Back to Archived Journals » Adolescent Health, Medicine and Therapeutics » Volume 8
Targeted therapy for soft tissue sarcomas in adolescents and young adults
Authors Steppan DA, Pratilas CA, Loeb DM
Received 2 October 2016
Accepted for publication 2 December 2016
Published 30 March 2017 Volume 2017:8 Pages 41—55
DOI https://doi.org/10.2147/AHMT.S70377
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Alastair Sutcliffe
Diana A Steppan, Christine A Pratilas, David M Loeb
Division of Pediatric Oncology, The Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University School of Medicine, Baltimore, MD, USA
Abstract: Soft tissue sarcomas (STSs) are a heterogeneous group of tumors originating from the mesenchyme. Even though they affect individuals in all age groups, the prevalence of subtypes of STSs changes significantly from childhood through adolescence into adulthood. The mainstay of therapy is surgery, with or without the addition of chemotherapy and/or radiation therapy. These treatment modalities are associated, in many cases, with significant morbidity and, given the heterogeneity of tumor histologies encompassed by the term “STS”, have not uniformly improved outcomes. Moreover, some subgroups of STSs appear to be more, and others less, responsive to conventional chemotherapy agents. Over the last two decades, our understanding of the biology of STSs is slowly increasing, allowing for the development of more targeted therapies. We review the new treatment modalities that have been tested on patients with STSs, with a special focus on adolescents and young adults, a group of patients that is often underrepresented in clinical trials and has not received the dedicated attention it deserves, given the significant differences in biology and treatment response in comparison to children and adults.
Keywords: synovial sarcoma, MPNST, soft tissue sarcoma, targeted therapy
Abbreviations
AJCC, American Joint Committee on Cancer; ASPS, alveolar soft part sarcoma; AYAs, adolescent and young adults; CAR, chimeric antigen receptor; COG, Children’s Oncology Group; CR, complete remission; DSRCT, desmoplastic small round cell tumor; EGFR, endothelial growth factor receptor; EORTC, European Organization for Research and Treatment of Cancer; FDA, Food and Drug Administration; FDG-PET, fludeoxyglucose - positron emission tomography; FNCLCC, Federation Nationales des Centres de Lutte Contre le Cancer; GIST, gastrointestinal stromal tumor; HDAC, histone deacetylase; Hsp90, heat-shock protein 90; IGF-1R, insulin-like growth factor-1 receptor; MPNST, malignant peripheral nerve sheath tumor; mTOR, mechanistic target of rapamycin; mTORC1, mTOR complex 1; mTORC2, mTOR complex 2; MTP-PE, liposomal muramyl tripeptide phosphatidylethanolamine; NRSTSs, nonrhabdomyosarcoma soft tissue sarcomas; ORR, overall response rate; OS, overall survival; PFS, progression-free survival; Ph+, Philadelphia chromosome-positive; PPTP, Pediatric Preclinical Testing Program; PR, partial response; RECIST, Response Evaluation Criteria in Solid Tumors; RMS, rhabdomyosarcoma; SARC, Sarcoma Alliance for Research through Collaboration; SD, stable disease; SEER, Surveillance, Epidemiology, and End Results; SINE, selective inhibitors of nuclear export; STSs, soft tissue sarcomas; SUVmax, standardized uptake value; TIL, tumor-infiltrating lymphocyte; TKIs, tyrosine kinase inhibitors; TNM, tumor–node–metastasis; UICC, Union for International Cancer Control; VEGF, vascular endothelial growth factor; VOIT, vincristine, oral irinotecan, and temozolomide; WHO, World Health Organization; XRT, radiation treatment.
Definition and demographics of STSs
STSs, as defined by the WHO, are soft tissue tumors with malignant potential, that is, a propensity for locally destructive growth, risk of recurrence, and risk of distant metastasis.1 A common feature of this group of tumors is that they derive from mesenchymal cells that normally give rise to connective tissue. Annually, ~900 children aged <20 years and 1500 AYAs between the ages of 15 and 29 years are diagnosed with STSs (excluding Kaposi’s sarcoma) in the USA.2,3 Historically, data regarding the prevalence of these tumors in AYAs have been sparse. Over the last decade, however, efforts to fill this gap have started to shed light on this age group. The AYA population is most often defined as those in the 15- to 29-year-old age group. The incidence of cancer in this group is 2.7 times higher than that in the first 15 years of life, and ~3% of tumors in the AYA group are STSs.4 It is important to note that the prevalence of STS types dramatically changes from childhood to the age of 30 years, and this holds true even in the short interval between 15 and 30 years.2,3 In children younger than 15 years, RMS constitutes the most common STS. Even though RMS occurs less frequently in the 15- to 29-year-old age group, the survival of AYA patients with RMS is significantly worse than that of younger patients with similar features. In contrast, in older adults (>29 years) with STS, high-grade pleomorphic sarcoma, liposarcoma, leiomyosarcoma, synovial sarcoma, and MPNST together comprise about three-fourths of tumors seen. Although AYA patients most commonly present with tumors in the ICCC (International Classification of Childhood Cancer) category of fibrosarcomas and related fibromatous entities (30%), patients in this age range can also present with tumors more common in young children and older adults.
