Back to Journals » OncoTargets and Therapy » Volume 15
Targeting KRASG12C-Mutated Advanced Colorectal Cancer: Research and Clinical Developments
Received 16 April 2022
Accepted for publication 24 June 2022
Published 7 July 2022 Volume 2022:15 Pages 747—756
DOI https://doi.org/10.2147/OTT.S340392
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Gaetano Romano
Jingran Ji, Chongkai Wang, Marwan Fakih
Department of Medical Oncology and Therapeutics Research, City of Hope Comprehensive Cancer Center, Duarte, CA, USA
Correspondence: Marwan Fakih, Department of Medical Oncology and Therapeutics Research, City of Hope Comprehensive Cancer Center, Pavillion, Room 3208, 1500 E Duarte St, Duarte, CA, 91010, USA, Tel +1 626-256-4673 extension 83087, Fax +1 626-218-8233, Email [email protected]
Abstract: Identifying mutations in the KRAS gene has become increasingly important in the treatment of colorectal cancer with many prognostic and therapeutic implications. However, efforts to develop drugs that target KRAS mutations have not been successful until more recently with the introduction of the KRASG12C inhibitors, sotorasib (AMG510) and adagrasib (MRTX849). Both agents have demonstrated safety and promising efficacy in preclinical studies and early phase trials, but it appears that not all tumor types harboring the KRASG12C mutation are sensitive to monotherapy approaches. In particular, patients with colorectal cancer (CRC) derive less benefit compared to those with non-small cell lung cancer (NSCLC), likely due to rapid treatment-induced resistance through increased epidermal growth factor receptor (EGFR) signaling. As a result, combination therapy trials with EGFR inhibitors are currently underway. Here, we will review the available clinical trial data on KRASG12C inhibitors in KRASG12C-mutated CRC, possible mechanisms of resistance to monotherapy, the research studying why available agents are proving to be less efficacious in CRC compared to NSCLC, and future directions for these promising new drugs.
Keywords: KRASG12C, sotorasib, adagrasib, colorectal cancer, targeted therapy
Introduction
KRAS mutations are found in approximately 45% of colorectal cancer (CRC) and are associated with resistance to targeted therapies such as anti-epidermal growth factor receptor (EGFR) inhibitors.1–3 The KRASG12C mutation is found in 14% of non-small cell lung cancer (NSCLC), 3% of CRC, and 1–3% of other solid tumors.4–7 Patients with metastatic KRASG12C-mutant CRC progress quickly on standard of care chemotherapy regimens and may have shorter overall survival (OS) compared to those with non-KRASG12C mutations.8
The glycine-to-cysteine substitution at position 12 leads to a KRAS protein that is predominantly in the GTP-bound state, which drives constitutive activation of oncogenic signaling.9–11 The mutant cysteine is located next to a pocket in the switch II region (S-IIP), which exists only in the inactive GDP-bound conformation of KRAS and has therefore been studied extensively to identify potential inhibitors of KRASG12C.12,13 The existing inhibitors bind to the mutant cysteine, which disrupts the switch I/II region. This drives KRAS to favor the GDP- over the GTP- bound state and traps the protein in an inactivated state.13,14 In addition, the occupation of the S-IIP region by these inhibitors disrupts downstream binding of effector proteins, such as RAF. In vitro studies found that these compounds lead to decreased viability and increased apoptosis in cancer cell lines harboring the KRASG12C mutation.12,14 After overcoming several challenges in drug development, sotorasib and adagrasib (developed by Amgen and Mirati Therapeutics, respectively) are the two KRASG12C inhibitors with the most promising clinical activity in solid tumors.15,16
Targeting KRASG12C with sotorasib and adagrasib in mice bearing KRASG12C-mutant NSCLC tumors reduced the phosphorylation of ERK and led to significant tumor regression.17,18 A Phase 1 clinical trial evaluating the safety and efficacy of sotorasib has demonstrated a tolerable safety profile as well as promising antitumor activity in patients with KRASG12C mutant solid tumors.19–21 Based on the significant clinical activity in patients with NSCLC in the CodeBreak100 trial, sotorasib was approved by the FDA for patients with locally advanced or metastatic NSCLC harboring the KRASG12C mutation who had progressed on prior systemic therapy.22 Similarly, based on data from a Phase 2 KRYSTAL-1 trial, adagrasib was approved in Europe for the same indications in NSCLC and is currently under review by the FDA.23 Herein, this review focuses on the current development of direct KRASG12C inhibitors and alternative strategies for targeting KRAS, particularly in CRC where the effect of monotherapy appears to be limited.
Monotherapy with Sotorasib or Adagrasib in KRASG12C-Mutated Tumors
Sotorasib (AMG510)
In the first in human phase 1 study, sotorasib was evaluated with a dose-escalation design in patients with refractory KRASG12C-mutated solid tumors (NCT 03600883).22 Doses were escalated from 160mg daily to 960mg daily. No dose limiting toxicities were noted in the escalation phase. The 960mg oral dose was selected for further development based on its safety and pharmacokinetics. Additional expansion cohorts of NSCLC, CRC, and other solid tumors with KRASG12C mutation were enrolled to include a total of 129 patients. Most patients enrolled in the study were NSCLC (59), followed by CRC (42) and other tumors (28). A total of 73 patients (56.6%) had treatment-related adverse events; only 15 (11.6%) patients experienced grade 3 or 4 events. Notable activity was noted in the NSCLC group with an objective response rate (ORR) of 32.2% and a disease control rate (DCR) of 88.1%. Median progression free survival (PFS) in this group was 6.3 months. Clinical activity was more modest in the CRC and other solid tumor groups. In the CRC cohort, the ORR was 7.1% and the DCR was 73.8%. The median PFS in this group was 4 months. Responses were also documented in the other solid tumors group, including patients with pancreatic, endometrial, and appendiceal cancers as well as one patient with melanoma.
