Back to Journals » International Journal of Nanomedicine » Volume 14
Targeting breast cancer stem cells by a self-assembled, aptamer-conjugated DNA nanotrain with preloading doxorubicin
Received 5 January 2019
Accepted for publication 15 May 2019
Published 27 August 2019 Volume 2019:14 Pages 6831—6842
DOI https://doi.org/10.2147/IJN.S200482
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lei Yang
Zhiyuan Xu,1 Ronghua Ni,1 Yun Chen1,2
1School of Pharmacy, Nanjing Medical University, Nanjing 211166, People’s Republic of China; 2State Key Laboratory of Reproductive Medicine, Nanjing 210029, People’s Republic of China
Correspondence: Yun Chen
School of Pharmacy, Nanjing Medical University, 818 Tian Yuan East Road
, Nanjing 211166, People’s Republic of China
Tel +86 258 686 8326
Fax +86 258 686 8467
Email [email protected]
Background: Cancer relapse and metastasis is an obstacle to the treatment of breast cancer. Breast cancer stem cells (BCSCs), which can evade the killing effect of traditional chemotherapies, such as doxorubicin (DOX), may contribute to cancer development. Therefore, it is necessary to develop novel drugs that can target and eliminate BCSCs. While multiple strategies have been conceived, they are normally limited by the low drug loading capacity.
Purpose: An aptamer-conjugated DNA nanotrain TA6NT-AKTin-DOX, which consists of a CD44 aptamer TA6, DNA building blocks M1 and M2 conjugated with an AKT inhibitor peptide AKTin individually and DOX, was designed.
Methods: This DNA nanotrain was prepared through hybridization chain reactionand this highly ordered DNA duplex has plenty of sites where DOX and AKTin can be intercalated or anchored. By performing on MCF-7 BCSCs and tumors by xenografting BCSCs into nude mice, efficacy of the newly prepared drug was evaluated and compared with that of free DOX and various DNA nanotrains.
Results: TA6NT-AKTin-DOX showed better efficacy both in vitro and in vivo. To some extent, the enhanced efficacy could be attributed to the targeting effect of TA6 and the high drug loading capacity of the nanotrain (∼20 DOX molecules). Besides, a synergistic response was demonstrated by combining DOX with AKTin, probably due to that the anchored AKTin can reverse the drug resistance of BCSCs including apoptosis resistance and ABC transporters overexpression via the AKT signaling pathway.
Conclusion: The aptamer-conjugated DNA nanotrain TA6NT-AKTin-DOX demonstrated its targeting capability to BCSCs.
Keywords: breast cancer stem cells (BCSCs), aptamer-conjugated DNA nanotrain, CD44 aptamer TA6, doxorubicin (DOX), AKT peptide inhibitor, hybridization chain reaction
Introduction
Breast cancer has the highest morbidity rate and death rate among females worldwide.1 There is evidence indicating that ~40% of breast cancer patients experience relapsed disease and 60–70% of the relapsed cases have metastasis, despite of administered treatment, such as chemotherapy and/or adjuvant therapies.2,3 Thus, cancer relapse and metastasis still serve as an obstacle in the treatment of breast cancer. Currently, BCSCs that can evade the killing effect of traditional chemotherapies, such as doxorubicin (DOX), may contribute to cancer relapse and metastasis.4 These cells are believed to act as a pivotal part in cancer establishment, progression, and most importantly in drug resistance due to their suppression of apoptosis and their increased expression of ATP-binding cassette (ABC) transporters, which pump drugs out of cells.5 Therefore, novel drugs targeting BCSCs could be helpful to cure breast cancer.
CD44 is a transmembrane glycoprotein and increasingly recognized as a marker for BCSCs.6 Recent studies have indicated that aptamers, which are a class of oligonucleotides with a three-dimensional structure, can bind to CD44 in nanomolar range.7 In addition, there is evidence indicating that aptamers can further mediate the transport of drugs into BCSCs via endocytosis.8,9 However, strategies based on direct conjugation of aptamers with drugs have own restrictions that could obstruct clinical translation, such as laborious and inefficient preparation of aptamer-drug conjugates and especially the low drug loading capacity.8,10,11 One potential solution is the aptamer-conjugated DNA nanotrain.12 It is a long linear double-stranded DNA nanostructure self-assembled through the hybridization chain reaction (HCR).13 HCR normally uses a pair of relatively short DNA building blocks to propagate a chain reaction of hybridization upon initiation of aptamer-tethered trigger probe.13 The product of HCR is highly ordered DNA duplex and drug molecules, such as DOX, can be intercalated or anchored on these helices, which allows high-capacity loading of drugs.14
In addition to targeting delivery of drugs to BCSCs, agents that can overcome drug resistance are also of great interest because apoptosis resistance and ABC transporters overexpression are key characteristics of BCSCs as mentioned earlier. Currently, much evidence has shown that AKT (protein kinase B) is a central node in these resistant events.15 For example, AKT can regulate the expression and function of ABC transporters.16 Additionally, the AKT signaling pathway controls the apoptosis of stem cells.17,18 Thus, treatment with AKT inhibitors should aid to reverse the drug resistance of BCSCs and further improve the clinical outcome. One of the inhibitors is NH2-AVTDHPDRLWAWEKFCOOH (AKTin), which can encompass the βA strand of human TCL1, and thus interact with AKT and specifically inhibit its kinase activity.19 Therefore, AKTin was often used to induce apoptosis via the AKT signaling pathway.20
In this study, an aptamer-conjugated DNA nanotrain TA6NT-AKTin-DOX, which consists of a CD44 aptamer TA6, DNA building blocks M1 and M2 conjugated with an AKT inhibitor peptide AKTin individually and DOX, was first designed (Figure 1). This DNA nanotrain was prepared upon initiation of TA6-tethered DNA trigger through HCR, followed by loading of DOX. Afterward, a comprehensive characterization of TA6NT-AKTin-DOX was performed. By performing on MCF-7 BCSCs and tumors by xenografting BCSCs into nude mice, efficacy of the newly prepared drug was evaluated and compared with that of free DOX and various DNA nanotrains. Furthermore, the effect of drug treatment on the apoptosis of cancer stem cell including the AKT signaling pathway and the reversal of drug resistance was specifically explored.