A multitude of diverse tumor histologies are grouped together within the class of STSs. Based on WHO recommendations, STSs are divided as follows: adipocytic tumors, fibroblastic/myofibroblastic, so-called fibrohistiocytic tumors, smooth muscle tumors, pericytic (perivascular) tumors, skeletal muscle tumors, vascular tumors of soft tissue, chondro-osseous tumors, GIST, nerve sheath tumors, tumors of uncertain differentiation, and undifferentiated/unclassified sarcomas.1 With increasing understanding of the molecular drivers of cancer, these histologic distinctions have undergone numerous changes over the past several decades. Following this trend, tumors are increasingly being classified based on molecular characteristics rather than histopathologic appearance, starting with the 2002 classification and then further more in the 2013 updated version.1,5 RMS is a prime example of this trend, as it has been classified historically as either embryonal or alveolar based on histopathology but more recently as either translocation positive or translocation negative, based on the presence or absence of a fusion transcript involving the FOXO1 gene. This change reflects recent data suggesting that this molecular characteristic more accurately predicts disease biology and outcomes, with translocation-negative patients usually having less aggressive disease and better outcomes regardless of histopathologic classification.6–9 In comparison to RMS, many NRSTSs are still lacking clearly defined molecular characteristics. NRSTSs in adults are usually classified based on the aforementioned WHO classification, but ICCC is traditionally used for the classification of childhood tumors. This classification system is used in the SEER program, and some of the STS tumors are not represented in this ICCC classification, because they do not appear in young children, even though they do occur in the AYA population, further complicating our ability to estimate STS incidence in the AYA population. Because of the diverse classification schemes and the overlapping age ranges in different epidemiologic studies, the exact distribution of STS subtypes in this age group is not known, but we have endeavored to compile the best available data, leaving out such entities as GIST and Kaposi’s sarcoma (Figure 1).
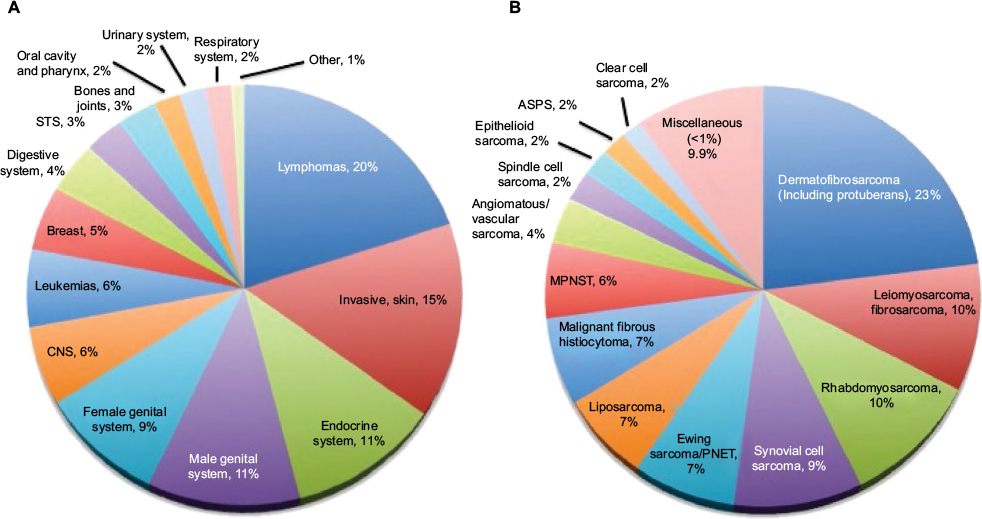 | Figure 1 Incidence of cancer (A) and soft tissue sarcomas (B) in the AYA population. Notes: (A) Cancer incidence in 15–29-year-olds in the USA based on SEER data, 1975–2000. (B) STS incidence (excluding Kaposi sarcoma) in 15–29-year olds in the USA based on SEER data, 1975–2000. Miscellaneous includes STS with <1% incidence of total; small cell sarcoma 0.9%, chondrosarcoma (soft tissue) 0.8%, giant cell sarcoma 0.6%, desmoplastic small round cell tumor 0.6%, and others 7%. Modified from Bleyer et al.3 Abbreviations: ASPS, alveolar soft part sarcoma; AYA, adolescent and young adults; CNS, central nervous system; MPNST, malignant peripheral nerve sheath tumor; PNET, peripheral neuroectodermal tumor; SEER, Surveillance, Epidemiology, and End Results; STS, soft tissue sarcoma. |
In addition to the variability in tumor classification schemas, there is also variability in the staging systems used for the AYA group of patients. In pediatric studies, STSs are classified as low-risk, intermediate-risk, or high-risk tumors. There is no consensus yet whether the COG or the FNCLCC system is more predictive of clinical outcome, hence this evaluation is part of the most recent COG study for NRSTS (ARST0332). In contrast, in adults, STSs are usually staged based on the TNM classification following the AJCC10 or the UICC.11 Given these diverse classification and staging systems, characterization of STS in the AYA population is far from uniform, which often makes comparison of results between different studies difficult.