The phase 2 CodeBreak 100 (NCT03600883)24 trial studied sotorasib in patients with metastatic KRASG12C-mutant CRC who had progressed on prior fluoropyrimidine, oxaliplatin, and irinotecan treatment, using the phase 1 dosing of 960mg daily.25 The ORR was 9.7% and the DCR was 82.3%. The median PFS in this group was 4 months, similar to the phase 1 trial. The adverse events profile was also similar with 7 (12%) patients experiencing a grade 3 or 4 event. A phase 2 trial also studied sotorasib in patients with KRASG12C-mutant advanced NSCLC who had progressed on prior platinum-based chemotherapy and programmed death 1 (PD-1) or programmed death ligand 1 (PD-L1) therapy. A total of 124 patients were evaluated for response, which resulted in an ORR of 37.1% with 4 (3.2%) patients who achieved a complete response. The DCR was 80.6%, the median PFS was 6.8 months, and the OS was 12.5 months.26
Adagrasib (MRTX849)
The KRYSTAL-1 study (NCT03785249)27 is a phase 1/2 study investigating adagrasib in patients with advanced or metastatic solid tumors harboring a KRASG12C mutation. Patients were all previously treated with chemotherapy, anti-PD-1/PD-L1 therapy, or both. The phase 1/1b dose expansion phase established a dose of 600mg twice daily. Of the 25 patients enrolled, 2 patients had CRC who received the phase 2 dose and were evaluable. One patient achieved a partial response with a duration of response of 4.2 months. Of the 15 patients with NSCLC, the median PFS was 11.1 months and median OS was not reached. The ORR was 53.3%. A total of 36% of patients experienced a grade 3–4 treatment-related adverse event with fatigue being the most common (15%) at the phase 2 dose.28 The phase 2 portion of this trial is ongoing. Interim analysis in August 2020 reported the data on 79 patients with pretreated NSCLC who received adagrasib at 600mg twice daily. Among the 51 evaluable patients (including those from the phase 1/1b cohort), the ORR was 45%. The DCR was 96%.23 Updated analysis of monotherapy in CRC patients in May 2021 included 45 evaluable patients with an ORR of 22% and a DCR of 87%. Median PFS was 5.6 months.29
Differences Between Sotorasib and Adagrasib
In addition to the maturing clinical data on sotorasib and adagrasib, there are several differences between the two agents. Both drugs share a chemical backbone and target the same S-IIP region of KRAS, but differences in chemical structure led to a mean half-maximum inhibitory concentration (IC50) of 47.9 nM for sotorasib and 89.9 nM for adagrasib. The half-life of adagrasib at 24.7 hours is also considerably longer than sotorasib, which is reported to be 5.5 hours.30,31 In mouse models, sotorasib had an oral bioavailability of 22–40% compared to 62.9% with adagrasib.32 Pre-clinical models also demonstrate different affinities for on-target resistance mutations between the two agents, as discussed below.33 While the clinical implications of these differences are not yet clear in human trials, there will likely be distinct characteristics of each drug with unique applications in select patient populations.
Mechanisms of Resistance to KRASG12C Inhibitors
Despite the early clinical data suggesting activity of the KRASG12C inhibitors in various cancer types, responses appear to be limited when used as monotherapy. There are several key mechanisms of resistance that have been identified in pathways both upstream and downstream of KRAS (Figure 1).34 In pre-clinical models of resistant KRASG12C-mutant cancer, secondary KRAS mutations were the most common. Specific mutations such as G13D, R68M, and A59S/T appeared to confer resistance to sotorasib while remaining sensitive to adagrasib. The Q99L alteration was resistant to adagrasib but sensitive to sotorasib.33 However, the most common mutation was Y96D/S, and this mutation conferred the strongest resistance against both sotorasib and adagrasib.33 Low allele frequency hotspot mutations in KRAS, NRAS, MRAS, and BRAF were also able to confer resistance. Single-cell sequencing identified that many cells with these secondary mutations still harbor KRASG12C, suggesting that ongoing inhibitor activity does not need to be disrupted to manifest resistance.35 An additional escape mechanism is the production of new KRASG12C protein in the GTP-bound state, which does not interact with existing inhibitors, thus avoiding inactivation.36
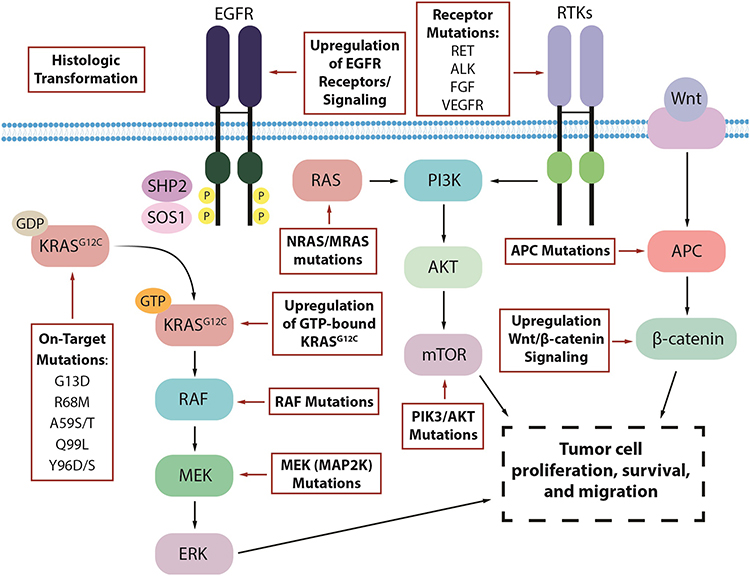 |
Figure 1 Proposed mechanisms of resistance to KRASG12C inhibitors in KRASG12C-mutated colorectal cancer. |