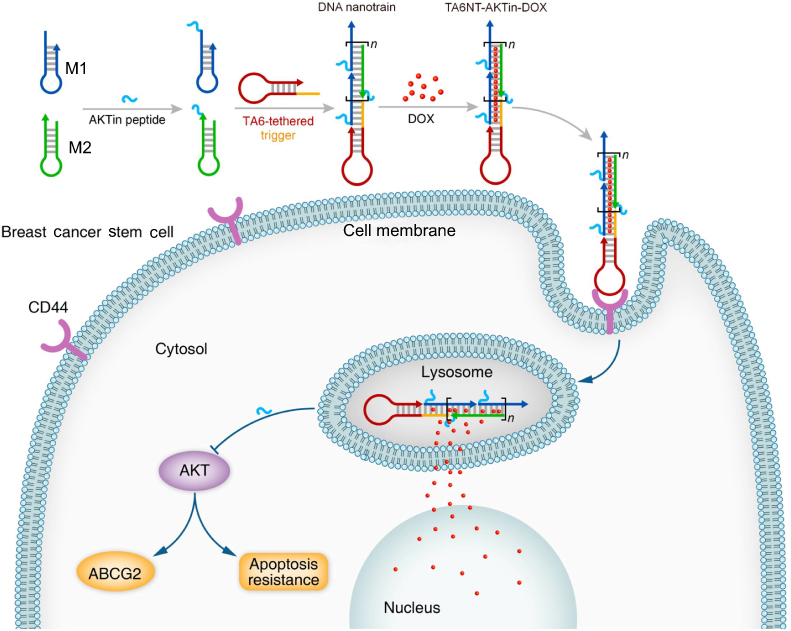 |
Figure 1 Schematic diagram of self-assembled TA6NT-AKTin-DOX. TA6NT-AKTin-DOX consists of a CD44 aptamer TA6, DNA building blocks M1 and M2 conjugated by an AKT inhibitor peptide AKTin individually, and DOX. This aptamer-conjugated DNA nanotrain was first taken up via targeted endocytosis. Following the release in acidic organelles, DOX diffused into the cell nucleus and the AKTin inhibited AKT. Drug resistance reversal can be achieved by interfering ABCG2-mediated drug efflux and inducing apoptosis. |
Experimental
Chemicals and reagents
Please see the Supplementary materials.
Preparation of DNA-peptide conjugates as building blocks
The DNA-peptide conjugates M1-AKTin and M2-AKTin were prepared according to the previous study.21 The disulfide modifications of M1 and M2 were first reduced prior to conjugation. Tris(2-carboxyethyl)-phosphine (TCEP) was used as the reducing agent. A total of 200 μL of 20 μmol/L M1 and M2 were separately incubated with 100 μL of TCEP reducing beads (Thermo Fisher Scientific, Chicago, IL, USA) at 37°C for 2 hrs with vigorous shaking. Afterward, the samples were centrifuged at 1,000× g for 5 mins. The supernatant containing the reduced M1 and M2 were separately collected and added with an equal volume of 200 μmol/L maleimide modified AKTin. The conjugation reaction was carried out at 37°C for 4 hrs with vigorous shaking, followed by immediate purification from the excess of M1, M2 and peptide using preparative HPLC. Specially, the samples were purified using an XBridge Prep C18 OBD column (5 μm, 19 mm×150 mm; Waters, USA) at room temperature. The mobile phase consisted of solvent A (50 mmol/L triethylamine, pH 7.6) and solvent B (ACN). A linear gradient with a flow rate of 10 mL/min was applied in the following manner: B 5% (0 min) → 20% (5 mins) → 30% (20 mins) → 5% (25 mins). The detection wavelength was set to 260 nm.
Preparation of aptamer-conjugated DNA nanotrain TA6NT-AKTin-DOX
TA6-tethered trigger, M1-AKTin and M2-AKTin were individually heated at 95°C for 5 mins, cooled immediately on ice for 5 mins and then left at room temperature for 1 hr. Then, TA6-tethered trigger, M1-AKTin and M2-AKTin were mixed and left at room temperature for 24 hrs with vigorous shaking. The HCR system was prepared in PBS supplemented with MgCl2 (5 mM). The HCR product TA6NT-AKTin was confirmed by agarose gel (3%) electrophoresis (80 V, 60 mins), followed by Gel Doc™ XR+ Gel Documentation System (Bio-Rad, Hercules, CA, USA).
Afterward, DOX and TA6NT-AKTin were incubated at room temperature for 2 hr in PBS supplemented with MgCl2 (5 mM). The drug loading capacity of DNA nanotrain was calculated by performing in a 96-well plate where each well contained 1 μM DOX and various amounts of TA6NT-AKTin, at DOX/TA6NT-AKTin molar ratios of 320, 160, 80, 40, 20, 0 and DOX only, respectively. Fluorescence was determined using TECAN reader infinite M200 pro (TECAN, Männedorf, Switzerland).
To determine the stability of the obtained aptamer-conjugated DNA nanotrain TA6NT-AKTin-DOX and its DOX release profile at different conditions, TA6NT-AKTin-DOX was prepared in PBS at pH 7.4, PBS at pH 7.4 containing 10% FBS, PBS at pH 5.0, PBS at pH 5.0 containing DNase II (0.1 U/µL) and incubated at 37°C. The amount of released DOX was determined using TECAN reader infinite M200 pro (TECAN). To determine the release of AKTin, the disassembly of this DNA nanotrain was confirmed by agarose gel (3%) electrophoresis (80 V, 60 mins), followed by Gel Doc™ XR+ Gel Documentation System (Bio-Rad).
Cell culture
Please see the Supplementary materials.
In vitro studies
Mammosphere formation assay was used to test the formation efficiency of BCSCs. BCSCs were treated with 2 µM free DOX and equivalent TA6NT-AKTin-DOX at 37°C for 12 hrs. 1×104 BCSCs were plated per well in ultra-low-attachment six-well plates (Corning, NY, USA) in Complete MammoCult™ Medium. Mammospheres were counted after 14 days of culture using Olympus IX2-SLP microscope (Olympus, Tokyo, Japan).
MTT assay was used to evaluate the cytotoxicity of TA6NT-AKTin-DOX. BCSCs and NIH-3T3 cells were seeded in a 96-well plate and incubated for 24 hrs. Then, cells were treated with a series of dilutions of free DOX, TA6NT-DOX (aptamer-conjugated DNA nanotrain TA6NT-DOX without AKTin anchoring) and TA6NT-AKTin (DNA nanotrain TA6NT-AKTin without DOX anchoring) together with DOX and TA6NT-AKTin-DOX at equivalent concentration for 48 hrs. After an addition of MTT solution (5 mg/mL in PBS) to each well, the cells were incubated for 4 hrs and lysed in 200 μL DMSO. Absorbance was read on an EL×800 absorbance microplate reader (Biotek, USA) at 490 nm. The IC50 values were calculated by GraphPad Prism 6 software (GraphPad Software, CA, USA).
For drug distribution study, BCSCs and NIH-3T3 cells were incubated with 2 µM free DOX and equivalent NT-AKTin-DOX and TA6NT-AKTin-DOX for 12 hrs at 37°C, respectively. Then, cells were washed with PBS and stained with 2 µg/mL DAPI for 5 mins. Finally, cells were imaged using inverted fluorescence microscope (Leica, DMI3000B, Wetzlar, Germany). The drug-treated cells were divided into two equal parts. One was used for whole cell extraction, the other was for subcellular studies. Cells for whole cell extraction were washed with PBS and resuspended in 1 mL of 0.2 M disodium hydrogen phosphate. Their counterpart was disrupted by the Dounce homogenizer with 0.5–2.5×10−3 inch clearance (Kontes, Vineland, NJ, USA). Following the disruption, the nuclear-enriched fraction was obtained by a centrifugation at 1,400× g for 5 mins and resuspended in 1 mL of 0.2 M disodium hydrogen phosphate. 4 mL extraction solvent (9:1 chloroform:heptanol) was added to cells and the nuclear-enriched fraction. Afterward, the samples were centrifuged at 4,000× g for 15 mins. Organic layer was removed and aqueous phase was re-extracted with 1 mL of extraction solvent for two more times. The solutions were combined and evaporated to dryness, and then resuspended in 100 μL of acetonitrile/ water 50:50 for LC-MS/MS analysis (please also see the Supplementary materials).22
BCSCs were exposed to 2 µM free DOX and equivalent TA6NT-DOX, TA6NT-AKTin together with DOX and TA6NT-AKTin-DOX for 24 hrs at 37°C. TUNEL assay was performed according to the DeadEnd Fluorometric TUNEL system (Promega, WI, USA). Cells with bright nuclear green fluorescence are regarded as apoptotic cells. Cells were imaged using an inverted fluorescence microscope (Leica, DMI3000B). Cells were counted with a hemocytometer (Qiujing, Shanghai, People's Republic of China).