The etiology of NRSTS in AYA patients is rarely known, and the majority of these tumors are thought to be sporadic. Germline cancer predisposition syndromes, such as Li–Fraumeni syndrome, neurofibromatosis, and Beckwith–Wiedeman syndrome, are sometimes associated with the development of NRSTS. In utero exposure to marijuana or cocaine has been associated with childhood RMS,12 but it is unclear if this applies to the AYA group, given the delay between exposure and tumor initiation. In rare cases, radiation therapy, viral infections, or immune deficiency may play a causative role in the development of STS.1 In addition to the diversity in STS etiology, histology, and staging in AYA patients, this age group encompasses a large ethnic/racial and gender diversity in cancer incidence and distribution. Despite all of the differences between children and AYA patients, and between adults and AYA patients, historically, AYA patients are usually merged in clinical trials with either children (1–18 years), or adults (>16 years). The overall treatment outcomes are worse in AYA patients than in the pediatric population, and the AYA group is underrepresented in clinical trials.13,14 Among the proposed explanations for this observation is that these patients are being treated by either adult or pediatric oncologists, who may vary in their familiarity/unfamiliarity with studies in the “other” field; alternatively, there may be differences in biology (some tumors may respond better to adult protocols, while some may respond better on pediatric protocols). Only recently has the AYA cancer population received dedicated attention in studying the incidence, biology, and outcomes of their cancers and their treatment. There is now an effort to specifically study the AYA population in clinical trials, but results are not yet readily available. Review of every STS histology in detail goes beyond the scope of this review. We review the therapies targeting the most common groups of STS (leaving out RMS and GIST, since treatment of these tumors differs radically from the treatment of most NRSTSs), keeping in mind that much of the data presented here is extrapolated either from pediatric trials or from adult trials, rarely from AYA-focused trials.
Current state of the field
Despite our increasing understanding of the biology of these tumors, treatment options for NRSTSs are still limited. For most STSs, resection with 1–2 cm negative margins is the primary treatment modality.15 In a pediatric multi-institutional study, patients with complete resection had the best outcome, with an OS of 85.1%, versus 35.3% for those who had incomplete resection. In this study, XRT increased OS in patients with incomplete resection to 68.7%.16 These findings are in agreement with adult Phase III studies where limb-sparing surgery in combination with XRT yields similar survival as amputation alone and, therefore, represents the current standard approach.17–19 Local recurrences can again be surgically approached.20 It has been shown that the likelihood of death is higher in unresectable tumors,21 nevertheless local control is not always achievable despite aggressive surgical therapy. Definitive radiation therapy is recommended for patients in whom the tumor cannot be surgically removed, but with radiation as the primary treatment modality, recurrence-free survival is only in the range of 30–40%.22–24 If the STS is unresectable, then neoadjuvant therapy with radiation therapy and/or chemotherapy is often recommended.25,26 Overall, it appears that some STS histologies are more chemosensitive than others. Clear classification regarding chemotherapy sensitivity and resistance has not been published, but, for example, synovial sarcoma is thought to be relatively chemotherapy sensitive, whereas MPNST is thought to be relatively chemotherapy resistant. More insight on this issue will hopefully emerge from the most recently completed COG trial for treatment of patients with NRSTS, ARST0332, since this was a primary aim of the study.
First-line chemotherapy regimens for treatment-naive patients usually consist of doxorubicin with or without addition of ifosfamide. Treatment of STS patients with doxorubicin alone showed similar OS but lower response rate and PFS compared to patients treated with doxorubicin in combination with ifosfamide.27,28 Similar studies showed higher antitumor activity, but not OS, with combination therapy. Based on the general notion that combination therapy is tolerable and potentially superior, the most recent NRSTS COG study, ARST0332, used doxorubicin with ifosfamide.29–32 As upfront therapy, docetaxel was found to be inferior to doxorubicin,33,34 but the combination of gemcitabine with docetaxel has been successfully used as therapy for recurrent disease with or without bevacizumab.35–39 Also high-dose ifosfamide- and cyclophosphamide-based therapies can be used in this setting; superiority of either has not yet been shown.40,41 In some cases of STS, even rechallenge with ifosfamide can be successful.42
Newer agents are being explored, including trabectedin, which causes DNA damage by binding to the N2 of guanine and affecting transcription regulation. Mechanisms of action are reviewed elsewhere.43,44 Trabectedin was approved in Europe in 2007 and by the US FDA in December 2015 for treatment of unresectable or metastatic liposarcoma and leiomyosarcoma, based on several studies that showed encouraging results in patients with STS.45–48 In a Phase III trial of patients with metastatic liposarcoma or leiomyosarcoma who failed conventional chemotherapy, trabectedin was superior to therapy with dacarbazine.49 Maintenance therapy has also been investigated but has not improved outcomes. Recently, a Phase III trial of the mTOR inhibitor (ridaforolimus) versus placebo given as maintenance therapy showed an improved PFS but not OS.50 Because current therapy is very toxic and nonspecific and patients with chemotherapy resistant STS histologies might not derive any benefit at all, while experiencing adverse effects, it is crucial to investigate drugs that are more selective, hopefully less toxic, and that improve the survival of our patients with STS.
Definition of targeted therapy
The National Cancer Institute defines targeted therapies as “drugs or other substances that block the growth and spread of cancer by interfering with specific molecules (ie, ‘molecular targets’) that are involved in the growth, progression, and spread of cancer”.51 Targeted therapy should not be mistaken as an invention of the past decade, even though agents have only been labeled “targeted” more often recently. Targeted therapies have been used in cancer therapy since the development of methotrexate, which specifically targets folate metabolism. Subsequently developed chemotherapeutics more directly induced cell death via DNA damage, acting on all fast-dividing cells. Although this approach to chemotherapy has been successful for many malignancies, it has unfortunately so far not translated into high cure rates in high-risk sarcomas and carries the risk for significant adverse events. Furthermore, AYA patients are more prone to toxicities from these nonspecific agents because they often require treatment intensification due to the biology of their disease and additionally experience more side effects with the same therapy than younger children.52 Moreover, AYA survivors of STS experience significant long-term side effects, including cardiotoxicity, infertility, and secondary malignancies.53–56 Therefore, new therapies are urgently needed to improve short-term as well as long-term outcomes of AYA patients with STS. For the purpose of this review, we discuss targeted therapies as any therapy that directly targets a specific cell mechanism or cell surface marker unique or at least more prominent in cancer cells compared with normal tissue.