Further investigation of the patients who progressed on the KRYSTAL-1 study revealed that 17 (45%) had a potential underlying cause of resistance as identified by next-generation sequencing of tissue or circulating tumor DNA (ctDNA). Nine of these patients had secondary mutations of the KRAS gene or amplification of the KRASG12C allele. Twelve had molecular alterations in non-KRAS genes that were key components of other parts of the RAS/MAPK signaling pathways such as activating mutations of BRAF, MAP2K1, NRAS, and RET. Gene fusions of RET, RAF1, BRAF, ALK, and FGFR3 were also observed. These mechanisms of resistance were not mutually exclusive as 7 patients had multiple concurrent mechanisms. Two NSCLC patients transformed to squamous cell histology.37 Similarly, post-treatment specimens from 43 patients in the CodeBreak 100 and CodeBreak 101 (NCT04185883)38 trials with sotorasib revealed new alterations in KRAS, NRAS, BRAF, EGFR, FGFR2, Myc, and others, in 27 patients. Six patients had undetectable KRASG12C alleles by ctDNA assessment.35 Histologic evaluation in a rapid-autopsy case in a patient with KRASG12C inhibitor resistant NSCLC also observed non-cell autonomous mechanisms of resistance including remodeling of the tumor microenvironment.39
KRASG12C Inhibitors are Less Effective in CRC Compared to NSCLC Due to Key Mechanisms of Resistance
Among the sotorasib outcomes to date, the efficacy of single agent KRASG12C inhibition in CRC has been much lower when compared to the efficacy seen in NSCLC (Table 1). This diminished response is in line with the experience using other MAPK pathway inhibitors. For example, BRAF inhibition in BRAFV600E-mutant colorectal cancer, with or without a MEK inhibitor, appears to be considerably less effective than what is seen in melanoma or NSCLC carrying the same alteration.40–42 This resistance is likely due to key differences in the biology and mechanism of oncogenesis of CRC tumors. For example, in BRAFV600E-mutant disease, acquired or pre-existing co-mutations in PIK3 and PTEN were more prevalent in CRC samples.43 In KRAS-mutant CRC, preclinical data suggests that oncogenesis is more heavily dependent on the KRAS signaling pathway, making primary resistance to KRAS inhibition less likely. Instead, rapid development of treatment-induced resistance appears to be the predominant issue.44 In contrast, KRAS-mutant NSCLC may have more primary resistance as there is a clear subgroup of disease which is less dependent on KRAS signaling alone despite harboring KRAS mutations.45 However, tumors that are sensitive appear to have a less rapid accumulation of resistance to KRAS inhibition compared to CRC.
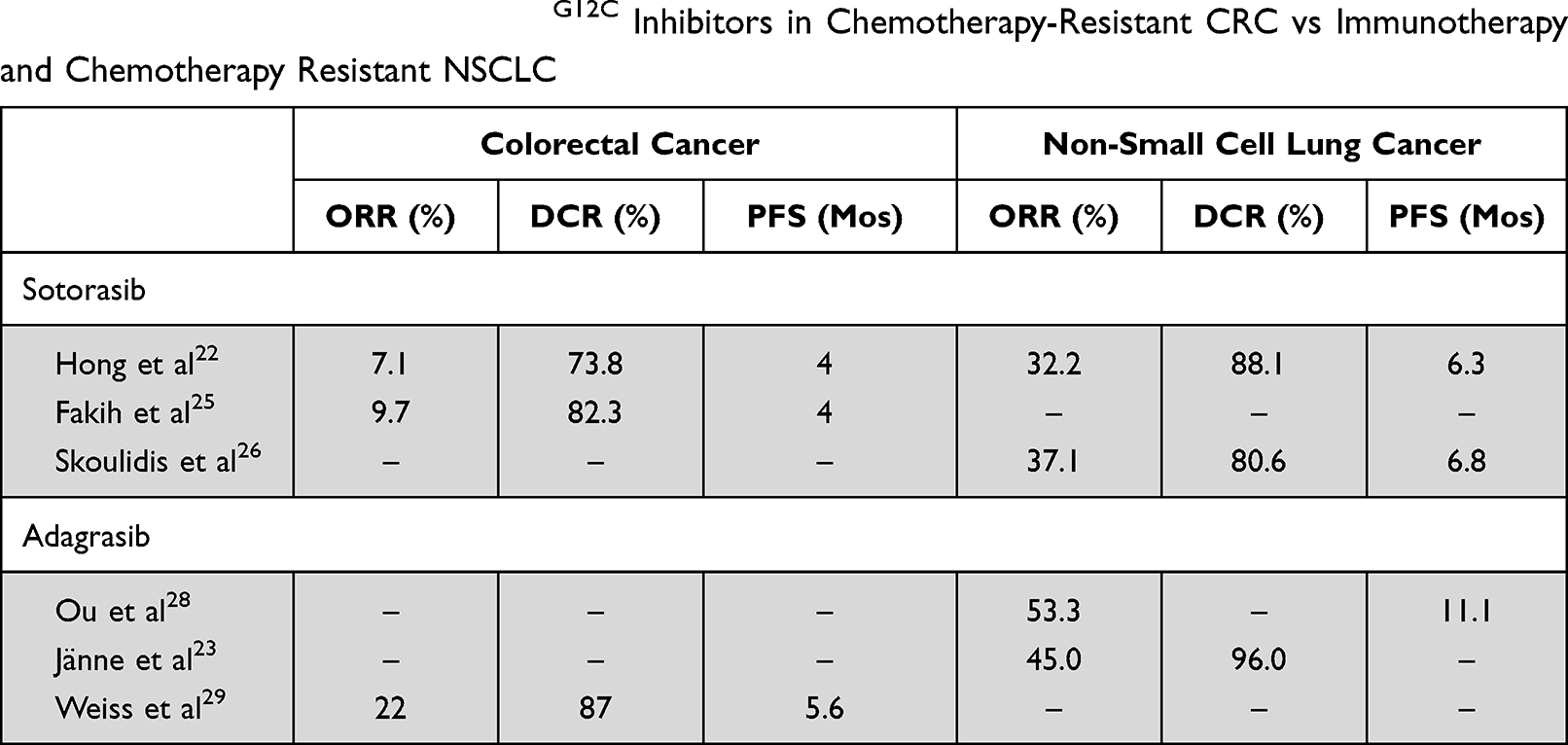 |
Table 1 Summary of Trials of KRASG12C Inhibitors in Chemotherapy-Resistant CRC vs Immunotherapy and Chemotherapy Resistant NSCLC |
While many of the mechanisms of resistance are shared among KRASG12C-mutant CRC and NSCLC, there are characteristic differences that may be drivers of the limited response to treatment in CRC. Amodio et al demonstrated that in KRASG12C-mutant CRC cell lines there is a much higher basal receptor tyrosine kinase (RTK) activation compared to NSCLC cell lines. This was also seen in clinical tissue samples where CRC tumors had more detectable phosphorylated RTKs. In particular, EGFR receptors appeared to be the primary activated subgroup. When treated with sotorasib, CRC cells showed initial response with concurrent down-regulation of ERK phosphorylation, but this was followed with a quick rebound in phosphorylated ERK levels within 24 hours of treatment. This rapid resistance was not seen in the NSCLC cell lines which showed further down-regulation of ERK phosphorylation over time. The CRC cells were also persistently sensitive to growth factor despite KRAS inhibition.46 This increased RTK signaling is coupled with other mechanisms of resistance such as increased GTP-bound KRASG12C protein to maintain active downstream signaling and thus leads to tumor progression.36 Alternative activated pathways more prevalent in CRC such as the Wnt/β-catenin pathway also interact with mutant KRAS signaling and degradation, promoting persistent oncogenic signaling and conferring resistance.47
Combination with Anti-EGFR Therapy to Improve Response of KRASG12C Inhibitors in CRC
In order to potentiate the effect of KRASG12C inhibitors and suppress early mechanisms of resistance, a combination approach of sotorasib or adagrasib with EGFR inhibitors such as panitumumab or cetuximab is an approach that is currently being tested. Preclinical work demonstrates that cetuximab sensitizes KRASG12C-mutated CRC cell lines to sotorasib and leads to sustained down-regulation of phosphorylated MEK and ERK proteins, which ultimately causes arrest of cell proliferation and cell death. This has been subsequently tested in patient-derived organoids and xenograft models, both showing resistance with single-agent therapy (either KRASG12C or anti-EGFR inhibition alone) versus significant synergistic effect when used in combination.46 The success of a combinatorial approach has been particularly well demonstrated with BRAFV600E-mutant colorectal cancer where single-agent BRAF inhibition alone only led to a 5% response rate.48 Combination therapy with EGFR inhibition in the BEACON trial showed a significant improvement in ORR to 26% as well as superior PFS and OS.49,50 Comparable response rates were also seen when using cetuximab, vemurafenib, and irinotecan in combination.51,52 This has led to the approval of the combination of encorafenib and cetuximab in pre-treated patients with BRAFV600E-mutated metastatic colorectal cancer.
