Back to Journals » OncoTargets and Therapy » Volume 7
Targeted treatment of head and neck squamous-cell carcinoma: potential of lapatinib
Received 13 November 2013
Accepted for publication 25 December 2013
Published 13 February 2014 Volume 2014:7 Pages 245—251
DOI https://doi.org/10.2147/OTT.S46933
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Mitul D Gandhi, Mark Agulnik
Division of Hematology and Oncology, Robert H Lurie Comprehensive Cancer Center, Feinberg School of Medicine, Northwestern University, Chicago, IL, USA
Abstract: Squamous-cell cancer of the head and neck is a heterogeneous malignancy with treatment predicated on a multimodality therapy involving surgery, chemotherapy, and radiation. However, this approach results in durable responses in only a subset of patients, and is associated with significant toxicity. In advanced disease, multi-agent platinum-based chemotherapy produces only modest improvements in survival. Increased insight into tumor biology has demonstrated several critical oncogenic pathways offering prospects for targeted therapy that may improve upon the existing treatment strategies. The epidermal growth factor receptor is one such target, and directed therapy with the monoclonal antibody cetuximab has been extensively studied. Lapatinib is an oral agent that targets multiple transmembrane receptors within the epidermal growth factor receptor family, and offers a promising new approach to treatment. This paper reviews the rationale for and clinical activity of lapatinib in squamous-cell cancer of the head and neck.
Keywords: epidermal growth factor receptor, targeted therapy, oral cavity, pharynx, larynx
Introduction
Squamous-cell cancer of the head and neck (SCCHN) broadly encompasses malignancies of the oral cavity, pharynx, and larynx that share common risk factors, clinical courses, and treatment paradigms, although increasing data suggest biological heterogeneity.1 There are geographic influences in incidence and prevalence, with an estimated 53,000 new cases expected to occur in the USA and approximately 11,000 deaths.2
Mortality rates have gradually improved with the use of modern protocols employing multimodality treatment predicated on combination chemotherapy and radiation therapy along with surgical intervention in select circumstances.3,4 However, this approach relies on platinum-based cytotoxic therapy functioning as a radiation sensitizer. Although the addition of chemotherapy to radiation confers an absolute survival advantage of approximately 8% at 5 years compared with radiotherapy alone,5 only 40% to 50% of patients achieve a durable remission.6–8 Moreover, toxicities associated with platinum agents – including neurotoxicity, ototoxicity, nephrotoxicity, and myelosuppression – can result in treatment-related morbidity and an impact on long-term quality of life.9 In the relapse or metastatic setting, outcomes are poor, with multi-agent platinum-based regimens associated with a median overall survival (OS) of 8 to 10 months.10–12 Thus, novel approaches formulated on the expanding knowledge of SCCHN molecular pathobiology are needed to improve clinical outcomes in locally advanced and metastatic settings, as well as to mitigate treatment-related toxicity.
Advances in the understanding of SCCHN tumor genomics and consequent proteomic aberrations have led to the recognition of a complex interplay between several pathways involved in its oncogenesis.13 Among the earliest mechanisms discovered to be involved were the epidermal growth factor receptor (EGFR) family of transmembrane tyrosine kinases.14 EGFR is highly expressed on malignant squamous cells,15 with incrementally increasing influence on the transition of healthy mucosa to invasive cancer.15,16 Several series have also suggested that increased expression predicts adverse clinical outcomes.17,18 Further, other members of the EGFR family, including HER2 and HER3, have been similarly implicated in the carcinogenesis of SCCHN.19 As such, the EGFR family of receptors represents a rational target for therapy, either in isolation or as an adjunct to cytotoxic and radiation therapy.
“Lapatinib” is a small molecule, irreversible inhibitor of EGFR and HER2 that has a potentially promising role in the treatment of SCCHN. The purpose of this paper is to review the scientific basis for and clinical activity of lapatinib as a targeted treatment for SCCHN.