Several reports have shown the importance of tumor-infiltrating macrophages in cancer in general and in STS in particular.57–61 Recently, the field of cancer therapy has broadened to not only target the tumor cell itself but also target the microenvironment that helps the tumor thrive. One of these agents is mifarmutide or MTP-PE, a macrophage activator that has been studied mostly in bone sarcomas. The addition of MTP-PE to standard chemotherapy did not result in a statistically significant improvement in PFS in the study design used in the Intergroup 0133 trial,62 although in a post hoc analysis, the drug did appear to improve OS with a trend for improved event-free survival in nonmetastatic osteosarcoma.63 The analysis of the group of metastatic patients followed the same trend but did not reach statistical significance.64 A Phase II study undertaken by the EORTC Soft Tissue and Bone Sarcoma Group did not show responses in patients with STS.65 Given the infancy of these tumor microenvironment-modifying agents in clinical studies, we will not include them in our review. The term “targeted therapy” could also be expanded to include nanoparticle and liposomal packaging of chemotherapeutic agents, as well as antibody-mediated drug delivery. The former has recently been reviewed in the context of osteosarcoma.66 Even though these groups of agents present interesting treatment approaches for patients with STS, discussion of these agents goes beyond the scope of this review. A summary of targeted therapies in STS can be found in Table 1.
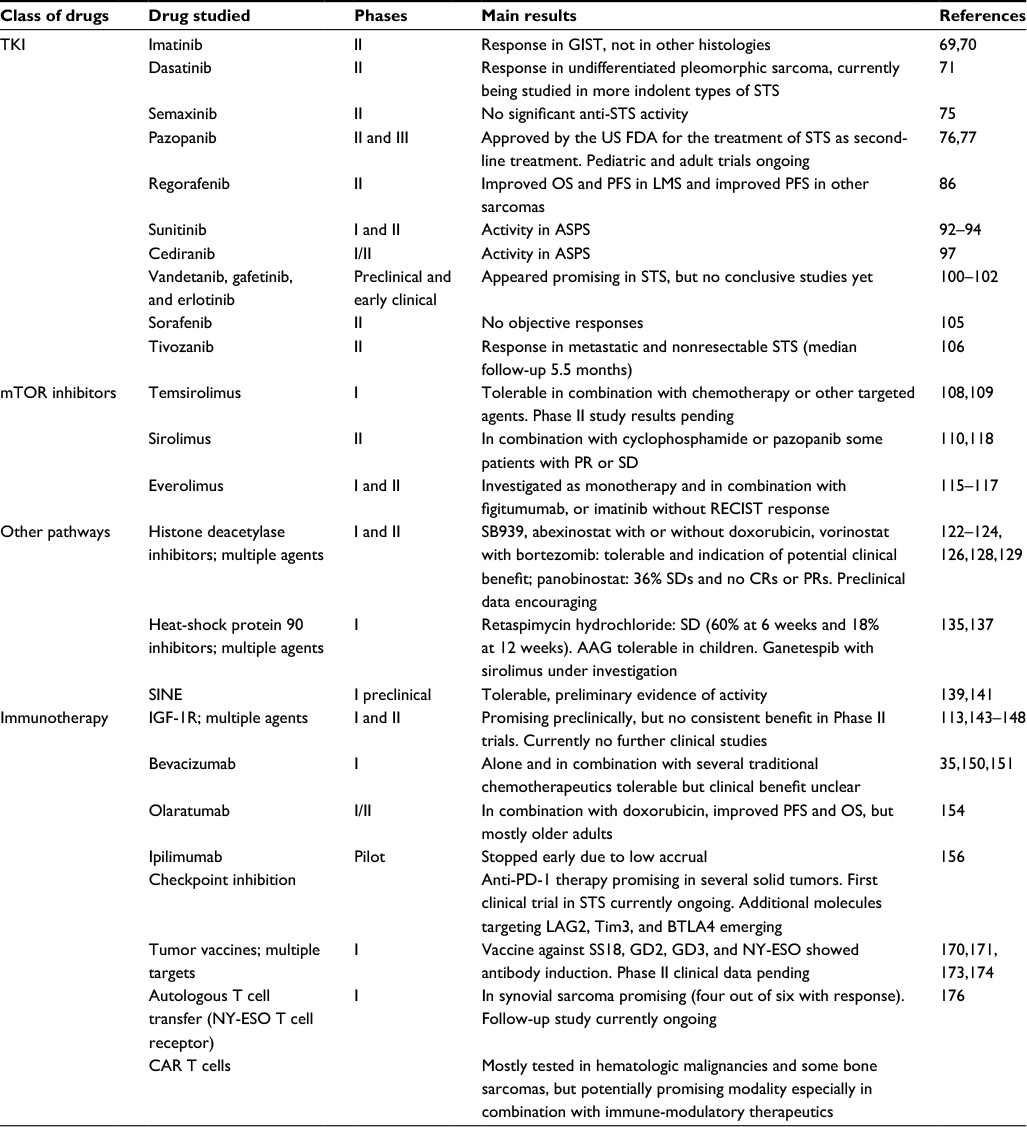 | Table 1 Summary of results of targeted therapies in STS Abbreviations: AAG, 17-N-allylamino-17-demethoxygeldanamycin; ASPS, alveolar soft part sarcoma; CAR, chimeric antigen receptor; CRs, complete remissions; FDA, Food and Drug Administration; GIST, gastrointestinal stromal tumor; IGF-1R, insulin-like growth factor-1 receptor; LMS, leiomyosarcomas; mTOR, mechanistic target of rapamycin; OS, overall survival; PFS, progression-free survival; PR, partial response; RECIST, Response Evaluation Criteria in Solid Tumors; SD, stable disease; SINE, selective inhibitors of nuclear export; STS, soft tissue sarcoma; TKI, tyrosine kinase inhibitor. |
TKIs
Tyrosine kinases are enzymes involved in signal transduction and regulation of many cellular processes. These enzymes can be overactive in cancers, often through mutational activation, genetic rearrangements, or amplification. TKIs have been used in the treatment of several cancers and are currently being tested for the treatment of STSs. Imatinib (a multitargeted TKI) is most well known for its effects in Ph+ chronic myelogenous leukemia;67 further promising results were then seen in patients with GIST.68 Similarly, the EORTC Soft Tissue and Bone Sarcoma Group conducted a Phase II trial of imatinib where responses were seen in patients with GIST but not in other types of STS.69 Later, imatinib was evaluated in ten histologic subtypes of sarcoma by SARC. Despite sufficient patient numbers in each group and some rare responses, evaluation via a Bayesian hierarchical model did not support further evaluation of imatinib as monotherapy for STS.70 Dasatinib is a TKI (with activity against BCR-ABL, Src, and others), which has also been evaluated in STS. Among the more aggressive STS histologies, responses were seen only in undifferentiated pleomorphic sarcoma,71 but results of a study investigating dasatinib in more indolent types of STS, including ASPS, chondrosarcoma, chordoma, epithelioid sarcoma, and solitary fibrous tumor, are expected to be available soon.
Angiogenesis plays an important role in tumor growth and metastasis,72 and VEGF has been shown to be overexpressed in some STSs.73,74 Therefore, VEGF inhibition through TKI and monoclonal antibodies (discussed below) has been pursued as one avenue to target STS. Even though SU415 (semaxinib) did not provide significant antitumor activity,75 pazopanib, a multikinase inhibitor with strong antiangiogenic effects, was found to have a more favorable profile. The PALETTE study, a Phase III study evaluating 369 adult patients with nonadipocytic STS who failed standard chemotherapy, showed prolonged median PFS and OS with pazopanib compared to placebo.76,77 Pazopanib was approved by the FDA in 2012 for the treatment of patients with STS who have received