Analysis of AKT-mediated drug resistance
BCSCs were exposed to 2 µM free DOX and TA6NT-AKTin-DOX for 24 hrs at 37°C. Protein was fractionated and extracted according to the previous study.23 For cytosol proteins, the cells were washed PBS and collected with 100 μL of RIPA buffer (Beyotime Institute of Biotechnology, People's Republic of China) containing a protease inhibitor cocktail (Sigma-Aldrich, MO, USA) and a phosphatase inhibitor cocktail (Sigma-Aldrich) by cell scraper (Corning, NY, USA). For membrane protein, cells were centrifuged at 1,480× g for 10 mins and resuspended in 500 µL 1% Triton X-114 extraction buffer (1 mM DTT, 2 mM EDTA in 50 mM Tris/HCl, pH 7.4) containing a protease inhibitor cocktail. After an incubation period of 30 mins on ice and 10 mins at 37°C, samples were centrifuged at 10,000× g for 5 mins to separate the detergent and aqueous phases. Protein was precipitated from the detergent in cold acetone. All protein pellets were dissolved in a 1% SDS solution. Finally, all the samples were subject to Western blotting.
In vivo study
Thirty-nine 6-week-old female BALB/c mice were used. The animal experiments were performed according to guidelines evaluated and approved by the ethics committee of Nanjing Medical University. An orthotopic mouse model of breast cancer was generated by the injection of 5×103 BCSCs suspended in 100 µL PBS at the mammary fat pad of each mouse.
When the average tumor volume was about 50 mm3, 30 mice were randomly divided into five groups. Animals were treated with 5 mg DOX equivalent/kg of either free DOX, TA6NT-DOX, TA6NT-AKTin together with DOX or TA6NT-AKTin-DOX once every 7 days for three times. Tumor growth and body weight were monitored every 3 days. Tumor volume was calculated based on the equation: TV=1/2×length×width2. To obtain biodistribution images, images were taken 4 hrs after intravenous injection with 5 mg DOX equivalent/kg of free DOX, NT-AKTin-DOX and TA6NT-AKTin-DOX on IVIS LuminaXR imaging system (Perkin Elmer, USA). Tumor, heart, liver, spleen, kidney and lung were isolated and stored in liquid nitrogen.
Approximately 50 mg tissue was weighed and resuspended in 1 mL of 0.2 M disodium hydrogen phosphate. Then, samples were homogenized using a Bio-Gen PRO200 homogenizer (PRO Scientific Inc., Oxford, CT, USA) for 1 min and vortexed for 5 mins. Extraction process is same as above. Samples were also analyzed for LC-MS/MS analysis.22
Bliss synergy calculations
Please see the Supplementary materials.
Results
Synthesis and characterization of DNA building blocks
To prepare DNA-peptide conjugates as building blocks, the thiol-maleimide linkage was used to link M1 and M2 with AKTin by Michael addition reaction.24 Briefly, the 5′ thiol-modified M1 and 3′ thiol-modified M2 (Figure 1 and Table S1) were first activated and then reacted with the maleimide group at the N-terminus of AKTin. As shown in the HPLC chromatogram (Figure S1), the peaks occurred at 10.6 mins for M1, 12.9 mins for M1-AKTin, 8.4 mins for M2 and 12.4 mins for M2-AKTin, respectively. The result of mass spectrometry also confirmed the formation of the DNA-peptide conjugates (Figure S1). The molecular mass increase of M1-AKTin and M2-AKTin matched the molecular weight of the added AKTin.
Construction and characterization of TA6NT-AKTin-DOX
In the assembly of TA6-conjugated DNA nanotrain TA6NT-AKTin, the aptamer TA6 that can bind to BCSCs but fail to bind to non-stem cells such as NIH-3T3 cells, was chosen as a targeting moiety (Figure S2 and Table S1).7 A DNA trigger was modified on the 5′-end of TA6 as TA6-tethered trigger to initiate the formation of TA6NT-AKTin self-assembly (Figure S2). Agarose gel electrophoresis indicated the assembly of long nanotrain (Figure S3). In addition, the specificity of TA6 was retained in its tethered form, as shown by flow cytometry (Figure S3). To maximize the subsequent drug loading, the molar ratio of TA6-tethered trigger to M1-AKTin and M2-AKTin in the initial reaction mixture was optimized. The result showed that 1:3 ratio of trigger: DNA building blocks can provide relatively higher number of “boxcar” compartments (Figure 2A).
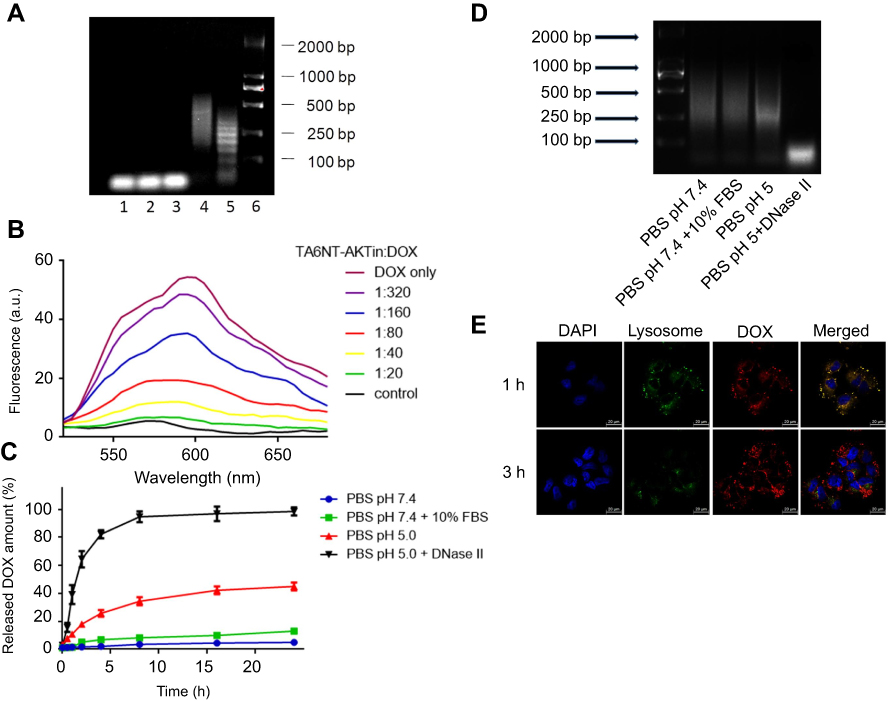 |