EGFR signaling in head and neck cancer
The EGFR family consists of four transmembrane receptors – ErbB1/EGFR, ErbB2/HER2, ErbB3, and ErbB4 – that share structural homology, with each receptor variably possessing a glycosylated extracellular domain, a hydrophobic transmembrane domain, an intracellular juxtamembrane segment, a protein tyrosine kinase domain, and a carboxyterminal tail.20 Notably, ErbB2 does not have a ligand-binding domain, and ErbB3 lacks protein kinase activity. Several ligands target EGFR, including transforming growth factor alpha (TGF-α) and epidermal growth factor,21 although many others have been reported.22 Ligand binding results in a conformational change allowing receptor homo- or heterodimerization, activating intrinsic tyrosine kinase activity and resulting in the autophosphorylation of a tyrosine residue within the cytoplasmic domain.22 The subsequent juxtaposition of two active sites and orientation of an activation loop facilitate substrate–receptor interaction, with the C-helix domain coordinating phosphatase activity and phosphate transfer.23,24 Consequent signal transduction ensues through several well-defined pathways (Figure 1).
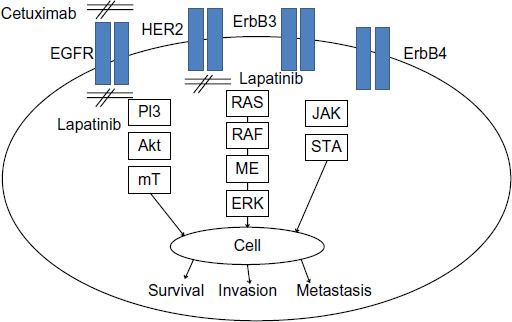 | Figure 1 Signal transduction in the epidermal growth factor receptor (EGFR) pathway. |
For example, activated EGFR phosphorylates and activates phosphoinositol triphosphate kinase (PI3K), generating phosphoinositol triphosphate (PIP3), which then serves as a cofactor in the recruitment and enzymatic activation of Akt. Activated Akt subsequently phosphorylates mammalian target of rapamycin (mTOR), which has wide-ranging substrates and influences several processes involved in cellular metabolism and survival.25 Alternatively, the adaptor protein Grb2 can bind to phosphotyrosine residues in ErbB1/2/3/4 leading to activation of the Ras/Raf/MET/ERK1/2 pathway.20 This similarly results in wide-ranging effects, with ERK1/2 simulating cell proliferation.26 The Ras/Raf/MET/ERK1/2 sequence can also be initiated through activation of phospholipase C, and subsequent generation of inositol triphosphate, diacylglycerol, and phosphokinase C. Through these pathways and the wide-ranging impacts on transcription, EGFR activation influences cellular proliferation,27 angiogenesis,28 cell survival, and migration.29
Experimental cell lines of SCCHN were first observed to have gene amplification along with protein overexpression of EGFR.30 Seminal work by Grandis and Tweardy31 demonstrated increased expression of EGFR mRNA in the majority of patients with SCCHN. Later studies elucidated early dysregulation of EGFR SCCHN in carcinogenesis, suggesting an active role in the transition from dysplasia to carcinoma.15 Similarly, overexpression of ErbB232 and ErbB3 in subsets of SCCHN has also been demonstrated, although the impact of this on clinical outcomes remains to be defined. Finally, more recent studies have suggested that EGFR may also play a role in epithelial to mesenchymal transition, conferring resistance to cytotoxic therapy and promoting migration.33
Inhibition of EGFR by monoclonal antibodies targeting the extracellular domain of the receptor, along with small-molecule tyrosine kinase inhibitors specific for the cytoplasmic activation site, has demonstrated significant activity in arresting growth of SCCHN cell lines.27,34 This model of growth arrest with EGFR inhibition has provided a platform for clinical testing.
Clinical evidence of EGFR inhibition
“Cetuximab” (Erbitux, Bristol-Myers Squibb, Princeton, NJ, USA) is a recombinant chimeric immunoglobulin G1 antibody that binds to the extracellular domain III of EGFR.35 As a consequence, native ligand binding is not permitted, thus precluding receptor dimerization and subsequent autophosphorylation, effectively dampening the downstream signal transduction of proliferative cytokines.