prior therapy. Based on these encouraging results, a Phase I study was performed by COG in pediatric patients; this study demonstrated tolerability in children with sarcoma.78 A Phase II study in pediatric patients with relapsed or refractory solid tumors is currently ongoing (NCT01956669). The combination of pazopanib with standard chemotherapy or radiation appears promising. Pazopanib in combination with paclitaxel in breast cancer patients in the neoadjuvant setting following doxorubicin and cyclophosphamide treatment showed activity but was associated with significant toxicity.79 A study evaluating pazopanib in combination with cyclophosphamide in ovarian cancer patients has been completed, and the results are currently pending.80 Pazopanib in combination with gemcitabine and docetaxel as neoadjuvant therapy for STS showed increased toxicity, but firm conclusions cannot be drawn since this study consisted of five patients only and closed early due to slow accrual.81 A randomized Phase II study treating STS patients with gemcitabine in combination with either pazopanib or docetaxel is ongoing and accruing well (NCT01593748). Furthermore, the addition of pazopanib to ifosfamide and doxorubicin in combination with radiation therapy is being studied in a COG trial enrolling pediatric and adult NRSTS patients (ARST1321). Finally, serum cytokines and angiogenic factors at baseline associated with pazopanib-specific toxicities and efficacy that might serve as biomarkers in future clinical trials or in routine clinical treatment of patients with STS have been reported.82
Regorafenib is another multitargeted TKI, with activity against angiogenic, stromal (VEGFR1–3, TIE2, FGFR1, and PDGFR-β), oncogenic (KIT and RET), and intracellular signaling (RAF1 and B-RAF) kinases among others, currently in Phase II trial (REGOSARC) for STS83 after showing activity in both colorectal cancer and GIST in Phase III trials.84,85 Preliminary results from REGOSARC demonstrated improved PFS and OS in patients with leiomyosarcoma and improved PFS in those with other sarcomas.86 Sunitinib is a TKI-targeting kinases in the PDGFR family, VEGFR and RET.87–89 Sunitinib was initially studied in renal cell carcinoma90 and GIST.91 It has activity as a single agent in a small group of patients with ASPS92,93 and in STS when combined with radiation.94,95
Cediranib is a TKI with activity against VEGFR1–3 and KIT that has been investigated in GIST and STS and caused reduced FDG-PET activity in four out of six patients with ASPS but no overall reductions in SUVmax.96 A larger Phase II prospective trial found an ORR of 35% and a disease control rate of 84% at 24 weeks in patients with ASPS treated with single-agent cediranib.97 Cediranib is tolerable in children and adolescents with solid tumors.98 In the PPTP, cediranib appeared to have an effect in combination with mTOR inhibition.99 Several other TKIs are in the early stages of evaluation. Vandetanib and gefitinib appeared promising in preclinical models.100 Gefitinib, a small molecule inhibitor of EGFR, was shown to be well tolerated in children in a Phase I study.101 Gefitinib tested in patients with solid tumors other than STS increased the bioavailability of oral irinotecan,102 which might benefit the group of patients with STS. Erlotinib, another small molecule inhibitor of EGFR, in combination with doxorubicin, seemed favorable in preclinical studies.103 Erlotinib as a single agent followed in combination with temozolomide was tolerated in a Phase I study that included 18 pediatric patients with STS.104 Sorafenib used in Wilms tumor and RMS was tolerated, but no objective responses were observed.105 Tivozanib, a TKI with activity against VEGFR1–3 was well tolerated and showed some responses in a Phase II study in patients with metastatic and nonresectable STSs.106 One has to keep in mind that targeted therapies might harbor side effects specific to the AYA group. Antiangiogenic agents, for example, in addition to their nonspecific side effects (skin, hepatic, and gastro-intestinal), were found to have an effect on the development of the growth plate.107 Although it is tempting to believe that these “targeted” agents are more tumor specific and therefore less toxic to patients, these hopes have not been borne out in clinical trials and in routine use, and toxicity remains a major concern.