Trials are already ongoing with sotorasib and adagrasib combined with panitumumab and cetuximab, respectively. CodeBreak 101 is an umbrella phase 1b trial studying sotorasib in combination with various agents, including panitumumab. As of April 2021, 26 patients have been treated with this combination with a promising ORR (confirmed and unconfirmed) of 33%.53,54 So far, no unexpected adverse events outside of those known for sotorasib and panitumumab have been seen. Similarly, the KRYSTAL-1 umbrella trial also had a cohort of patients who received adagrasib in combination with cetuximab and 32 patients have been enrolled as of July 2021. Among the 28 evaluable patients, the confirmed and unconfirmed ORR was 43% with a DCR of 100%. Again, the adverse events have been limited to those expected from the individual agents, with only 16% experiencing grade 3–4 toxicity.29 These preliminary results are promising and further data from these trials are eagerly awaited as more patients are enrolled. Larger, confirmatory randomized trials in the second- and third-line settings are being conducted to further define the role of these combinations in metastatic colorectal cancer.
Emerging Strategies to Overcome Resistance to KRASG12C Inhibitors in CRC
While trials with the combination of KRASG12C inhibitors and EGFR inhibitors are ongoing, the advent of a new targeted therapy for CRC opens many new avenues of investigation and inquiry regarding the efficacy of these inhibitors in combination with existing therapies as well as new agents in the drug development pipeline (Figure 2). Additional cohorts in the CodeBreak 101 umbrella trial combine sotorasib with other approved agents including a MEK inhibitor, PD1/PD-L1 inhibitors, a CDK 4/6 inhibitor, an mTOR inhibitor, a VEGF inhibitor with various chemotherapies, as well as with other investigational agents. The KRYSTAL-1 umbrella trial is also exploring similar strategies.
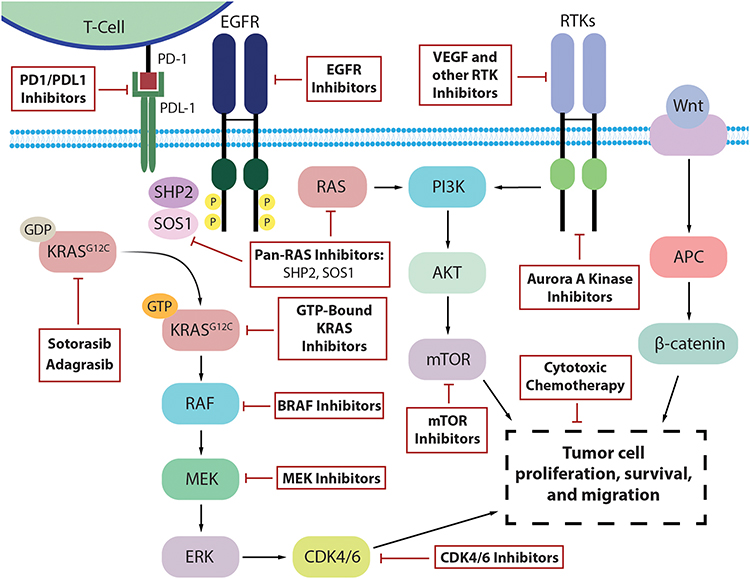 |
Figure 2 Existing and novel targets to potentially bypass resistance to KRASG12C inhibition in KRASG12C-mutated colorectal cancer. |
The rationale is to potentiate the effect and preemptively target mechanisms of resistance. Several pre-clinical studies have provided strong rationales to move these combinations to the clinical setting. For example, the combination with trametinib, a MEK inhibitor, would inhibit the MAPK pathway and avoid early resistance through a bypass tract, thus deepening the response.33 Early data from a phase 1 trial of sotorasib plus trametinib, including 18 patients with CRC, has demonstrated safety and some signals of efficacy with a median treatment duration of 84 days and no new or unexpected toxicity.55
Similarly, combination with CDK4/6 inhibitors such as palbociclib would block cell cycle entry despite activation of down-stream signaling pathways.56 In vivo, when KRASG12C inhibitors were used alone, cell cycle progression was only modestly inhibited. Similarly, CDK4/6 inhibitors were unable to suppress RAS pathway signaling. However, adding Palbociclib to KRASG12C inhibitors demonstrated significantly more down-regulation of RAS pathway phosphorylation, cell division genes, and cell cycle progression.57 Another strategy of adding everolimus, an mTOR inhibitor, has been shown to prevent resistance through the PI3K/mTOR pathway. In KRAS-mutant NSCLC mouse models, everolimus in combination with KRASG12C inhibitors was better at reducing cell viability when compared to either agent alone. The effect was further compounded by the addition of linsitinib, an IFG1 receptor inhibitor upstream of PI3K/mTOR, leading to complete suppression of cell proliferation.58
Beyond intracellular signaling mechanisms, synergistic combinations of KRASG12C inhibitors with PD1/PDL-1 agents such as pembrolizumab and atezolizumab could lead to enhanced immune-related anti-tumor activity. Preclinical data with sotorasib showed increased CD8+ T cell infiltration, macrophages, and dendritic cells in the tumor microenvironment. PD-1 expression on the associated CD8+ T cells was also moderately increased. When sotorasib was used in combination with immune-checkpoint inhibitors, there was a significant increase in the number of mice with complete regression (nine of ten) compared to the agents used as monotherapy (one of ten).17 Furthermore, combinations with chemotherapy such as FOLFIRI or FOLFOX as well as VEGF inhibitors such as bevacizumab can optimize anti-tumor inflammation and immunity and thus further increase efficacy.
