MI, Trubetskoy VS, Whiteman K, Milstein AM. Amphiphilic vinyl polymers effectively prolong liposome circulation time in vivo. Biochim Biophys Acta. 1994;1195:181–184. | ||
Torchilin VP, Omelyanenko VG, Papisov MI, et al. Poly(ethylene glycol) on the liposome surface: on the mechanism of polymer-coated liposome longevity. Biochim Biophys Acta. 1994;1195:11–20. | ||
Maeda H. Macromolecular therapeutics in cancer treatment: the EPR effect and beyond. J Control Release. 2012;164:138–144. | ||
Iden DL, Allen TM. In vitro and in vivo comparison of immunoliposomes made by conventional coupling techniques with those made by a new post-insertion approach. Biochim Biophys Acta. 2001;1513:207–216. | ||
Allen TM. Ligand-targeted therapeutics in anticancer therapy. Nat Rev Cancer. 2002;2:750–763. | ||
Torchilin VP. Immunoliposomes and PEGylated immunoliposomes: possible use for targeted delivery of imaging agents. Immunomethods. 1994;4:244–258. | ||
Shmeeda H, Tzemach D, Mak L, Gabizon A. Her2-targeted pegylated liposomal doxorubicin: retention of target-specific binding and cytotoxicity after in vivo passage. J Control Release. 2009;136:155–160. | ||
Hofmann O, Caballero OL, Stevenson BJ, et al. Genome-wide analysis of cancer/testis gene expression. Proc Natl Acad Sci U S A. 2008;105:20422–20427. | ||
Chen YT, Stockert E, Chen Y, et al. Identification of the MAGE-1 gene product by monoclonal and polyclonal antibodies. Proc Natl Acad Sci U S A. 1994;91:1004–1008. | ||
De Smet C, Lurquin C, van der Bruggen P, De Plaen E, Brasseur F, Boon T. Sequence and expression pattern of the human MAGE2 gene. Immunogenetics. 1994;39:121–129. | ||
van der Bruggen P, Traversari C, Chomez P, et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science. 1991;254:1643–1647. | ||
Lucas S, De Plaen E, Boon T. MAGE-B5, MAGE-B6, MAGE-C2, and MAGE-C3: four new members of the MAGE family with tumor-specific expression. Int J Cancer. 2000;87:55–60. | ||
Caballero OL, Chen YT. Cancer/testis (CT) antigens: potential targets for immunotherapy. Cancer Sci. 2009;100:2014–2021. | ||
Chames P, Hufton SE, Coulie PG, Uchanska-Ziegler B, Hoogenboom HR. Direct selection of a human antibody fragment directed against the tumor T-cell epitope HLA-A1-MAGE-A1 from a nonimmunized phage-Fab library. Proc Natl Acad Sci U S A. 2000;97:7969–7974. | ||