mTOR inhibitors
Additional agents target specific pathways that are known to be upregulated in STS. mTOR is a part of two multiprotein complexes, mTORC1 and mTORC2, that signals downstream of PI3K/AKT, affecting many processes important for cancer cell survival. Rapamycin and its analogs (rapalogs) inhibit mainly mTORC1, and only after long-term treatment, mTORC2 as well. A full review of this pathway and its inherent complexity, including feedback mechanisms, goes beyond the scope of this review. A Phase I study of temsirolimus in combination with irinotecan and temozolomide in children and adolescents with relapsed and refractory solid tumors included RMS patients. The combination was tolerable in children and adolescents.108 Temsirolimus was further tested in combination with liposomal doxorubicin in patients with recurrent and refractory bone sarcomas and STSs. Here, increased exposure to sirolimus (active metabolite of temsirolimus) was seen when patients were concurrently treated with liposomal doxorubicin.109 Results of the Phase II study are forthcoming. Another Phase II study evaluated sirolimus and cyclophosphamide in the treatment of advanced sarcoma. A majority of patients had STS. Though the regimen resulted in PR or SD in some patients, it was overall deemed not superior to other “active” regimens.110 Based on the notion that mTORC2 is minimally affected by rapalogs and that negative feedback mechanisms exist, combinations with upstream signaling pathways have been tested. Temsirolimus was tested in a Phase I trial in combination with EKB-569, an EGFR inhibitor, in patients with unresectable tumors. This study included one patient with ASPS who reached a PR for three cycles. Overall, no CRs were seen and the combination caused significant toxicity.111 Temsirolimus in combination with cixutumumab (an antibody against IGF-1R) was first tested in an adult Phase I trial112 and subsequently in pediatric and AYA patients with recurrent and refractory sarcomas and showed tolerability.113
Another mTOR inhibitor, everolimus, was tested in combination with figitumumab, a monoclonal antibody against IGF-1R, in advanced sarcomas and other solid tumors. Similar to other IGF-1R studies, overall response was limited, without CRs, but strong PRs were seen in a small number of patients.114 Everolimus monotherapy again was tolerable in sarcoma patients after the failure of anthracyclines and ifosfamide but had only moderate antitumor activity.115 Combining everolimus with imatinib appeared beneficial in a case report in synovial sarcoma,116 but a Phase Ib/II study failed to achieve responses based on RECIST in synovial and other STS patients (though three patients had SD), so this combination might be effective in a subgroup of patients.117 When sirolimus was added to pazopanib in nine patients with metastatic unresectable high-grade STS who had previously failed pazopanib monotherapy, one PR, four SDs, and four PDs were seen, with a median PFS of 5.5 months. This combination might prolong the effective phase of pazopanib but has to be evaluated in larger clinical trials.118 These trials are currently ongoing in solid tumors (NCT01072890 and NCT01184326). Ridaforolimus was tested as monotherapy in patients with STS and showed clinical benefit responses (defined as CR, PR, or SD) in 21.1% of patients, but only one patient in the cohort of other STS showed a PR and there were no CRs observed.119
Drugs that target other pathways
AYA sarcomas are distinct from adult carcinomas in their genetic and epigenetic characteristics. While adult carcinomas often have a significant mutational burden, gene mutations in sarcomas appear to be less frequent,120 and therefore, epigenetic alterations might have a stronger influence on sarcomagenesis.121 HDAC inhibitors are a particularly promising group of epigenetic modifiers. These drugs have been used more in the setting of hematologic malignancies but are increasingly being evaluated in patients with solid tumors. A Phase II trial using SB939 in patients with recurrent or metastatic translocation-associated STS showed tolerability, but efficacy could not be evaluated due to unavailability of the drug.122 The same drug was tested in a Phase I pediatric trial in patients with refractory solid tumors. The majority of patients had Ewing’s sarcoma, but the drug appeared to be well tolerated.123 Another Phase I study again showed tolerability of abexinostat in combination with doxorubicin.124 The combination of vorinostat with bortezomib (a proteasome inhibitor) was tested in a Phase I study of patients with advanced solid tumors and appeared to have a favorable side effect profile, with some clinical responses.125 Panobinostat was evaluated in a Phase II trial in patients with advanced previously treated STS. A total of 36% of patients achieved SD, while no PRs were seen.126 General tolerability of HDAC inhibitors in the pediatric population has been shown in a Phase I study of vorinostat.127 Preclinical studies in epithelioid sarcoma128 and MPNST129 are encouraging, providing further impetus to explore these drugs in the clinical setting.