Cetuximab gained regulatory approval following Phase II data demonstrating efficacy in platinum refractory metastatic SCCHN. A total of 96 patients with advanced SCCHN who had progressed on platinum-based chemotherapy were treated with cetuximab in combination with the same platinum agent at time of progression.36 A 10% overall response rate (ORR) was observed and a median progression-free survival (PFS) and an OS of 85 and 183 days, respectively, were reported. Another Phase II study demonstrated similar efficacy in setting of progression on platinum-based treatment.37
Subsequently, cetuximab was evaluated as part of first-line therapy for metastatic disease. In combination with a platinum agent and 5 fluorouracil, it was noted to confer a survival advantage compared with the same regimen without EGFR inhibition.38 In the randomized trial of 442 patients, the cohort who received cetuximab-based chemo-immunotherapy had a median OS of 10.1 months compared with 7.4 months in the control arm (P=0.04), and improved PFS from 3.3 months to 5.6 months (P<0.001). Rates of grade 3 and 4 toxicity were not appreciably higher in the cetuximab arm, although there was a significantly higher incidence of sepsis compared with control (9.0 versus [vs] 1.0, P=0.02). Several other Phase II studies have suggested the combination of cetuximab and cytotoxic chemotherapy to be efficacious, although superiority over conventional treatment was not conclusive.39–41
“Panitumumab,” a fully humanized anti-EGFR monoclonal antibody, has also been investigated in advanced SCCHN. A total of 657 patients were randomized to panitumumab or placebo in combination with a platinum agent and 5 fluorouracil.42 There was an improvement in PFS from 4.6 to 5.8 months (P=0.003), although no OS benefit was detected when the entire study population as a whole was analyzed. However, in the subset of patients who were human papilloma virus (HPV) negative, an OS benefit was observed (11.7 vs 8.6 months, P-0.00115), whereas there was no difference in the HPV-positive cohort. Taken together, clinical experience with cetuximab and panitumumab suggests inhibition of EGFR signaling is synergistic with conventional cytotoxic therapy and improves clinical outcomes.
Lapatinib
Lapatinib (Tykerb, GlaxoSmithKline, Brentford, England) is a quinazoline-class small-molecule tyrosine kinase inhibitor that can reversibly bind with high affinity to the adenosine-triphosphate catalytic binding sites of both EGFR and HER2.43 While a crystal structure demonstrating lapatinib bound to EGFR, ErbB2 is unavailable, it is clear that on its binding to the intracytoplasmic domain of EGFR, auto-phosphorylation of tyrosine is prevented.44 Conduction of the down-signaling cascade is aborted, as cell lines treated with lapatinib have demonstrated decreased levels of phosphorylated levels of Akt and ERK1/2.45 Subsequently, the proliferative signals are terminated, resulting in growth arrest and apoptosis.
Lapatinib is available only as an oral formula, achieving peak serum concentration in approximately 3 to 4 hours; has a 14-hour half-life on once-daily dosing; and demonstrates a wide volume of distribution.46 Metabolism is primarily hepatic through CYP3A4 and subsequent fecal elimination, with less than 2% of the drug recovered in the urine.
Preclinical experiments have demonstrated that EGFR and HER2 can be potently inhibited in SCCHN cell lines along with in xenograft mouse models.47–49 The additional inhibition of ErbB2/HER2 may circumvent a resistance mechanism observed in EGFR-directed therapy, involving signal transduction through related receptors.50 A pilot study of lapatinib in humans with advanced solid malignancies corroborated decreased detection of phosphorylated ErbB2, Akt, and ERK1/2 with response or stable disease, prompting further clinical investigation in prospective trials.51
Interestingly, lapatinib has been found to exert effects through alternative pathways as well, as emerging research has demonstrated several off-target effects. One study has suggested lapatinib-mediated upregulation of tumor necrosis factor-related apoptosis-inducing ligand receptors, subsequently potentiating cell death in the presence of recombinant antibody agonists of the tumor necrosis factor-related apoptosis-inducing ligand receptors.52 Another study has suggested that lapatinib may activate nuclear factor-kappa B in malignant breast-cancer cells, potentially making them susceptible to protesome inhibitors such as bortezomib.53 To date, however, the non-EGFR-based action of lapatinib has not been described in SCCHN, and clinical activity appears to be predicated on this pathway.
Clinical evidence for lapatinib
Lapatinib has been observed to have efficacy in several other tumor types including breast cancer54 and patient small subset of gastric cancers, malignancies that are associated with HER2 overexpression. Smaller pilot studies have also demonstrated efficacy in gallbladder cancer, endometrial cancer,55 and salivary-gland cancer56 although experience is less robust. Based on the preclinical data noted here and the drug’s efficacy noted in other cancers, further clinical investigation in SCCHN was undertaken.