Chames P, Willemsen RA, Rojas G, et al. TCR-like human antibodies expressed on human CTLs mediate antibody affinity-dependent cytolytic activity. J Immunol. 2002;169:1110–1118. | ||
Deshpande PP, Biswas S, Torchilin VP. Current trends in the use of liposomes for tumor targeting. Nanomedicine (Lond). 2013;8:1509–1528. | ||
Messerschmidt SK, Beuttler J, Rothdiener M. Immunoliposomes. In: Kontermann R, Dubel S, editors. Antibody Engineering. New York, NY: Springer; 2010:401–428. | ||
Skerra A, Pfitzinger I, Plückthun A. The functional expression of antibody Fv fragments in Escherichia coli: improved vectors and a generally applicable purification technique. Biotechnology (N Y). 1991;9: 273–278. | ||
Hochuli E. Large-scale chromatography of recombinant proteins. J Chromatogr. 1988;444:293–302. | ||
Messerschmidt SK, Kolbe A, Muller D, Knoll M, Pleiss J, Kontermann RE. Novel single-chain Fv’ formats for the generation of immunoliposomes by site-directed coupling. Bioconjug Chem. 2008;19:362–369. | ||
Baum P, Muller D, Ruger R, Kontermann RE. Single-chain Fv immunoliposomes for the targeting of fibroblast activation protein-expressing tumor stromal cells. J Drug Target. 2007;15:399–406. | ||
Ruger R, Muller D, Fahr A, Kontermann RE. Generation of immunoliposomes using recombinant single-chain Fv fragments bound to Ni-NTA-liposomes. J Drug Target. 2005;13:399–406. | ||
Rothdiener M, Beuttler J, Messerschmidt SK, Kontermann RE. Antibody targeting of nanoparticles to tumor-specific receptors: immunoliposomes. Methods Mol Biol. 2010;624:295–308. | ||
Willemsen RA, Ronteltap C, Chames P, Debets R, Bolhuis RL. T cell retargeting with MHC class I-restricted antibodies: the CD28 costimulatory domain enhances antigen-specific cytotoxicity and cytokine production. J Immunol. 2005;174:7853–7858. | ||
Schaft N, Lankiewicz B, Drexhage J, et al. T cell retargeting to EBV antigens following TCR gene transfer: CD28-containing receptors mediate enhanced antigen-specific IFNγ production. Int Immunol. 2006;18:591–601. | ||
Abraham SA, Waterhouse DN, Mayer LD, Cullis PR, Madden TD, Bally MB. The liposomal formulation of doxorubicin. Methods Enzymol. 2005;391:71–97. | ||