Additionally, there are many investigational agents with strong pre-clinical data now moving into the clinical setting in combination with KRASG12C inhibition. For instance, the strategy of simultaneously targeting other components of the KRAS scaffold shows promise. One target is SHP2, a tyrosine phosphatase that has been shown to promote KRAS signaling and progression in CRC when activated.59 Inhibition of SHP2 increases GDP-bound KRASG12C and there is in vitro evidence of synergism with KRASG12C inhibitors.60 Novel SHP2 inhibitors such as TNO155, BBP-398, and RMC-4630 are in ongoing phase 1 trials with concurrent plans to test in combination with KRASG12C inhibition.61–63 SOS1 is another component of the scaffold integral to KRAS function, and BI-3406 is an SOS1 inhibitor that has demonstrated activity in resistant KRASY96D/S cells in combination with trametinib.33 Another agent is VIC-1911, an Aurora A Kinase (AURKA) inhibitor, which has pre-clinical data showing efficacy in resistant KRASG12C-mutant NSCLC cells when used in combination with WEE1 inhibition.64 Finally, targeting KRASG12C in its GTP-bound state with inhibitors such as RM-032 would bypass mechanisms of resistance such as overexpression of active GTP-bound KRASG12C protein, which are not targetable with the current agents.65
Future Directions
Beyond early phase trials with the aforementioned combinations, randomized Phase 3 trials will be necessary to establish the efficacy of both sotorasib and adagrasib and move them forward as standard of care. Efforts are already underway. For instance, KRYSTAL-10 (NCT04793958)66 is an open-label, randomized phase 3 trial comparing adagrasib plus cetuximab versus chemotherapy in the second-line setting for patients with KRASG12C metastatic CRC. NCT0519893467 is a phase 3 multicenter, randomized trial of sotorasib and panitumumab versus investigator’s choice (trifluridine and tipiracil or regorafenib) in previously treated metastatic KRASG12C-mutant CRC. In NSCLC, CodeBreak200 (NCT04303780)68 is a randomized phase 3 trial comparing sotorasib with docetaxel in previously treated patients with locally advance or metastatic disease. Other planned trials will study sotorasib as first-line therapy for those with KRASG12C-mutant metastatic disease (NCT04933695)69 as well as using sotorasib in conjunction with chemotherapy in the neoadjuvant setting for stage IIA-IIIB KRASG12C-mutant NSCLC (NCT05118854).70
Beyond KRASG12C inhibition, there still remains limited options for patients harboring other KRAS mutations found in the remaining 97% of KRAS-mutant CRC. However, there are agents on the horizon targeting mutational subtypes G12F, G12V, and G12R with RMC-6236 and G12D with MRTX1133.71,72 Furthermore, targeting SOS1 and disrupting the KRAS scaffold may not be limited to KRASG12C and agents such as BI-1701963 are being tested as monotherapy in patients with any KRAS mutant cancer.73
Conclusion
After years of drug development, there are now finally targeted agents for a subgroup of KRAS-mutant cancers. Early phase 1/2 data has shown sotorasib and adagrasib to be safe and efficacious in the clinical setting. While KRASG12C-mutant NSCLC appears to have the best and most durable response to therapy, patients with KRASG12C-mutant CRC are also seeing some benefit. For those who have been through several lines of therapy, the benefit of a well-tolerated targeted agent can be meaningful even if the duration of response is limited. Yet the development of KRASG12C inhibitors in CRC has only just begun and there is a need for more data using combination therapy, larger randomized trials, and ultimately novel inhibitors for other, more prevalent, KRAS mutations.