Monotherapy
Lapatinib was initially investigated in a Phase I trial involving 67 patients with advanced staged refractory solid tumors, five of whom had SCCHN.57 Eligible patients were enrolled if their tumor demonstrated expression of ErbB1 by immunohistochemistry and/or overexpression of ErbB2 by immunohistochemistry or fluorescent in situ hybridization. Six dosing cohorts – 500, 650, 900, 1,000, 1,200, and 1,600 mg daily – were investigated. At the conclusion of follow-up, 66% of patients were found to have experienced an adverse event (AE), the most frequent of which were diarrhea (42%) and rash (31%). The majority of the toxicity was mild, with 96% of reported AEs being grade 1 or 2. Subsequently, the maximal tolerated dose was determined to be 1,600 mg per day. Notably, three of the five patients with SCCHN demonstrated stable disease.
Two subsequent Phase II studies evaluated the efficacy of lapatinib as monotherapy in advanced SCCHN. The first examined 45 previously treated patients who had either relapsed after radiation-based treatment or progressed after one to two systemic treatments.58 Patients were enrolled in two cohorts, based on whether they had had prior exposure to EGFR-directed therapy (cohort A) or not (cohort B) and treated with lapatinib at 1,500 mg daily until disease progression or intolerability secondary to AEs occurred. In an intention-to-treat analysis, no complete or partial responses were observed in either group, and stable disease was noted in 17% of patients in cohort A and 41% in cohort B. Median PFS was 52 days among all patients, with no difference noted based on prior EGFR-directed therapy; median OS was 155 and 288 days for cohorts A and B, respectively. The study had a biological correlative as well, with tissue sampled pre- and post-treatment. Remarkably, while phosphorylated ErbB2 was significantly decreased following lapatinib treatment compared with pre-treatment assessment (P=0.0048), this was not observed with EGFR, ERK, or Akt. This suggests receptor blockade through HER2 inhibition but continued signal transduction through EGFR or alternative pathways.
A second study investigated the role of lapatinib as induction therapy, prior to definitive chemoradiation (CRT).59 In that study, 107 treatment-naïve patients were randomized in a 2:1 fashion to receive either lapatinib (n=71) or placebo (n=36) 2–6 weeks prior to CRT. The primary endpoint was apoptotic index, calculated as a ratio of apoptotic cells pre- and post-lapatinib exposure. Secondary endpoints included proliferation index, ORRs, assessment of AEs, and biological correlatives. On average, patients were on either lapatinib or placebo for 25 days prior to the start of CRT.
No statistically significant difference was observed in the apoptotic index in patients who received lapatinib compared with those who received placebo (P=0.394), although a more pronounced reduction in the proliferative index was observed (P=0.030). Prior to CRT, a 17% ORR was observed in the lapatinib cohort compared with 0% in the placebo. Further, following completion of CRT, a higher ORR was observed in those receiving lapatinib (70% vs 53%), despite the placebo cohort having had a higher prevalence of tumors positive for HPV. The authors concluded that despite the unremarkable change in apoptotic index, lapatinib demonstrated clinical activity, particularly in HPV-negative patients.
Combination radiotherapy
Given the observed efficacy and acceptable toxicity for lapatinib as monotherapy for relapsed disease, its use in the frontline setting with radiotherapy for locally advanced disease was investigated. In an initial Phase I study,60 three cohorts with escalating doses of lapatinib – seven patients treated with 500 mg daily, seven patients treated with 1,000 mg daily, and 17 patients treated with 1,500 mg daily – were combined with cisplatin (100 mg/m2) and radiation therapy for Stage IVA SCCHN. No dose-limiting toxicities were observed, and a dose of 1,500 mg was selected for the Phase II study. A 64% ORR was observed in the 1,500 mg cohort.
The subsequent Phase II study61 randomized patients with Stage III to IVB SCCHN to cisplatin-based CRT, with or without concurrent lapatinib 1,500 mg daily, followed by maintenance lapatinib or placebo. A total of 67 patients were randomized – 34 to the lapatinib arm and 33 to the placebo. The primary endpoint of complete response rate trended toward significance in the lapatinib cohort compared with in the placebo (53% vs 36%, P=0.093) on independent assessment; however, complete response rate was found to be significant on investigator assessment (50% vs 24%, P=0.009). With respect to clinical outcomes, the PFS was observed to be 35.3 vs 12.1 months, which resulted in a hazard ratio of 0.74 (P=0.184); the corresponding hazard ratio for OS was 0.9 (P=0.382). Notably, there was a significant improvement in PFS in the HPV-negative cohort in favor of lapatinib (median PFS not reached vs 10.3 months). The authors identified this population as a subset that may warrant further investigation, given the otherwise poor outcomes observed with CRT alone.