Figure 2 Construction and characterization of TA6NT-AKTin-DOX. (A) Agarose gel electrophoresis showing the effect of TA6-tethered trigger concentration on HCR amplification. Lanes 1–5: five different concentrations of TA6-tethered trigger (0.00, 0.10, 0.20, 0.33 and 1.00 μM) in a 1 μM mixture of M1-AKTin and M2-AKTin. Lane 6: DL2000 DNA marker. (B) Fluorescence spectra of DOX (1 μM) with increasing equivalents of TA6NT-AKTin, at DOX/TA6NT-AKTin molar ratios of 320, 160, 80, 40, 20, 0 and DOX only, respectively. The fluorescence quenching indicates DOX loading into TA6NT-AKTin. (C) In vitro release profiles of DOX at pH 7.4 containing 10% FBS, pH 7.4, 5.0 and 5.0 with DNase II. Data are mean±SD (n=3). (D) Agarose gel electrophoresis of TA6NT-AKTin-DOX at pH 7.4 containing 10% FBS, pH 7.4, 5.0 and 5.0 with DNase II for 24 hrs. (E) Fluorescence images of BCSCs treated with both LysoTracker Green and TA6NT-AKTin-DOX. |
As is well known to all, DOX can emit a fluorescence, which can be quenched when it intercalates into DNA structure.14,25 Taking advantage of this phenomenon, the formation of the TA6NT-AKTin-DOX can be evaluated. Specifically, DOX at a fixed concentration was incubated with TA6NT-AKTin at varying concentrations. The native fluorescence intensity of DOX decreased with increasing concentration of TA6NT-AKTin (Figure 2B). The fluorescence of DOX was completely gone when TA6NT-AKTin/DOX molar ratio achieved 1:20. This fluorescence can be subsequently recovered after the release of DOX in cells.26
To further examine the stability of TA6NT-AKTin-DOX, this DNA nanotrain was incubated with PBS at pH 7.4 containing 10% FBS. The complex was apparently stable with about 10% release after 24 h. To ensure the intracellular triggered release of DOX once inside acidic organelles such as endosomes and lysosomes (pH~4.5–6.0), we examined the drug release profile at pH =5 and with DNase II. The pH of 5 simulates the acidic features of acidic organelles and DNase II, which is an acidic endonuclease, is usually found within intracellular compartments, most notably lysosomes. The data revealed that the release of DOX was much faster at pH 5.0 compared with that at pH 7.4, with approximately 40% after 24 hrs, respectively (Figure 2C). This speed of release was even faster after the addition of DNase II (~100%), which plays a role in the degradation of exogenous DNA encountered by endocytosis.27 Moreover, the degradation of TA6NT-AKTin-DOX was also confirmed by the existence of broken DNA strands using agarose gel electrophoresis (Figure 2D). The cytosol and lysosomal distribution of DOX suggested a successful lysosomal escape of the drug (Figure 2E).
Inhibition of mammosphere formation
Mammospheres formation of stem cells in serial non-adherent passage is up to its self-renewal ability.28,29 Thus, mammosphere formation assay was used in this study to evaluate whether TA6NT-AKTin-DOX could suppress the activity of stem cells. After examination of CD24 and CD44 expression of BCSCs at both early and last passages (Figure S4), the result of the assay indicated there was no significant difference in the number of mammospheres between the control group and the DOX group, whereas TA6NT-AKTin-DOX had a significant inhibition effect (Figure 3A). The number of spheres declined by 57.1±1.9% (p<0.01).
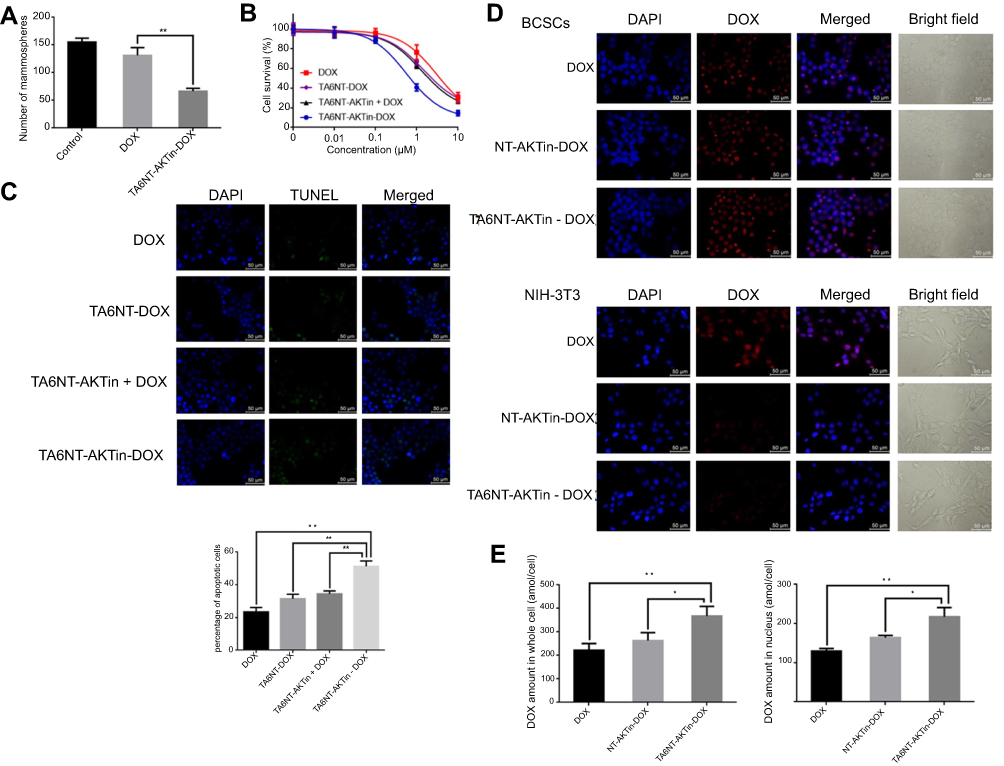 |
Figure 3 (A) Quantification of the ability to regenerate mammosphere of BCSCs after treating 2 µM DOX and TA6NT-AKTin-DOX for 12 hrs. (B) Cytotoxicity profiles of free DOX, TA6NT-DOX, TA6NT-AKTin together with DOX and TA6NT-AKTin-DOX for BCSCs (n=3). (C) Cellular apoptosis of BCSCs detected using TUNEL assay. Nuclei were stained with DAPI, and merged images were considered as apoptotic cells. (D) Fluorescence images of BCSCs and NIH-3T3 cells after the treatment of 2 µM free DOX, NT-AKTin-DOX and TA6NT-AKTin-DOX for 12 hrs, and (E) the measured DOX amounts in the whole cell and nucleus isolated from BCSCs (n=3). Two-tailed Student’s t-test was used. *p<0.05, **p<0.01. |
Cytotoxicity of TA6NT-AKTin-DOX
The cells were treated with increasing concentrations of free DOX, TA6NT-DOX and TA6NT-AKTin together with DOX and TA6NT-AKTin-DOX, respectively. The result indicated that TA6NT-AKTin-DOX inhibited the growth of BCSCs in a dose-dependent manner (Figure 3B). The IC50 value of TA6NT-AKTin-DOX was 764.6±98.9 nM. This value was significantly lower than that of free DOX (3862.0±507.3 nM), 2779.0±259.8 nM for TA6NT-DOX (aptamer-conjugated DNA nanotrain TA6NT-DOX without AKTin anchoring) and 2331.0±183.7 nM for TA6NT-AKTin (DNA nanotrain TA6NT-AKTin without DOX intercalation) together with DOX (n=3, p<0.01), which demonstrated that BCSCs were more sensitive to TA6NT-AKTin-DOX. The cytotoxicity profiles of these compounds in MCF-7 cells and TA6NT-AKTin in BCSCs were also provided in Figure S5. As shown, TA6NT-AKTin-DOX has some cytotoxic effect on MCF-7 cells.