Hsp90 is a member of the heat shock group of molecular chaperones involved in the maintenance of protein folding and assembly.130,131 Due to the ubiquitous requirement for heat shock protein function in signal transduction pathways, and in the proliferation and maintenance of cancer cells, agents targeting Hsp90 have been developed.132–134 One Phase I study of an Hsp90 inhibitor, retaspimycin hydrochloride (IPI-504), included STS patients. A total of 60% of patients with STS showed SD for at least 6 weeks, but only 18% at 12 weeks.135 Severe hepatic toxicity was noted in a Phase III trial of the same drug (NCT00688766), which prompted change in dosing.136 The Hsp90 inhibitor 17-AAG was found to be well tolerated in pediatric patients, though the study included only one patient with STS (DSRCT).137 Ganetespib in combination with the mTOR inhibitor sirolimus is currently under investigation for patients with unresectable sarcomas and MPNST, in a SARC Phase I/II study (NCT02008877).
SINE inhibitors are a new group of small molecules that target nucleocytoplasmic transport.138 Selinexor has shown preclinical activity in sarcomas,139 with preliminary evidence of activity in STS patients,140 and furthermore appears to be tolerable in children.141
Immunotherapy
Immunotherapy to treat cancer has been explored for several decades and was initially restricted to immunogenic tumors. In the early stages, IL-2 therapy was considered as a breakthrough in melanoma therapy. When applied to sarcomas, some responses were seen in osteosarcoma but not in STSs.142 Cancer immunotherapy has since expanded, and antitumor immunity can be achieved by several means: transfer of preformed antibodies (which can either be directly therapeutic, for example, trastuzumab, or can indirectly target cancer cells, for example, immune checkpoint inhibitors), vaccine strategies that activate host T cells in vivo, or adoptive transfer of in vitro activated T cells. Recently, several new immunotherapeutic targets were found in STS that may allow more widespread application of immunotherapy to sarcomas.
Monoclonal antibodies
Therapy with monoclonal antibodies, such as alemtuzumab (CD52), trastuzumab (HER2), brentuximab vedotin (CD30), rituximab (CD20), and blinatumomab (CD19 and CD3), has revolutionized the treatment of many cancers. IGF-1R is a target that was thought to be ideal in bone sarcomas and was therefore tested with much enthusiasm in STSs as well. One antibody targeting IGF-1R, cixutumumab, was used in a Phase II trial for children with refractory or recurrent solid tumors. Of the 20 patients with RMS, one patient had a PR, and none of the ten patients with synovial sarcomas responded.143 Since it had previously been seen that cixutumumab had better efficacy in combination with mTOR inhibitors,144,145 it was combined with temsirolimus in an adult Phase II study enrolling AYA patients with chemotherapy-refractory sarcomas. Even though the primary endpoint of PFS at 12 weeks was reached in 31–39% of patients, no CRs were seen and only 2–3% of STS patients achieved a PR.112 A Phase II COG study of the same combination showed equally disappointing results, and based on limited drug availability (related to the failure of these agents in clinical trials treating a variety of carcinomas), no further studies of this combination are currently underway.113
Based on these initial exciting preclinical data, several other agents targeting IGF-1R were tested as single agents in Phases I and II clinical trials. Most of them showed great responses in singular patients, but no benefit overall in larger cohorts. These agents include R1507,146 AVE1642,147 MK-0646,148 and ganitumab alone or in combination with conatumumab (death receptor 5 agonist).149 These results suggest that agents targeting IGF-1R might benefit a small subset of patients, but our current understanding of the biology of these tumors does not allow for selection of these patients yet.
Additional monoclonal antibodies with different targets have also been tested. As discussed earlier, inhibition of angiogenesis is thought to be a promising approach to treating STS. Bevacizumab, an anti-VEGF antibody, was tested in combination with docetaxel and gemcitabine in chemotherapy-naive patients. It was well tolerated, but its clinical effect was unclear in this Phase Ib study.35 Pediatric studies have evaluated bevacizumab in combination with irinotecan alone,150 or in combination with VOIT (vincristine, oral irinotecan, and temozolomide).151 In these studies, the treatments were tolerable, but benefit was unclear. Among STS, HER2 expression was found in synovial sarcomas152 and trastuzumab was found to be tolerable in the pediatric age group in a Phase II study of patients with Ewing’s sarcoma and osteosarcoma, but efficacy in STS remains controversial153 and there have been no other prospective clinical trials published in STS patients.
In a Phase Ib/II study, olaratumab, a PDGFR-α inactivating monoclonal antibody, in combination with doxorubicin was compared with doxorubicin monotherapy in patients with unresectable or metastatic STS without prior anthracycline treatment. The olaratumab arm showed improved median PFS and median OS.154 Although encouraging, these results might be difficult to apply to children and AYA patients since the youngest patient in the study was 22 years old, with a median age of 58.5 years, with a slightly different spectrum of STS histologies than one would expect in the AYA population.