The efficacy of lapatinib in the HPV-negative cohort – similarly observed when lapatinib was used as induction therapy prior to CRT – is of particular interest. Over the past three decades, there has been an epidemiological shift, with the majority of SCCHNs found to be related to HPV infection rather than to tobacco or alcohol use, risk factors traditionally implicated in carcinogenesis.62 Landmark analyses have observed significantly better outcomes with CRT in patients with HPV-positive tumors,63,64 prompting studies investigating de-intensification of therapy in this favorable-risk subset. Subsequently, while declining in prevalence, HPV-negative SCCHN still accounts for a significant proportion of all patients and represents a higher-risk cohort. Thus, the observed benefit of lapatinib in this population may result in an important treatment advance.
Active trials
Three trials involving lapatinib in SCCHN are currently active and recruiting patients. The first is a Phase II study investigating use of lapatinib in combination with cisplatin-based CRT in HPV-negative SCCHN.65 The second is a Phase II study examining the use of lapatinib in combination with carboplatin and paclitaxel as induction therapy prior to transoral surgery and risk-adapted adjuvant therapy.66 The final trial is a Phase II study assessing PFS with combination lapatinib and capecitabine in patients with recurrent or metastatic disease.67 Several other studies have completed accrual and are pending maturation of data.
Conclusion
SCCHN is a heterogeneous malignancy with evolving treatment paradigms increasingly incorporating targeted therapy. Within this context, preclinical data lends support to EGFR inhibition as a rational approach to increase response rates and improve outcomes. The clinical data currently available suggest a marginal benefit with the use of lapatinib, either as monotherapy or in combination with platinum-based CRT therapy. However, recognition of increased response in distinct biological subsets, such as HPV-negative tumors that have an otherwise poor prognosis, may allow enriching of future trials with appropriately selected patients to detect those who will derive the greatest clinical benefit. Finally, combination therapy with other small-molecule inhibition of critical proteins on separate pathways may potentiate the efficacy of lapatinib.
Disclosure
The authors declare no conflicts of interest in this work.
References
Bose P, Brockton NT, Dort JC. Head and neck cancer: from anatomy to biology. Int J Cancer. 2013;133(9):2013–2023. | |
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11–30. | |
Chen AY, Zhu J, Fedewa S. Temporal trends in oropharyngeal cancer treatment and survival, 1998–2009. Laryngoscope. Epub July 6, 2013. | |
Chen AY, Fedewa S, Zhu J. Temporal trends in the treatment of early- and advanced-stage laryngeal cancer in the United States, 1985–2007. Arch Otorlaryngol Head Neck Surg. 2011;137(10):1017–1024. | |
Furness S, Glenny AM, Worthington HV, et al; CSROC Expert Panel. Interventions for the treatment of oral cavity and oropharyngeal cancer: chemotherapy. Cochrane Database Syst Rev. 2010;(9):CD006386. | |
Forastiere AA, Goepfert H, Maor M, et al. Concurrent chemotherapy and radiotherapy for organ preservation in advanced laryngeal cancer. N Engl J Med. 2003;349(22):2091–2098. | |
Newlin HE, Amdur RJ, Riggs CE, Morris CG, Kirwan JM, Mendenhall WM. Concomitant weekly cisplatin and altered fractionation radiotherapy in locally advanced head and neck cancer. Cancer. 2010;116:4533–4540. | |
Huguenin P, Beer KT, Allal A, et al. Concomitant cisplatin significantly improves locoregional control in advanced head and neck cancers treated with hyperfractionated radiotherapy. J Clin Oncol. 2004;22(23):4665–4673. | |
Machtay M, Moughan J, Trotti A, et al. Factors associated with severe late toxicity after concurrent chemoradiation for locally advanced head and neck cancer: an RTOG analysis. J Clin Oncol. 2008;26(21):3582–3589. | |