DOX and AKTin are two active drug components in TA6NT-AKTin-DOX. To further assess the efficacy of combining DOX with AKTin, the Bliss independence method was adopted. As shown by the analysis, TA6NT-AKTin-DOX (observed) had a better efficacy than either agent alone. What is more, TA6NT-AKTin-DOX generated more fractional inhibition compared with the predicted inhibition by the simple combination of DOX with AKTin, which implied the presence of a synergistic interaction between DOX and AKTin (Table S2).
Effects of TA6NT-AKTin-DOX on apoptosis, DNA damage and intracellular drug accumulation
TUNEL assay was used to show the DNA damage in nuclei, which was indicated by counterstaining with DAPI. TUNEL-positive or apoptotic cells are defined as cells with nuclear green fluorescence labeling.30 As shown in Figure 3C, the number of TUNEL-positive cells significantly increased after 24 hrs treatment. The percentage of apoptotic cells was 51.2±2.0% for TA6NT-AKTin-DOX and much higher than 23.5±1.5% for DOX, 31.5±1.6% for TA6NT-DOX and 34.5±1.0% for TA6NT-AKTin together with DOX (n=3, p<0.01).
The intracellular DOX distribution in BCSCs was imaged by inverted fluorescence microscope (Figure 3D). The amounts of DOX in whole cell and nuclei were also determined by LC-MS/MS (Figure 3E). As shown in the images, TA6NT-AKTin-DOX was internalized and concentrated most heavily in the nuclear envelope, which was consistent with the previous observations of the released DOX from the acidic organelles.31,32 Furthermore, free DOX that enters cells via simple diffusion can be easily pumped out by ABC transporters. Therefore, the greater intracellular accumulation of TA6NT-AKTin-DOX than free DOX implied that TA6NT-AKTin-DOX bypassed drug efflux pumps and achieved the targeted delivery. Moreover, the DNA nanotrain NT-AKTin-DOX without TA6 moiety can also be taken into the cells via endocytosis, but was not efficient and active as targeted or site-specific delivery in this study. The targeting of TA6NT-AKTin-DOX to BCSCs was also confirmed by its lower accumulation and higher IC50 in CD44− NIH-3T3 cells (Figure S6).
Effect of TA6NT-AKTin-DOX on reversal of drug resistance
Previous studies have demonstrated that the AKT signaling pathway acts as a pivotal part in drug resistance, including the resistance of apoptosis and the overexpression of ABC transporters.16–18 Notably, AKT is activated through phosphorylation mechanism, which can lead to repressing apoptosis involving the downstream cleavage of caspase-3. In addition, there is evidence indicating that ABC transporters, such as ABCG2, overexpressed after the activation of AKT. Therefore, the expression levels of p-AKT, cleaved caspase-3 and ABCG2 were evaluated in this study. As shown in Figure 4, decrease of p-AKT and ABCG2 was observed in BCSCs treated with TA6NT-AKTin-DOX in comparison with free DOX alone, whereas greater extent of active caspase-3 increase was observed in the group of TA6NT-AKTin-DOX. These findings provide preliminary support for that TA6NT-AKTin-DOX can reverse the drug resistance of BCSCs via the AKT signaling pathway.
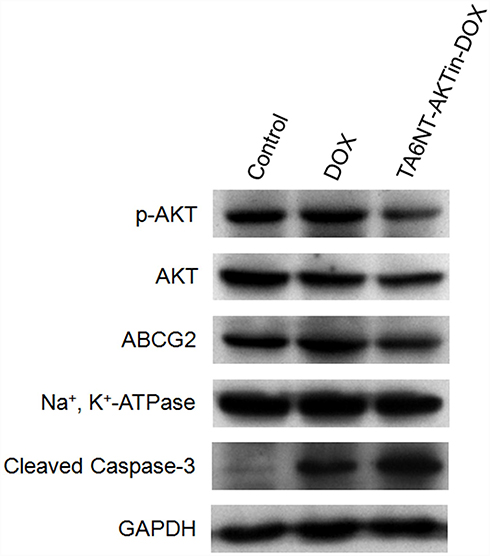 |
Figure 4 Western blotting of p-AKT, AKT, ABCG2 and cleaved caspase-3 in BCSCs with the treatment of DOX and TA6NT-AKTin-DOX. |
In vivo anti-tumor effect and biodistribution study
BCSCs tumor-bearing mice were treated with 5 mg DOX equivalent/kg of either TA6NT-AKTin-DOX, TA6NT-DOX, TA6NT-AKTin together with DOX, free DOX and saline once every 7 days for three times via vein injection.33 Tumor curves of five groups have been shown in Figure 5A. On day 15 after treatment, TA6NT-AKTin-DOX inhibited tumor growth by 64.8±2.8%. Although all drug-treated groups inhibited tumor growth, the extent of inhibition was quite different. TA6NT-AKTin-DOX had the most evident efficacy. In addition, TA6NT-AKTin-DOX had a tumor weight inhibition that was 40.8±1.6%, which was higher compared with that of free DOX (Figure 5B and C). Toxicity of drugs was evaluated by weight change of mice. Obviously, TA6NT-AKTin-DOX had less influence on mice weight compared with free DOX and TA6NT-AKTin together with DOX (Figure 5D). Complete blood count testing was also performed and the result is shown in Figure 5E.
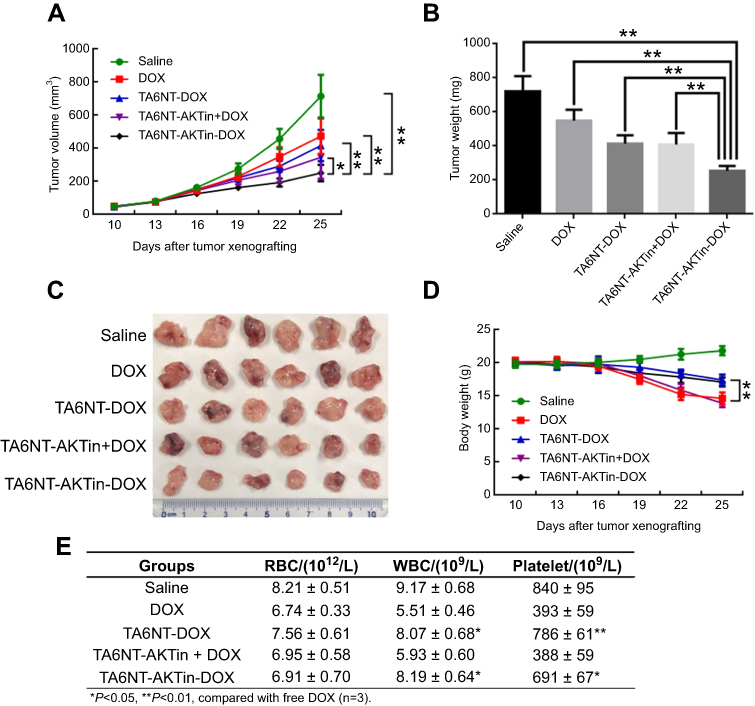 |
Figure 5 (A) Tumor volume change, (B) tumor weight on day 25, (C) excised tumor and (D) body weight change of mice bearing BCSCs xenografts after the treatment of free DOX, TA6NT-DOX, TA6NT-AKTin together with DOX, TA6NT-AKTin-DOX and saline. *p<0.05, **p<0.01. (E) The result of CBC testing. |
Furthermore, biodistribution study exhibited the tumor accumulation of DOX, NT-AKTin-DOX and TA6NT-AKTin-DOX. DOX amount in tumors significantly increased in the TA6NT-AKTin-DOX group compared with other groups (Figure 6A). This result was further confirmed by LC-MS/MS (Figure 6B). The amount of DOX in other organs was also imaged and analyzed. Comparatively, lower accumulation was found in the TA6NT-AKTin-DOX group (Figure S7, p<0.05).