Disclosure
Prof. Dr. Marwan Fakih reports grants and/or personal fees from Amgen, AstraZeneca, Novartis, advisory for Array, Bayer, GSK, Taiho, Incyte, and Pfizer, during the conduct of the study; grants and/or personal fees from Amgen, AstraZeneca, Novartis, advisory for Array, Bayer, GSK, Taiho, Incyte, and Pfizer, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Karapetis CS, Khambata-Ford S, Jonker DJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359(17):1757–1765. doi:10.1056/NEJMoa0804385
2. Amado RG, Wolf M, Peeters M, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26(10):1626–1634. doi:10.1200/JCO.2007.14.7116
3. Van Cutsem E, Cervantes A, Adam R, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol. 2016;27(8):1386–1422. doi:10.1093/annonc/mdw235
4. Sanchez-Vega F, Mina M, Armenia J, et al. Oncogenic signaling pathways in the cancer genome atlas. Cell. 2018;173(2):321–337.e10. doi:10.1016/j.cell.2018.03.035
5. Ou SHI, Sokol ES, Madison R, et al. Comprehensive pan-cancer analysis of KRAS genomic alterations (GA) including potentially targetable subsets. Ann Oncol. 2019;30:v26. doi:10.1093/annonc/mdz239.003
6. Neumann J, Zeindl-Eberhart E, Kirchner T, Jung A. Frequency and type of KRAS mutations in routine diagnostic analysis of metastatic colorectal cancer. Pathol Res Pract. 2009;205(12):858–862. doi:10.1016/j.prp.2009.07.010
7. Ouerhani S, Elgaaied ABA. The mutational spectrum of HRAS, KRAS, NRAS and FGFR3 genes in bladder cancer. Cancer Biomark. 2011;10(6):259–266. doi:10.3233/CBM-2012-0254
8. Henry JT, Coker O, Chowdhury S, et al. Comprehensive clinical and molecular characterization of KRAS G12C-mutant colorectal cancer. JCO Precis Oncol. 2021;5:
9. Muñoz-Maldonado C, Zimmer Y, Medová M. A comparative analysis of individual RAS mutations in cancer biology. Front Oncol. 2019;9:1088. doi:10.3389/fonc.2019.01088
10. Khan AQ, Kuttikrishnan S, Siveen KS, et al. RAS-mediated oncogenic signaling pathways in human malignancies. Semin Cancer Biol. 2019;54:1–13. doi:10.1016/j.semcancer.2018.03.001
11. McCormick F. Targeting KRAS directly. Annu Rev Cancer Biol. 2018;2(1):81–90. doi:10.1146/annurev-cancerbio-050216-122010
12. Patricelli MP, Janes MR, Li LS, et al. Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discov. 2016;6(3):316–329. doi:10.1158/2159-8290.CD-15-1105
13. Lito P, Solomon M, Li LS, Hansen R, Rosen N. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science. 2016;351(6273):604–608. doi:10.1126/science.aad6204
14. Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503(7477):548–551. doi:10.1038/nature12796
15. Fell JB, Fischer JP, Baer BR, et al. Identification of the clinical development candidate MRTX849, a covalent KRASG12C inhibitor for the treatment of cancer. J Med Chem. 2020;63(13):6679–6693. doi:10.1021/acs.jmedchem.9b02052
16. Janes MR, Zhang J, Li LS, et al. Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell. 2018;172(3):578–589.e17. doi:10.1016/j.cell.2018.01.006
17. Canon J, Rex K, Saiki AY, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575(7781):217–223. doi:10.1038/s41586-019-1694-1
18. Hallin J, Engstrom LD, Hargis L, et al. The KRASG12C inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov. 2020;10(1):54–71. doi:10.1158/2159-8290.CD-19-1167
19. Fakih M, O’Neil B, Price TJ, et al. Phase 1 study evaluating the safety, tolerability, pharmacokinetics (PK), and efficacy of AMG 510, a novel small molecule KRAS G12C inhibitor, in advanced solid tumors. J Clin Oncol. 2019;37(15_suppl):3003. doi:10.1200/JCO.2019.37.15_suppl.3003
20. Fakih M, Desai J, Kuboki Y, et al. CodeBreak 100: activity of AMG 510, a novel small molecule inhibitor of KRAS G12C, in patients with advanced colorectal cancer. J Clin Oncol. 2020;38(15_suppl):4018. doi:10.1200/JCO.2020.38.15_suppl.4018
21. Hong DS, Kuo J, Sacher AG, et al. CodeBreak 100: Phase I study of AMG 510, a novel KRAS G12C inhibitor, in patients (pts) with advanced solid tumors other than non-small cell lung cancer (NSCLC) and colorectal cancer (CRC). J Clin Oncol. 2020;38(15_suppl):3511. doi:10.1200/JCO.2020.38.15_suppl.3511
22. Hong DS, Fakih MG, Strickler JH, et al. KRAS G12C inhibition with sotorasib in advanced solid tumors. N Engl J Med. 2020;383(13):1207–1217. doi:10.1056/NEJMoa1917239
23. Jänne P, Rybkin II, Spira A, et al. KRYSTAL-1: activity and safety of adagrasib (MRTX849) in advanced/ metastatic Non–Small-Cell Lung Cancer (NSCLC) harboring KRAS G12C mutation. Eur J Cancer. 2020;138(Suppl2):S1–2.
24. NCT03600883. A Phase 1/2, study evaluating the safety, tolerability, PK, and efficacy of sotorasib (AMG 510) in Subjects with solid tumors with a specific KRAS mutation (CodeBreaK 100). Available from: https://clinicaltrials.gov/ct2/show/NCT03600883.
25. Fakih MG, Kopetz S, Kuboki Y, et al. Sotorasib for previously treated colorectal cancers with KRASG12C mutation (CodeBreaK100): a prespecified analysis of a single-arm, phase 2 trial. Lancet Oncol. 2022;23(1):115–124. doi:10.1016/S1470-2045(21)00605-7
26. Skoulidis F, Li BT, Dy GK, et al. Sotorasib for lung cancers with KRAS p.G12C mutation. N Engl J Med. 2021;384(25):2371–2381. doi:10.1056/NEJMoa2103695
27. NCT03785249. Phase 1/2 study of MRTX849 in patients with cancer having a KRAS G12C mutation KRYSTAL-1. Available from: https://clinicaltrials.gov/ct2/show/NCT03785249.