Worden FP, Moon J, Samlowski W, et al; Southwest Oncology Group, Head and Neck Working Group. A phase II evaluation of a 3-hour infusion of paclitaxel, cisplatin, and 5-fluorouracil in patients with advanced or recurrent squamous cell carcinoma of the head and neck: Southwest Oncology Group study 0007. Cancer. 2006;107(2):319–327. | |
Gibson MK, Li Y, Murphy B, et al; Eastern Cooperative Oncology Group. Randomized phase III evaluation of cisplatin plus fluorouracil versus cisplatin plus paclitaxel in advanced head and neck cancer (E1395): an intergroup trial of the Eastern Cooperative Oncology Group. J Clin Oncol. 2005;23(15):3562–3567. | |
Benasso M, Ponzanelli A, Merlano M, et al. Paclitaxel, cisplatin and 5-fluorouracil in recurrent squamous cell carcinoma of the head and neck: a phase II trial from an Italian cooperative group. Acta Oncol. 2006;45(2):168–174. | |
Stransky N, Egloff AM, Tward AD, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333(6046):1157–1160. | |
Kalyankrishna S, Grandis JR. Epidermal growth factor receptor biology in head and neck cancer. J Clin Oncol. 2006;24(17):2666–2672. | |
Shin DM, Ro JY, Hong WK, Hittelman WN. Dysregulation of epidermal growth factor receptor expression in premalignant lesions during head and neck tumorigenesis. Cancer Res. 1994;54(12):3153–3159. | |
Rubin Grandis J, Melhem MF, Barnes EL, Tweardy DJ. Quantitative immunohistochemical analysis of transforming growth factor-alpha and epidermal growth factor receptor in patients with squamous cell carcinoma of the head and neck. Cancer. 1996;78(6):1284–1292. | |
Temam S, Kawaguchi H, El-Naggar AK, et al. Epidermal growth factor receptor copy number alterations correlate with poor clinical outcome in patients with head and neck squamous cancer. J Clin Oncol. 2007;25(16):2164–2170. | |
Zhu X, Zhang F, Zhang W, He J, Zhao Y, Chen X. Prognostic role of epidermal growth factor receptor in head and neck cancer: a meta-analysis. J Surg Oncol. 2013;108(6):387–397. | |
Rao VH, Kandel A, Lynch D, et al. A positive feedback loop between HER2 and ADAM12 in human head and neck cancer cells increases migration and invasion. Oncogene. 2012;31(23):2888–2898. | |
Roskoski R Jr. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol Res. 2013;79C:34–74. | |
Rubin Grandis J, Melhem MF, Gooding WE, et al. Levels of TGF-alpha and EGFR protein in head and neck squamous cell carcinoma and patient survival. J Natl Cancer Inst. 1998;90(11):824–832. | |
Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2(2):127–137. | |
Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125(6):1137–1149. | |
Kumar A, Petri ET, Halmos B, Boggon TJ. Structure and clinical relevance of the epidermal growth factor receptor in human cancer. J Clin Oncol. 2008;26(10):1742–1751. | |
Freudlsperger C, Burnett JR, Friedman JA, Kannabiran VR, Chen Z, Van Waes C. EGFR-PI3K-AKT-mTOR signaling in head and neck squamous cell carcinomas: attractive targets for molecular-oriented therapy. Expert Opin Ther Targets. 2011;15(1):63–74. | |
Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26(22):3291–3310. | |
Rubin Grandis J, Chakraborty A, Melhem MF, Zeng Q, Tweardy DJ. Inhibition of epidermal growth factor receptor gene expression and function decreases proliferation of head and neck squamous carcinoma but not normal mucosal epithelial cells. Oncogene. 1997;15(4):409–416. | |
Lionello M, Lovato A, Staffieri A, et al. The EGFR-mTOR pathway and laryngeal cancer angiogenesis. Eur Arch Otorhinolaryngol. Epub September 25, 2013. | |
Zuo JH, Zhu W, Li MY, et al. Activation of EGFR promotes squamous carcinoma SCC10A cell migration and invasion via inducing EMT-like phenotype change and MMP-9-mediated degradation of E-cadherin. J Cell Biochem. 2011;112(9):2508–2517. | |
Weichselbaum RR, Dunphy EJ, Beckett MA, et al. Epidermal growth factor receptor gene amplification and expression in head and neck cancer cell lines. Head Neck. 1989;11(5):437–442. | |
Grandis JR, Tweardy DJ. Elevated levels of transforming growth factor alpha and epidermal growth factor receptor messenger RNA are early markers of carcinogenesis in head and neck cancer. Cancer Res. 1993;53(15):3579–3584. | |
Cavalot A, Martone T, Roggero N, Brondino G, Pagano M, Cortesina G. Prognostic impact of HER-2/neu expression on squamous head and neck carcinomas. Head Neck. 2007;29(7):655–664. | |