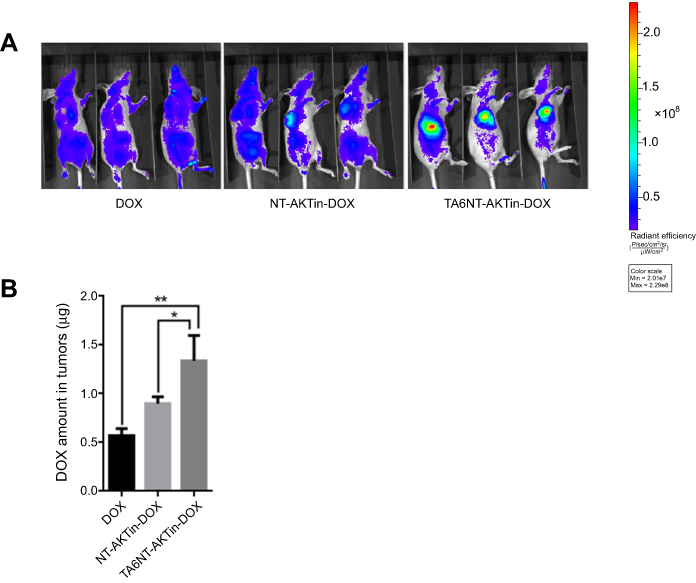 |
Figure 6 (A) In vivo images of mice treated with 5 mg DOX equivalent/kg of free DOX, NT-AKTin-DOX and TA6NT-AKTin-DOX for 4 hrs. (B) The measured DOX amounts in tumors. Two-tailed Student’s t-test was used. *p<0.05, **p<0.01. |
Discussion
In tumor progression and therapeutic drug resistance, CSCs play an important role due to its features of self-renewal and differentiation. More importantly, CSCs are resistant to the killing effects of chemotherapeutic agents and even can be enriched in the remaining or recurrent tumor. To achieve better treatment outcomes, elimination of CSCs is of great importance. Recent identification of surface markers and understanding of molecular feature of CSC phenotype contributed to the design of an effective drug delivery system.
To date, multiple strategies have been conceived, such as targeting specific surface markers, modulation of signaling pathways, inhibiting drug-efflux and inducing apoptosis and differentiation of CSCs.5,34,35 In these strategies, targeting CD44 appears as a good strategy to eliminate BCSCs.4 Several previous studies exploited the interaction of CD44-hyaluronic acid (HA) for targeted delivery of anti-cancer agents.36–38 However, numerous transcripts of CD44 are formed in gene transcription by alternative splicing.39 Standard CD44 (CD44S) is comprised of the constant, non-variant exon products, whereas the variant isoforms arise by splicing of additional exon products into a single site within the membrane-proximal region of the ectodomain.39 CD44 variants rather than CD44S may contribute to tumor progression, which increase in carcinomas.39,40 There is evidence indicating that HA interactions with the CD44 splice variants are diminished compared to those with CD44S.41 Therefore, the targeting effect on BCSCs relying on HA-interactions could be relieved.
Alternatively, aptamers are termed as “chemical antibodies” due to their ability to fold into unique tertiary structures and bind to targets with high affinity and specificity.42,43 In this case, TA6 has a significantly higher affinity than that of HA.7 Moreover, aptamers have many advantages, such as low toxicity and non-immunogenicity in comparison with conventional antibodies, which make aptamers ideal for the targeted delivery of drugs.43 Nowadays, some aptamers targeting surface markers of CSC have been developed and medicine systems based on aptamers have obtained great anti-CSCs effects in preclinical studies.44,45 However, the direct conjugation of drug with aptamer is restricted due to their low drug loading capacity.10 For TA6, it has only one drug attachment site and the turnover rate of CD44 was found to be ~8 hrs, which could significantly limit the efficacy of conjugated drug.46 In addition, low drug loading can also lead to undesired systemic toxicity and excretion concern.47,48 Therefore, self-assembled, aptamer-tethered DNA nanotrain was employed here for the targeted transport of DOX.
HCR was employed to build the DNA nanotrain. This reaction has many advantages, such as enzyme-free nature, efficient isothermal amplification, ultrahigh sensitivity and structural flexibility.13 To date, HCR has been widely used as a prominent vehicle for biosensing and bioimaging, whereas few studies have been reported for its application in targeted drug delivery until most recently.49 Indeed, the periodically aligned boxcar segments provide plenty of spatially addressable sites for drugs. Originally, maximum 10 DNA blocks can be constructed into HCR products.12 In this study, the shorter product may be due to the AKTin anchored at the DNA building blocks. On the other side, AKTin can help reverse the drug resistance of BCSCs and synergistic response was observed after combining AKTin with DOX. As an excellent molecular biotechnology vehicle, HCR has broad applications in nanomedicine.
The result of this work indicated that each TA6NT-AKTin-DOX molecule has ~20 DOX molecules. Compared with free DOX, the cytotoxicity of TA6NT-AKTin-DOX is higher due to the targeting effect of TA6 and the high drug loading capacity. Both in vitro and in vivo data demonstrated that TA6 as a targeting moiety has potential in enhancing internalization to appropriate target cells. The anchoring of AKTin and the intercalation of DOX can significantly improve the drug efficacy. Besides, rapid metabolic degradation of DOX during circulation in bloodstream weakens its efficacy.50 Drug intercalating into DNA nanotrain can shield the molecules from the immune system and enzymatic degradation.51 Moreover, the length of nanotrain was at a range of 100 nm. In general, the objects with such size have been reported to accumulate well in tumor tissues via the enhanced permeability and retention effect, which may also contribute to the high antitumor efficacy and reduce the side effect of TA6NT-AKTin-DOX in mouse xenograft model.51,52
Finally, CD44 is found at low levels on epithelial, hemopoietic, and neuronal cells and at elevated levels in various carcinoma, melanoma, lymphoma, breast, colorectal and lung tumor cells.53 Although CD44 may be expressed on a number of cell types in normal tissues, it turns out that these cell types are either not in direct contact with the blood or require activation.54 Because the vascular system is primarily leaky in tumors,55 CD44 may be a suitable surface receptor for targeted chemotherapy of cancers that express this receptor. But it does not mean that TA6NT-AKTin-DOX can be simply applied to other tumors due to the complexity and specificity of individual cancer.