28. Ou SHI, Jänne PA, Leal TA, et al. First-in-human phase I/IB dose-finding study of adagrasib (MRTX849) in patients with advanced KRAS G12C solid tumors (KRYSTAL-1). J Clin Oncol. 2022. doi:10.1200/JCO.21.02752
29. Weiss J, Yaeger RD, Johnson ML, et al. LBA6 KRYSTAL-1: adagrasib (MRTX849) as monotherapy or combined with cetuximab (Cetux) in patients (Pts) with colorectal cancer (CRC) harboring a KRASG12C mutation. Ann Oncol. 2021;32:S1294. doi:10.1016/j.annonc.2021.08.2093
30. Corral de la Fuente E, Olmedo Garcia ME, Gomez Rueda A, Lage Y, Garrido P. Targeting KRAS in Non-Small Cell Lung Cancer. Front Oncol. 2022;11:792635. doi:10.3389/fonc.2021.792635
31. Lanman BA, Allen JR, Allen JG, et al. Discovery of a covalent inhibitor of KRASG12C (AMG 510) for the treatment of solid tumors. J Med Chem. 2020;63(1):52–65. doi:10.1021/acs.jmedchem.9b01180
32. Goebel L, Müller MP, Goody RS, Rauh D. KRasG12C inhibitors in clinical trials: a short historical perspective. RSC Med Chem. 2020;11(7):760–770. doi:10.1039/D0MD00096E
33. Koga T, Suda K, Fujino T, et al. KRAS secondary mutations that confer acquired resistance to KRAS G12C inhibitors, sotorasib and adagrasib, and overcoming strategies: insights from in vitro experiments. J Thorac Oncol. 2021;16(8):1321–1332. doi:10.1016/j.jtho.2021.04.015
34. Zhang SS, Nagasaka M. Spotlight on Sotorasib (AMG 510) for KRAS G12C positive Non-Small Cell Lung Cancer. Lung Cancer. 2021;12:115–122. doi:10.2147/LCTT.S334623
35. Zhao Y, Murciano-Goroff YR, Xue JY, et al. Diverse alterations associated with resistance to KRAS(G12C) inhibition. Nature. 2021;599(7886):679–683. doi:10.1038/s41586-021-04065-2
36. Xue JY, Zhao Y, Aronowitz J, et al. Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature. 2020;577(7790):421–425. doi:10.1038/s41586-019-1884-x
37. Awad MM, Liu S, Rybkin II, et al. Acquired resistance to KRAS G12C inhibition in cancer. N Engl J Med. 2021;384(25):2382–2393. doi:10.1056/NEJMoa2105281
38. NCT04185883. Sotorasib activity in subjects with advanced solid tumors with KRAS p.G12C mutation (CodeBreak 101). Available from: https://www.clinicaltrials.gov/ct2/show/NCT04185883.
39. Tsai YS, Woodcock MG, Azam SH, et al. Rapid idiosyncratic mechanisms of clinical resistance to KRAS G12C inhibition. J Clin Invest. 2022;132(4):e155523. doi:10.1172/JCI155523
40. Robert C, Grob JJ, Stroyakovskiy D, et al. Five-year outcomes with dabrafenib plus trametinib in metastatic melanoma. N Engl J Med. 2019;381(7):626–636. doi:10.1056/NEJMoa1904059
41. Planchard D, Besse B, Groen HJM, et al. Dabrafenib plus trametinib in patients with previously treated BRAF(V600E)-mutant metastatic non-small cell lung cancer: an open-label, multicentre phase 2 trial. Lancet Oncol. 2016;17(7):984–993. doi:10.1016/S1470-2045(16)30146-2
42. Corcoran RB, André T, Atreya CE, et al. Combined BRAF, EGFR, and MEK inhibition in patients with BRAFV600E-mutant colorectal cancer. Cancer Discov. 2018;8(4):428–443. doi:10.1158/2159-8290.CD-17-1226
43. Planchard D, Smit EF, Groen HJM, et al. Dabrafenib plus trametinib in patients with previously untreated BRAFV600E-mutant metastatic non-small-cell lung cancer: an open-label, phase 2 trial. Lancet Oncol. 2017;18(10):1307–1316. doi:10.1016/S1470-2045(17)30679-4
44. Dunnett-Kane V, Nicola P, Blackhall F, Lindsay C. Mechanisms of resistance to KRASG12C inhibitors. Cancers. 2021;13(1):E151. doi:10.3390/cancers13010151
45. Singh A, Greninger P, Rhodes D, et al. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell. 2009;15(6):489–500. doi:10.1016/j.ccr.2009.03.022
46. Amodio V, Yaeger R, Arcella P, et al. EGFR blockade reverts resistance to KRAS G12C inhibition in colorectal cancer. Cancer Discov. 2020;10(8):1129–1139. doi:10.1158/2159-8290.CD-20-0187
47. Lee S, Jeong W, Cho Y, et al. β‐Catenin‐
48. Kopetz S, Desai J, Chan E, et al. Phase II pilot study of Vemurafenib in patients with metastatic BRAF-mutated colorectal cancer. J Clin Oncol. 2015;33(34):4032–4038. doi:10.1200/JCO.2015.63.2497
49. Van Cutsem E, Huijberts S, Grothey A, et al. Binimetinib, Encorafenib, and Cetuximab triplet therapy for patients with BRAF V600E–mutant metastatic colorectal cancer: safety Lead-in results from the Phase III BEACON colorectal cancer study. J Clin Oncol. 2019;37(17):1460–1469. doi:10.1200/JCO.18.02459
50. Kopetz S, Grothey A, Yaeger R, et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E-mutated colorectal cancer. N Engl J Med. 2019;381(17):1632–1643. doi:10.1056/NEJMoa1908075
51. Hong DS, Morris VK, El Osta B, et al. Phase IB study of Vemurafenib in combination with Irinotecan and Cetuximab in patients with metastatic colorectal cancer with BRAFV600E mutation. Cancer Discov. 2016;6(12):1352–1365. doi:10.1158/2159-8290.CD-16-0050
52. Kopetz S, Guthrie KA, Morris VK, et al. Randomized trial of Irinotecan and Cetuximab with or without Vemurafenib in BRAF-mutant metastatic colorectal cancer (SWOG S1406). J Clin Oncol. 2021;39(4):285–294. doi:10.1200/JCO.20.01994
53. Fakih M, Falchook GS, Hong DS, et al. 434P CodeBreaK 101 subprotocol H: phase Ib study evaluating combination of sotorasib (Soto), a KRASG12C inhibitor, and panitumumab (PMab), an EGFR inhibitor, in advanced KRAS p.G12C-mutated colorectal cancer (CRC). Ann Oncol. 2021;32:S551. doi:10.1016/j.annonc.2021.08.955