Voon DC, Wang H, Koo JK, et al. EMT-induced stemness and tumorigenicity are fueled by the EGFR/Ras pathway. PloS One. 2013;8(8):e70427. | |
Modjtahedi H, Affleck K, Stubberfield C, Dean C. EGFR blockade by tyrosine kinase inhibitor or monoclonal antibody inhibits growth, directs terminal differentiation and induces apoptosis in the human squamous cell carcinoma HN5. Int J Oncol. 1998;13(2):335–342. | |
Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P, Ferguson KM. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell. 2005;7(4):301–311. | |
Baselga J, Trigo JM, Bourhis J, et al. Phase II multicenter study of the antiepidermal growth factor receptor monoclonal antibody cetuximab in combination with platinum-based chemotherapy in patients with platinum-refractory metastatic and/or recurrent squamous cell carcinoma of the head and neck. J Clin Oncol. 2005;23(24):5568–5577. | |
Herbst RS, Arquette M, Shin DM, et al. Phase II multicenter study of the epidermal growth factor receptor antibody cetuximab and cisplatin for recurrent and refractory squamous cell carcinoma of the head and neck. J Clin Oncol. 2005;23(24):5578–5587. | |
Vermorken JB, Mesia R, Rivera F, et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med. 2008;359(11):1116–1127. | |
Knoedler M, Gauler TC, Gruenwald V, et al. Phase II study of cetuximab in combination with docetaxel in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck after platinum-containing therapy: a multicenter study of the Arbeitsgemeinschaft Internistische Onkologie. Oncology. 2013;84(5):284–289. | |
Yoshino T, Hasegawa Y, Takahashi S, et al. Platinum-based chemotherapy plus cetuximab for the first-line treatment of Japanese patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck: results of a phase II trial. Japanese J Clin Oncol. 2013;43(5):524–531. | |
Vermorken JB, Licitra L, Stöhlmacher-Williams J, et al. Phase II study of pemetrexed in combination with cisplatin and cetuximab in recurrent or metastatic squamous cell carcinoma of the head and neck. Eur J Cancer. 2013;49(13):2877–2883. | |
Vermorken JB, Stöhlmacher-Williams J, Davidenko I, et al; SPECTRUM investigators. Cisplatin and fluorouracil with or without panitumumab in patients with recurrent or metastatic squamous-cell carcinoma of the head and neck (SPECTRUM): an open-label phase 3 randomised trial. Lancet Oncol. 2013;14(8):697–710. | |
Medina PJ, Goodin S. Lapatinib: a dual inhibitor of human epidermal growth factor receptor tyrosine kinases. Clin Ther. 2008;30(8):1426–1447. | |
Wood ER, Truesdale AT, McDonald OB, et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004;64(18):6652–6659. | |
Hegde PS, Rusnak D, Bertiaux M, et al. Delineation of molecular mechanisms of sensitivity to lapatinib in breast cancer cell lines using global gene expression profiles. Mol Can Ther. 2007;6(5):1629–1640. | |
Bence AK, Anderson EB, Halepota MA, et al. Phase I pharmacokinetic studies evaluating single and multiple doses of oral GW572016, a dual EGFR-ErbB2 inhibitor, in healthy subjects. Invest New Drugs. 2005;23(1):39–49. | |
Xia W, Mullin RJ, Keith BR, et al. Anti-tumor activity of GW572016: a dual tyrosine kinase inhibitor blocks EGF activation of EGFR/erbB2 and downstream Erk1/2 and AKT pathways. Oncogene. 2002;21(41):6255–6263. | |
Rusnak DW, Lackey K, Affleck K, et al. The effects of the novel, reversible epidermal growth factor receptor/ErbB-2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Mol Cancer Ther. 2001;1(2):85–94. | |
Kondo N, Tsukuda M, Ishiguro Y, et al. Antitumor effects of lapatinib (GW572016), a dual inhibitor of EGFR and HER-2, in combination with cisplatin or paclitaxel on head and neck squamous cell carcinoma. Oncol Rep. 2010;23(4):957–963. | |
Wheeler DL, Huang S, Kruser TJ, et al. Mechanisms of acquired resistance to cetuximab: role of HER (ErbB) family members. Oncogene. 2008;27(28):3944–3956. | |
Spector NL, Xia W, Burris H 3rd, et al. Study of the biologic effects of lapatinib, a reversible inhibitor of ErbB1 and ErbB2 tyrosine kinases, on tumor growth and survival pathways in patients with advanced malignancies. J Clin Oncol. 2005;23(11):2502–2512. | |