Messerschmidt SK, Musyanovych A, Altvater M, et al. Targeted lipid-coated nanoparticles: delivery of tumor necrosis factor-functionalized particles to tumor cells. J Control Release. 2009;137:69–77. | ||
Ruger R, Muller D, Fahr A, Kontermann RE. In vitro characterization of binding and stability of single-chain Fv Ni-NTA-liposomes. J Drug Target. 2006;14:576–582. | ||
Mei L, Liu Y, Zhang Q, Gao H, Zhang Z, He Q. Enhanced antitumor and anti-metastasis efficiency via combined treatment with CXCR4 antagonist and liposomal doxorubicin. J Control Release. 2014;196:324–331. | ||
Rouser G, Fkeischer S, Yamamoto A. Two dimensional then layer chromatographic separation of polar lipids and determination of phospholipids by phosphorus analysis of spots. Lipids. 1970;5:494–496. | ||
Kontermann RE. Immunoliposomes for cancer therapy. Curr Opin Mol Ther. 2006;8:39–45. | ||
Peterson RC. Application of lowry protein determination to influenza vaccine. J Pharm Sci. 1969;58:141–142. | ||
Mommaas AM, Wijsman MC, Verduijn W, Vermeer BJ, Claas FM. Internalization of MHC class I molecules is a prerequisite for endocytosis of endorphin by lymphocytes. Clin Exp Immunol. 1991;84:170–174. | ||
Bangham AD. A correlation between surface charge and coagulant action of phospholipids. Nature. 1961;192:1197–1198. | ||
Seifert O, Pollak N, Nusser A, et al. Immuno-LipoTRAIL: targeted delivery of TRAIL-functionalized liposomal nanoparticles. Bioconjug Chem. 2014;25:879–887. | ||
Pernis B. Internalization of lymphocyte membrane components. Immunol Today. 1985;6:45–49. | ||
Koning GA, Kamps JA, Scherphof GL. Efficient intracellular delivery of 5-fluorodeoxyuridine into colon cancer cells by targeted immunoliposomes. Cancer Detect Prev. 2002;26:299–307. | ||
Koning GA, Morselt HW, Gorter A, et al. Interaction of differently designed immunoliposomes with colon cancer cells and Kupffer cells. An in vitro comparison. Pharm Res. 2003;20:1249–1257. | ||
Cheng WW, Allen TM. Targeted delivery of anti-CD19 liposomal doxorubicin in B-cell lymphoma: a comparison of whole monoclonal antibody, Fab’ fragments and single chain Fv. J Control Release. 2008;126:50–58. | ||
Moghimi SM. The effect of methoxy-PEG chain length and molecular architecture on lymph node targeting of immuno-PEG liposomes. Biomaterials. 2006;27:136–144. | ||