54. Pfeiffer P, Qvortrup C. KRASG12C inhibition in colorectal cancer. Lancet Oncol. 2022;23(1):10–11. doi:10.1016/S1470-2045(21)00652-5
55. Ramalingam S, Fakih M, Strickler J, et al. Abstract P05-01: a phase 1b study evaluating the safety and efficacy of sotorasib, a KRAS G12C inhibitor, in combination with trametinib, a MEK inhibitor, in KRAS p.G12C-mutated solid tumors. In: Oral Presentations - Late-Breaking Proffered Abstracts. American Association for Cancer Research; 2021. doi:10.1158/1535-7163.TARG-21-P05-01
56. Santana-Codina N, Chandhoke AS, Yu Q, et al. Defining and targeting adaptations to oncogenic KRASG12C inhibition using quantitative temporal proteomics. Cell Rep. 2020;30(13):4584–4599.e4. doi:10.1016/j.celrep.2020.03.021
57. Lou K, Steri V, Ge AY, et al. KRASG12C inhibition produces a driver-limited state revealing collateral dependencies. Sci Signal. 2019;12(583):eaaw9450. doi:10.1126/scisignal.aaw9450
58. Molina-Arcas M, Moore C, Rana S, et al. Development of combination therapies to maximize the impact of KRAS-G12C inhibitors in lung cancer. Sci Transl Med. 2019;11(510):eaaw7999. doi:10.1126/scitranslmed.aaw7999
59. Huang Y, Wang J, Cao F, et al. SHP2 associates with nuclear localization of STAT3: significance in progression and prognosis of colorectal cancer. Sci Rep. 2017;7(1):17597. doi:10.1038/s41598-017-17604-7
60. Fedele C, Li S, Teng KW, et al. SHP2 inhibition diminishes KRASG12C cycling and promotes tumor microenvironment remodeling. J Exp Med. 2021;218(1):e20201414. doi:10.1084/jem.20201414
61. Ou SI, Koczywas M, Ulahannan S, et al. A12 the SHP2 inhibitor RMC-4630 in patients with KRAS-mutant non-small cell lung cancer: preliminary evaluation of a first-in-man phase 1 clinical trial. J Thorac Oncol. 2020;15(2):S15–S16. doi:10.1016/j.jtho.2019.12.041
62. Stice JP, Donovan S, Sun Y, et al. Abstract P207: BBP-398, a potent, small molecule inhibitor of SHP2, enhances the response of established NSCLC xenografts to KRAS G12C and mutEGFR inhibitors. In: Poster Presentations - Proffered Abstracts. American Association for Cancer Research; 2021:P207–P207. doi:10.1158/1535-7163.TARG-21-P207
63. Liu C, Lu H, Wang H, et al. Combinations with allosteric SHP2 inhibitor TNO155 to block receptor tyrosine kinase signaling. Clin Cancer Res. 2021;27(1):342–354. doi:10.1158/1078-0432.CCR-20-2718
64. Lee JW, Kim S, Yang C, Burtness B. Abstract P078: aurora A kinase inhibition with VIC-1911 overcomes intrinsic and acquired resistance to KRAS G12C inhibition in KRAS(G12C) -mutated lung cancer. In: Poster Presentations - Proffered Abstracts. American Association for Cancer Research; 2021:P078–P078. doi:10.1158/1535-7163.TARG-21-P078
65. Nichols RJ, Cregg J, Schulze CJ, et al. Abstract 1261: a next generation tri-complex KRASG12C(ON) inhibitor directly targets the active, GTP-bound state of mutant RAS and may overcome resistance to KRASG12C(OFF) inhibition. Cancer Res. 2021;81(13_Supplement):1261. doi:10.1158/1538-7445.AM2021-1261
66. NCT04793958. Phase 3 study of MRTX849 with Cetuximab vs Chemotherapy in patients with advanced colorectal Cancer with KRAS G12C mutation (KRYSTAL-10). Available from: https://clinicaltrials.gov/ct2/show/NCT04793958.
67. NCT05198934. Sotorasib and panitumumab versus investigator’s choice for participants with Kirsten Rat Sarcoma (KRAS) p.G12C mutation (CodeBreak 300). Available from: https://clinicaltrials.gov/ct2/show/NCT05198934.
68. NCT04303780. Study to compare AMG 510 “Proposed INN Sotorasib” with docetaxel in non small cell lung cancer (NSCLC) (CodeBreak 200). Available from: https://www.clinicaltrials.gov/ct2/show/NCT04303780.
69. NCT04933695. A study of Sotorasib (AMG 510) in participants with stage IV NSCLC whose tumors harbor a KRAS p.G12C mutation in need of first-line treatment (CodeBreaK201). Available from: https://clinicaltrials.gov/ct2/show/NCT04933695.
70. NCT05118854. A Phase II study of Neoadjuvant Sotorasib in combination with Cisplatin or Carboplatin and pemetrexed for surgically resectable stage IIA-IIIB non-squamous Non-Small Cell Lung Cancer With a KRAS p.G12C mutation. Available from: https://clinicaltrials.gov/ct2/show/NCT05118854.
71. Wang X, Allen S, Blake JF, et al. Identification of MRTX1133, a noncovalent, potent, and selective KRASG12D inhibitor. J Med Chem. 2022;65(4):3123–3133. doi:10.1021/acs.jmedchem.1c01688
72. Gustafson WC, Wildes D, Rice MA, et al. Direct targeting of RAS in pancreatic ductal adenocarcinoma with RMC-6236, a first-in-class, RAS-selective, orally bioavailable, tri-complex RAS MULTI (ON) inhibitor. J Clin Oncol. 2022;40(4_suppl):591. doi:10.1200/JCO.2022.40.4_suppl.591
73. Gort E, Johnson ML, Hwang JJ, et al. A Phase I, open-label, dose-escalation trial of BI 1701963 as monotherapy and in combination with trametinib in patients with KRAS mutated advanced or metastatic solid tumors. J Clin Oncol. 2020;38(15_suppl):TPS3651–TPS3651. doi:10.1200/JCO.2020.38.15_suppl.TPS3651
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.