Dolloff NG, Mayes PA, Hart LS, Dicker DT, Humphreys R, El-Deiry WS. Off-target lapatinib activity sensitizes colon cancer cells through TRAIL death receptor up-regulation. Sci Transl Med. 2011; 3(86):86ra50. | |
Chen YJ, Yeh MH, Yu MC, et al. Lapatinib – induced NF-kappaB activation sensitizes triple-negative breast cancer cells to proteasome inhibitors. Breast Cancer Res. 2013;15(6):R108. | |
Geyer CE, Forster J, Lindquist D, et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med. 2006;355(26):2733–2743. | |
Leslie KK, Sill MW, Lankes HA, et al. Lapatinib and potential prognostic value of EGFR mutations in a Gynecologic Oncology Group phase II trial of persistent or recurrent endometrial cancer. Gynecol Oncol. 2012;127(2):345–350. | |
Agulnik M, Cohen EW, Cohen RB, et al. Phase II study of lapatinib in recurrent or metastatic epidermal growth factor receptor and/or erbB2 expressing adenoid cystic carcinoma and non adenoid cystic carcinoma malignant tumors of the salivary glands. J Clin Oncol. 2007;25(25):3978–3984. | |
Burris HA 3rd, Hurwitz HI, Dees EC, et al. Phase I safety, pharmacokinetics, and clinical activity study of lapatinib (GW572016), a reversible dual inhibitor of epidermal growth factor receptor tyrosine kinases, in heavily pretreated patients with metastatic carcinomas. J Clin Oncol. 2005;23(23):5305–5313. | |
de Souza JA, Davis DW, Zhang Y, et al. A phase II study of lapatinib in recurrent/metastatic squamous cell carcinoma of the head and neck. Clin Cancer Res. 2012;18(8):2336–2343. | |
Del Campo JM, Hitt R, Sebastian P, et al. Effects of lapatinib monotherapy: results of a randomised phase II study in therapy-naive patients with locally advanced squamous cell carcinoma of the head and neck. Br J Cancer. 2011;105(5):618–627. | |
Harrington KJ, El-Hariry IA, Holford CS, et al. Phase I study of lapatinib in combination with chemoradiation in patients with locally advanced squamous cell carcinoma of the head and neck. J Clin Oncol. 2009;27(7):1100–1107. | |
Harrington K, Berrier A, Robinson M, et al. Randomised Phase II study of oral lapatinib combined with chemoradiotherapy in patients with advanced squamous cell carcinoma of the head and neck: rationale for future randomised trials in human papilloma virus-negative disease. Eur J Cancer. 2013;49(7):1609–1618. | |
Mehanna H, Beech T, Nicholson T, et al. Prevalence of human papillomavirus in oropharyngeal and nonoropharyngeal head and neck cancer – systematic review and meta-analysis of trends by time and region. Head Neck. 2013;35(5):747–755. | |
Ang KK, Harris J, Wheeler R, et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N Engl J Med. 2010;363(1):24–35. | |
Fakhry C, Westra WH, Li S, et al. Improved survival of patients with human papillomavirus-positive head and neck squamous cell carcinoma in a prospective clinical trial. J Natl Cancer Inst. 2008;100(4):261–269. | |
Radiation Therapy Oncology Group. TRYHARD: Radiation therapy plus cisplatin with or without lapatinib in treating patients with head and neck cancer. In: ClinicalTrials.gov [website on the Internet]. Bethseda, MD: US National Library of Medicine; 2012 [updated December 23, 2013]. Available from: http://clinicaltrials.gov/show/NCT01711658. NLM identifier: NCT01711658. Accessed December 28, 2013. | |
UNC Lineberger Comprehensive Cancer Centre. Multimodality risk adapted tx including induction chemo for SCCHN amenable to transoral surgery. In: ClinicalTrials.gov [website on the Internet]. Bethseda, MD: US National Library of Medicine; 2012 [updated February 15, 2013]. Available from: http://clinicaltrials.gov/show/NCT01612351. NLM identifier: NCT01612351. Accessed December 28, 2013. | |
Abramson Cancer Center of the University of Pennsylvania. Capecitabine and lapatinib ditosylate in treating patients with squamous cell cancer of the head and neck. In: ClinicalTrials.gov [website on the Internet]. Bethseda, MD: US National Library of Medicine; 2009 [updated February 25, 2013]. Available from: http://clinicaltrials.gov/show/NCT01044433. NLM identifier: NCT01044433. Accessed December 28, 2013. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.