Conclusion
In this study, the aptamer-tethered DNA nanotrain TA6NT-AKTin-DOX has demonstrated its efficacy in BCSCs. Extensive preclinical toxicological and pharmacological studies of TA6NT-AKTin-DOX will be performed in our lab to support further clinical trials. In addition, nanodrugs generally have attractive pharmacokinetic advantages, such as enhanced bioavailability of orally taken drugs, prolonged half-life of injected drugs (by reducing immunogenicity) and targeted delivery to specific tissues.56 In near future, we will also take an effort to analyze the pharmacokinetic properties of TA6NT-AKTin-DOX. Indeed, HCR that is used to construct the aptamer-tethered DNA nanotrain has been applied in a variety of fields. To date, the HCR products have shown a number of excellent features, such as easy design and preparation, high affinity, shielding effect, good biodegradability, specific targeting and especially high loading capacity. However, rare studies have used this strategy in drug delivery and nanomedicine. From our point of view, the aptamer-tethered DNA nanotrain can use a relatively low amount of DNA to targeted deliver a specific amount of drug. Most importantly, this design can be applied to different cell types and drugs by substituting the aptamer or drug.
Copyright/ethics
The use of the gifted cells was approved by the Institutional Review Board of Nanjing Medical University, Nanjing, People's Republic of China. This study was approved by our institutional ethics committee.
Acknowledgments
The National Natural Science Fund (21722504, 21675089), the SEU-NJMU-CPU cooperation project (2242019K3DNZ2), the SEU-NJMU cooperation project (2242017K3DN12), the Primary Research & Development Plan of Jiangsu Province (BE2018725) and the Open Foundation of State Key Laboratory of Reproductive Medicine [SKLRM-GA201804] awarded to Dr Chen are gratefully acknowledged.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. Ca-Cancer J Clin. 2015;65(2):87–108. doi:10.3322/caac.21294
2. Haffty BG, Yang Q, Reiss M, et al. Locoregional relapse and distant metastasis in conservatively managed triple negative early-stage breast cancer. J Clin Oncol. 2006;24(36):5652–5657. doi:10.1200/JCO.2006.06.5664
3. Voduc KD, Cheang MS, Gelmon K, Nielsen TO, Kennecke H. Breast cancer subtypes and the risk of local and regional relapse. J Clin Oncol. 2010;28(10):1684–1691. doi:10.1200/JCO.2009.24.9284
4. Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8(10):755–768. doi:10.1038/nrc2499
5. Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5(4):275–284. doi:10.1038/nrc1590
6. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. P Natl Acad Sci USA. 2003;100(7):3983–3988. doi:10.1073/pnas.0530291100
7. Somasunderam A, Thiviyanathan V, Tanaka T, et al. Combinatorial selection of DNA thioaptamers targeted to the HA binding domain of human CD44. Biochemistry. 2010;49(42):9106–9112. doi:10.1021/bi1009503
8. Sun H, Zhu X, Lu PY, Rosato RR, Tan W, Zu Y. Oligonucleotide aptamers: new tools for targeted cancer therapy. Mol Ther Nucl Acids. 2014;3(8):e182. doi:10.1038/mtna.2014.32
9. Zhang Z, Ali MM, Eckert MA, et al. A polyvalent aptamer system for targeted drug delivery. Biomaterials. 2013;34(37):9728–9735. doi:10.1016/j.biomaterials.2013.08.079
10. Huang YF, Shangguan D, Liu H, et al. Molecular assembly of an aptamer-drug conjugate for targeted drug delivery to tumor cells. Chembiochem. 2009;10(5):862–868. doi:10.1002/cbic.200900172
11. Subramanian N, Raghunathan V, Kanwar JR, et al. Target-specific delivery of doxorubicin to retinoblastoma using epithelial cell adhesion molecule aptamer. Mol Vis. 2012;18(283):2783–2795.
12. Zhu G, Zheng J, Song E, et al. Self-assembled, aptamer-tethered DNA nanotrains for targeted transport of molecular drugs in cancer theranostics. P Natl Acad Sci USA. 2013;110(20):7998–8003. doi:10.1073/pnas.1220817110
13. Dirks RM, Pierce NA. Triggered amplification by hybridization chain reaction. P Natl Acad Sci USA. 2004;101(43):15275–15278. doi:10.1073/pnas.0407024101
14. Bagalkot V, Farokhzad OC, Langer R, Jon S. An aptamer-doxorubicin physical conjugate as a novel targeted drug-delivery platform. Angew Chem Int Edit. 2006;45(48):8149–8152. doi:10.1002/anie.200602251
15. Mcdermott SP, Wicha MS. Targeting breast cancer stem cells. Mol Oncol. 2010;4(5):404–419. doi:10.1016/j.molonc.2010.06.005
16. Bleau AM, Hambardzumyan D, Ozawa T, et al. PTEN/PI3K/AKT pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell. 2009;4(3):226–235. doi:10.1016/j.stem.2009.01.007
17. Gargini R, Cerliani JP, Escoll M, Anton IM, Wandosell F. Cancer stem cell-like phenotype and survival are coordinately regulated by Akt/FoxO/Bim pathway. Stem Cells. 2015;33(3):646–660. doi:10.1002/stem.1904
18. Kumar D, Shankar S, Srivastava RK, Srivastava RK. Rottlerin induces autophagy and apoptosis in prostate cancer stem cells via PI3K/AKT/mTOR signaling pathway. Cancer Lett. 2014;343(2):179–189. doi:10.1016/j.canlet.2013.10.003
19. Hiromura M, Okada F, Obata T, et al. Inhibition of Akt kinase activity by a peptide spanning the βA strand of the proto-oncogene TCL1. J Biol Chem. 2004;279(51):53407–53418. doi:10.1074/jbc.M403775200
20. Davoudi Z, Akbarzadeh A, Rahmatiyamchi M, Movassaghpour AA, Alipour M, Nejatikoshki K. Molecular target therapy of AKT and NF-kB signaling pathways and multidrug resistance by specific cell penetrating inhibitor peptides in HL-60 cells. Asian Pac J Cancer P. 2014;15(10):4353–4358. doi:10.7314/APJCP.2014.15.10.4353
21. Xu F, Yang T, Chen Y. Quantification of microRNA by DNA-peptide probe and liquid chromatography-tandem mass spectrometry-based quasi-targeted proteomics. Anal Chem. 2016;88(1):754–763. doi:10.1021/acs.analchem.5b03056
22. Xu J, Liu Y, Yu Y, Ni Q, Chen Y. Subcellular quantification of doxorubicin and its metabolite in cultured human leukemia cells using liquid chromatography-tandem mass spectrometry. Anal Lett. 2012;45(14):1980–1994. doi:10.1080/00032719.2012.680056
23. Yang T, Xu F, Xu J, Fang D, Yu Y, Chen Y. Comparison of liquid chromatography-tandem mass spectrometry-based targeted proteomics and conventional analytical methods for the determination of P-glycoprotein in human breast cancer cells. J Chromatogr B. 2013;936(1):18–24. doi:10.1016/j.jchromb.2013.07.023