Mareeva T, Martinez-Hackert E, Sykulev Y. How a T cell receptor-like antibody recognizes major histocompatibility complex-bound peptide. J Biol Chem. 2008;283:29053–29059. | ||
Hulsmeyer M, Chames P, Hillig RC, et al. A major histocompatibility complex-peptide-restricted antibody and T cell receptor molecules recognize their target by distinct binding modes: crystal structure of human leukocyte antigen (HLA)-A1-MAGE-A1 in complex with FAB-HYB3. J Biol Chem. 2005;280:2972–2980. | ||
Linette GP, Stadtmauer EA, Maus MV, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. 2013;122:863–871. | ||
Oren R, Hod-Marco M, Haus-Cohen M, et al. Functional comparison of engineered T cells carrying a native TCR versus TCR-like antibody-based chimeric antigen receptors indicates affinity/avidity thresholds. J Immunol. 2014;193:5733–5743. | ||
Fujimori K, Covell DG, Fletcher JE, Weinstein JN. A modeling analysis of monoclonal antibody percolation through tumors: a binding-site barrier. J Nucl Med. 1990;31:1191–1198. | ||
Adams GP, Schier R, McCall AM, et al. High affinity restricts the localization and tumor penetration of single-chain Fv antibody molecules. Cancer Res. 2001;61:4750–4755. | ||
Parsa AT, Waldron JS, Panner A, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. 2007;13:84–88. | ||
Chen J, Zhang XD, Proud C. Dissecting the signaling pathways that mediate cancer in PTEN and LKB1 double-knockout mice. Sci Signal. 2015;8:pe 1. | ||
Sun M, Yang C, Zheng J, et al. Enhanced efficacy of chemotherapy for breast cancer stem cells by simultaneous suppression of multidrug resistance and antiapoptotic cellular defense. Acta Biomater. 2015;28: 171–182. |
Supplementary materials
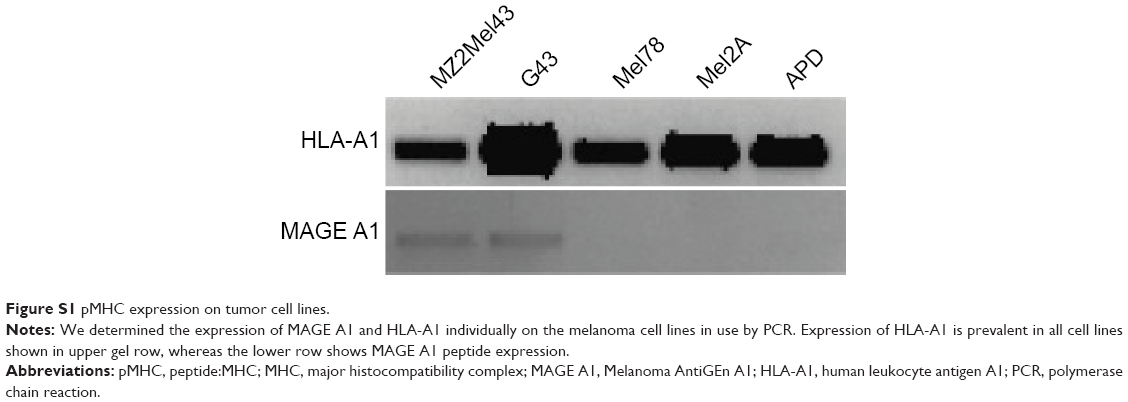 | Figure S1 pMHC expression on tumor cell lines. |
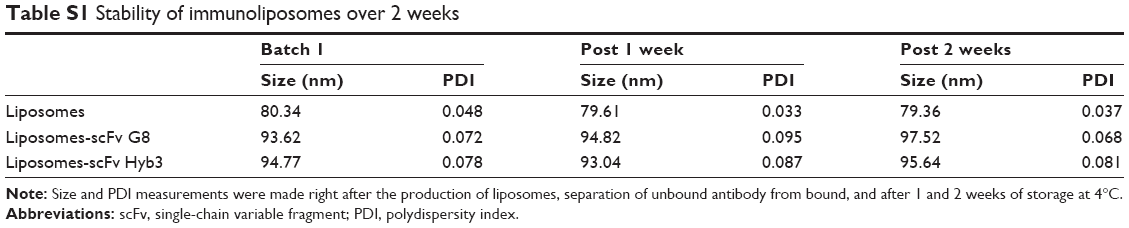 | Table S1 Stability of immunoliposomes over 2 weeks |
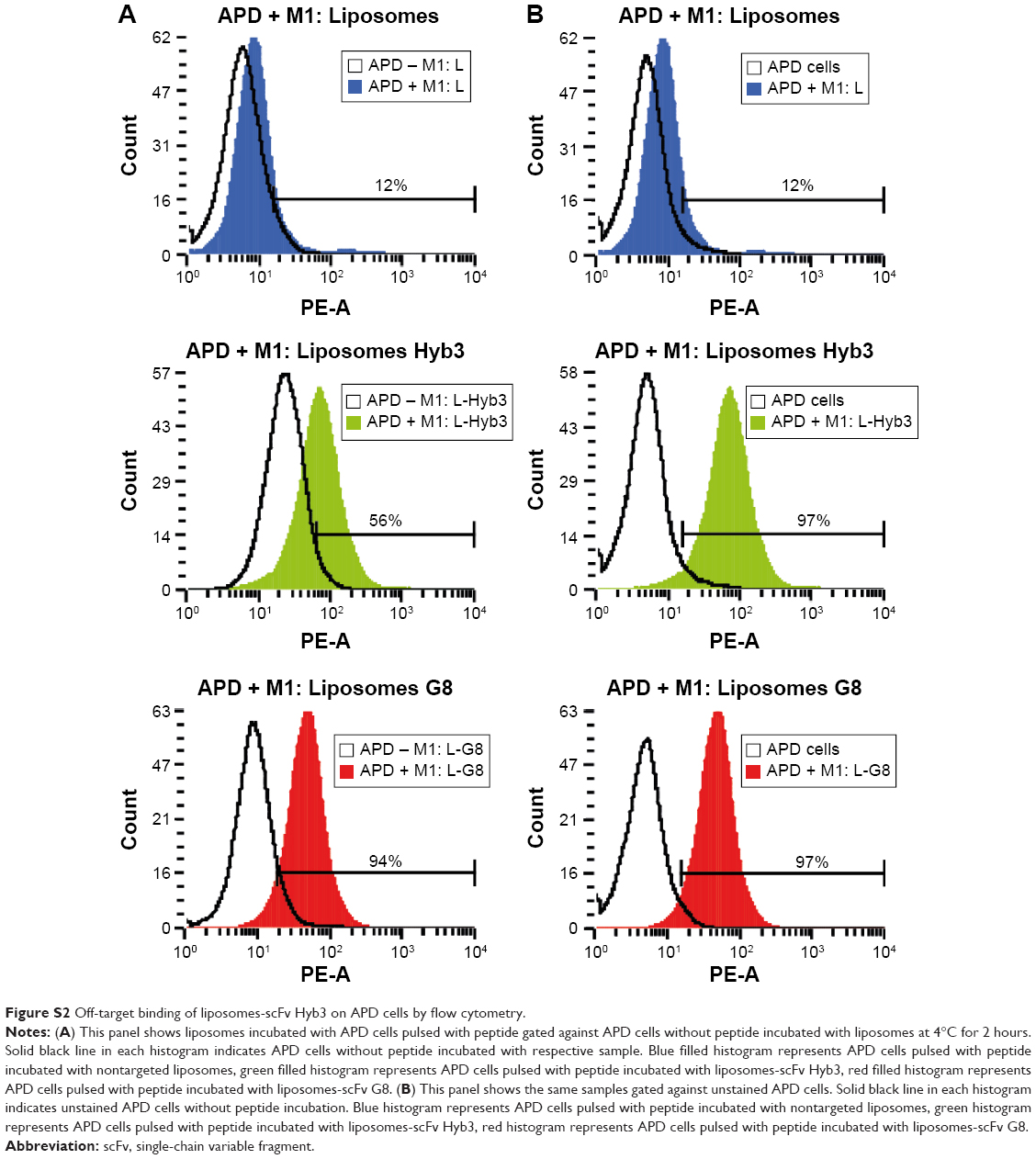 | Figure S2 Off-target binding of liposomes-scFv Hyb3 on APD cells by flow cytometry. |
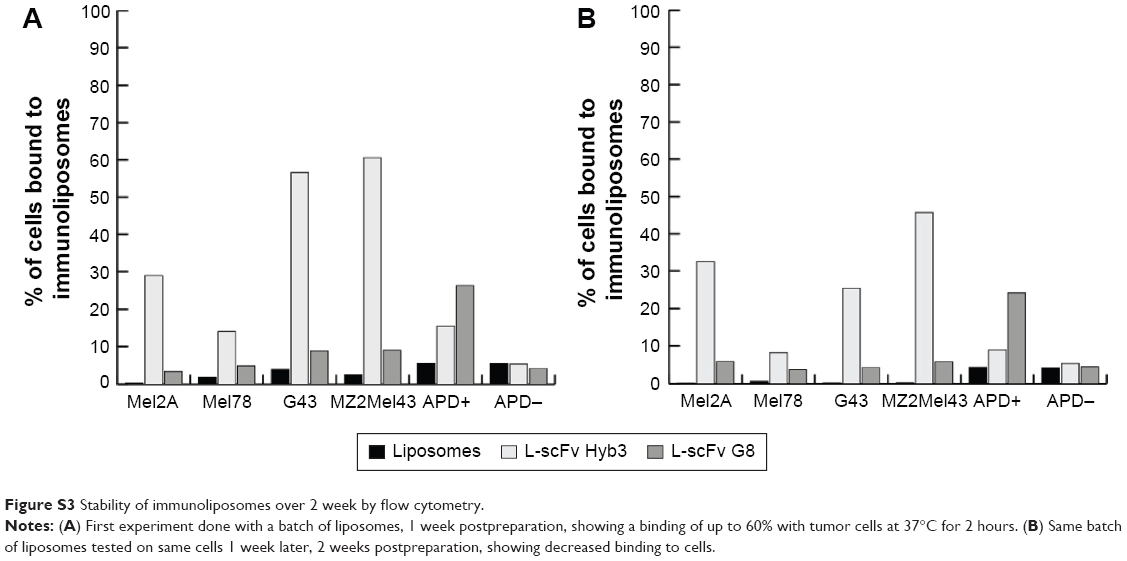 | Figure S3 Stability of immunoliposomes over 2 week by flow cytometry. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.