24. Buck E, Eyzaguirre A, Brown E, et al. Rapamycin synergizes with the epidermal growth factor receptor inhibitor erlotinib in non-small-cell lung, pancreatic, colon, and breast tumors. Mol Cancer Ther. 2006;5(11):2676–2684. doi:10.1158/1535-7163.MCT-05-0235
25. Mohan P, Rapoport N. Doxorubicin as a molecular nanotheranostic agent: effect of doxorubicin encapsulation in micelles or nanoemulsions on the ultrasound-mediated intracellular delivery and nuclear trafficking. Mol Pharmaceut. 2010;7(6):1959–1973. doi:10.1021/mp100269f
26. Guertin AD, Martin MM, Roberts B, et al. Unique functions of CHK1 and WEE1 underlie synergistic anti-tumor activity upon pharmacologic inhibition. Cancer Cell Int. 2012;12(1):45–56. doi:10.1186/1475-2867-12-45
27. Nitin N, Santangelo PJ, Kim G, Nie S, Bao G. Peptide-linked molecular beacons for efficient delivery and rapid mRNA detection in living cells. Nucleic Acids Res. 2004;32(6):e58. doi:10.1093/nar/gnh063
28. Yu F, Yao H, Zhu P, et al. Let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007;131(6):1109–1123. doi:10.1016/j.cell.2007.10.054
29. Sun TM, Wang YC, Wang F, et al. Cancer stem cell therapy using doxorubicin conjugated to gold nanoparticles via hydrazone bonds. Biomaterials. 2014;35(2):836–845. doi:10.1016/j.biomaterials.2014.01.026
30. Kyaw T, Winship A, Tay C, et al. Cytotoxic and proinflammatory CD8+ T lymphocytes promote development of vulnerable atherosclerotic plaques in ApoE-deficient mice. Circulation. 2013;127(9):1028–1039. doi:10.1161/CIRCULATIONAHA.112.001347
31. Wu C, Han D, Chen T, et al. Building a multifunctional aptamer-based DNA nanoassembly for targeted cancer therapy. J Am Chem Soc. 2013;135(49):18644–18650. doi:10.1021/ja4094617
32. Wu XA, Choi CH, Zhang C, Hao L, Mirkin CA. Intracellular fate of spherical nucleic acid nanoparticle conjugates. J Am Chem Soc. 2014;136(21):7726–7733. doi:10.1021/ja503010a
33. Alibolandi M, Taghdisi SM, Ramezani P, et al. Smart AS1411-aptamer conjugated pegylated PAMAM dendrimer for the superior delivery of camptothecin to colon adenocarcinoma in vitro and in vivo. Int J Pharmaceut. 2017;519(1):352–364. doi:10.1016/j.ijpharm.2017.01.044
34. Hu Y, Fu L. Targeting cancer stem cells: a new therapy to cure cancer patients. Am J Cancer Res. 2012;2(3):340–356.
35. Morrison BJ, Schmidt CW, Lakhani SR, Reynolds BA, Lopez JA. Breast cancer stem cells: implications for therapy of breast cancer. Breast Cancer Res. 2008;10(4):210–223. doi:10.1186/bcr2111
36. Eliaz RE
37. Peer D, Margalit R. Loading mitomycin C inside long circulating hyaluronan targeted nano-liposomes increases its antitumor activity in three mice tumor models. Int J Cancer. 2004;108(5):780–789. doi:10.1002/ijc.11459
38. Dan P, Margalit R. Tumor-targeted hyaluronan nanoliposomes increase the antitumor activity of liposomal doxorubicin in syngeneic and human xenograft mouse tumor models. Neoplasia. 2004;6(4):343–353. doi:10.1593/neo.03460
39. Ponta H, Sherman L, Herrlich PA. CD44: from adhesion molecules to signalling regulators. Nat Rev Mol Cell Bio. 2003;4(1):33–45. doi:10.1038/nrm1004
40. Skotheim RI, Nees M. Alternative splicing in cancer: noise, functional, or systematic? Int J Biochem Cell B. 2007;39(7):1432–1449. doi:10.1016/j.biocel.2007.02.016
41. Jackson DG, Bell JI, Dickinson R, Timans J, Shields J, Whittle N. Proteoglycan forms of the lymphocyte homing receptor CD44 are alternatively spliced variants containing the V3 exon. J Cell Biol. 1995;128(4):673–685. doi:10.1083/jcb.128.4.673
42. Radom F, Jurek PM, Mazurek MP, Otlewski J, Jeleń F. Aptamers: molecules of great potential. Biotechnol Adv. 2013;31(8):1260–1274. doi:10.1016/j.biotechadv.2013.04.007
43. Tan W, Wang H, Chen Y, et al. Molecular aptamers for drug delivery. Trends Biotechnol. 2011;29(12):634–640. doi:10.1016/j.tibtech.2011.06.009
44. Tao W, Gantier MP, Xiang D, et al. EpCAM aptamer-mediated survivin silencing sensitized cancer stem cells to doxorubicin in a breast cancer model. Theranostics. 2015;5(12):1456–1472. doi:10.7150/thno.11692
45. Wang J, Sefah K, Altman MB, et al. Aptamer-conjugated nanorods for targeted photothermal therapy of prostate cancer stem cells. Chem Asian J. 2013;8(10):2417–2422. doi:10.1002/asia.201300375
46. Naor D, Sionov RV, Ish-Shalom D. CD44: structure, function, and association with the malignant process. Adv Cancer Res. 1997;71:241–319.
47. Roa WH, Azarmi S, Al-Hallak MHDK, Finlay WH, Magliocco AM, Löbenberg R. Inhalable nanoparticles, a non-invasive approach to treat lung cancer in a mouse model. J Control Release. 2011;150(1):49–55. doi:10.1016/j.jconrel.2010.10.035
48. Maeda H, Nakamura H, Fang J. The EPR effect for macromolecular drug delivery to solid tumors: improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv Drug Deliver Rev. 2013;65(1):71–79. doi:10.1016/j.addr.2012.10.002
49. Bi S, Yue S, Zhang S. Hybridization chain reaction: a versatile molecular tool for biosensing, bioimaging, and biomedicine. Chem Soc Rev. 2017;46(14):4281–4298. doi:10.1039/C7CS00055C
50. Bachur NR, Gee M. Daunorubicin metabolism by rat tissue preparations. J Pharmacol Exp Ther. 1971;177(3):567–572.
51. Dan P, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2(12):751–760. doi:10.1038/nnano.2007.387
52. Petros RA, Desimone JM. Strategies in the design of nanoparticles for therapeutic applications. Nat Rev Drug Discov. 2010;9(8):615–627. doi:10.1038/nrd2591
53. Yu M, Jambhrunkar S, Thorn P, Chen J, Gu W, Yu C. Hyaluronic acid modified mesoporous silica nanoparticles for targeted drug delivery to CD44-overexpressing cancer cells. Nanoscale. 2013;5(1):178–183. doi:10.1039/C2NR32145A
54. Misra S, Heldin P, Hascall VC, et al. Hyaluronan–CD44 interactions as potential targets for cancer therapy. Febs J. 2011;278(9):1429–1443. doi:10.1111/j.1742-4658.2011.08071.x
55. Cun X, Chen J, Ruan S, et al. A novel strategy through combining iRGD peptide with tumor-microenvironment-responsive and multistage nanoparticles for deep tumor penetration. ACS Appl Mater Inter. 2015;7(49):27458–27466. doi:10.1021/acsami.5b09391
56. Onoue S, Yamada S, Chan H. Nanodrugs: pharmacokinetics and safety. Int J Nanomed. 2014;9(1):1025–1037. doi:10.2147/IJN.S38378
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.