PD-1 and CTLA-4 mediate T-cell-inhibiting signals and have been found to be present in tumors, impairing the natural immunity against cancer. Inhibition of these pathways leads to T-cell activation. This approach showed promising results in melanoma and was translated successfully to several other solid tumors. Initially, ipilimumab, an antibody against CTLA-4, was developed for therapy in metastatic melanoma.155 Ipilimumab was then tested in synovial sarcoma patients in a pilot study, but the study was stopped due to slow accrual, no response based on RECIST, and lack of an immune response (only six patients enrolled).156
TILs have been demonstrated in sarcomas, and while PD-L1 expression did not correlate with aggressive features or clinical outcome,157 targeting this pathway might prove useful in STS. Monoclonal antibodies were designed that block the inhibitory signal of PD-1 on T cells, thereby activating T-cell responses. Pembrolizumab, an anti-PD-1 antibody, demonstrated activity in melanoma and was approved by the FDA in September 2014.158 The results of the first trial of anti-PD-1 monotherapy utilizing pembrolizumab in patients with advanced sarcomas (SARC028) were presented at the annual meeting of the American Society for Clinical Oncology in 2016 (Chicago, IL, USA) and showed limited activity, with infrequent responses seen in patients with osteosarcoma, pleomorphic sarcoma, liposarcoma, and chondrosarcoma. Another PD-1 inhibitor, nivolumab, was initially approved for the treatment of melanoma by the FDA in 2014.159,160 Several trials show activity in other solid tumors,161,162 and it gained approval in non-small-cell lung cancer, renal cell carcinoma, and Hodgkin lymphoma shortly thereafter. In melanoma, it has been shown that the combination of anti-PD-1 and anti-CTLA-4 therapy is superior to monotherapy with either agent and also caused increased toxicity.163 Chan et al found that responders and nonresponders to anti-PD-1 therapy in melanoma can be grouped based on their mutation and neoepitope signatures.164 This approach might increase our ability to select patients with higher likelihood of responding to immune checkpoint therapy. New checkpoint inhibitors are currently under investigation, including molecules targeting LAG3, Tim3, and BTLA4. One could also envision the combination of immune checkpoint blockade and vaccine trials (decrease immune evasion and increase cytotoxic T cells),165 conventional chemotherapy (eg, doxorubicin, which is thought to result in immunogenic cell death and already proven to be useful in many STSs), HDAC inhibitors,166 or radiation therapy.167
Tumor vaccines
Synovial sarcoma is an attractive tumor for immunotherapeutic approaches because it expresses the cancer testis antigen NY-ESO-1 at a frequency approaching 100%.168,169 Other tumor antigens overexpressed in STSs are SSX2/3, MAGE, GAGE, and WT1. Also, fusion protein expression in STS (such as SS18-SSX in synovial sarcoma, PAX3/7-FOXO1 in RMS, TLS-CHOP in myxoid liposarcoma, and EWSR1 in clear cell sarcoma, myxoid chondrosarcoma, DSRCT, and others) can potentially be used as selective immunotherapy targets. Vaccine trials targeting SS18 in patients with synovial sarcoma showed immunologic responses,170 and a subsequent clinical trial showed SD in vaccination groups.171 Gangliosides GD2 and GD3 are expressed in many sarcomas,172 and vaccine trials in sarcoma patients targeting these antigens elicit immune responses.173 A randomized Phase II clinical trial examining a trivalent GM2, GD2, and GD3 vaccine in patients with metastatic disease, rendered disease-free after surgery, showed antibody induction but no change in PFS. Follow-up for OS is ongoing, and results so far have only been published as an abstract.174 Dendritic cell vaccines have been tested in a Phase I clinical trial in children with solid tumors and sarcomas and appear to be well tolerated and result in responses in some of the children.175 As an alternative to relying on active immunization, autologous T cells transduced with an NY-ESO-1 T-cell receptor have been adoptively transferred to six patients with synovial cell sarcoma. Four had a response and one had a durable response for 18 months.176 A follow-up study is currently recruiting patients with sarcomas (NCT02319824).
CAR T cells
Another targeted therapy approach pioneered in hematologic malignancies is the use of CAR T cells. These are T cells that have been transduced with a genetically engineered T-cell receptor with a specific, targeted extracellular antigen recognition domain, a transmembrane domain, and an intracellular signaling domain.177 Even though CAR T cells have shown great promise in hematologic malignancies, this success has not yet been translated to solid tumors or sarcomas. Thus far, the CAR T cell experience in STS is limited. CAR T cells directed at the fetal acetylcholine receptor to target RMS have been developed but have not gone beyond the preclinical testing phase.178–180 CAR T cells are under investigation in bone sarcomas targeting GD2 in Ewing’s sarcoma and osteosarcoma and HER2 in osteosarcoma.181,182 One Phase I trial that included mostly patients with bone sarcomas, but one patient with DSRCT, showed tolerability of HER2 CAR T cells.183 In solid tumor CAR T cell therapy, the tumor microenvironment seems to interfere with proper T cell function184 and this might also apply to sarcomas.179 New approaches incorporating immune-modulatory strategies, such as PD-1 or gamma delta T cells into CAR T cell therapeutic approaches,185–187 are being investigated.
Conclusion
STSs in pediatric and AYA patients remain a therapeutic challenge. Many targeted agents are on the horizon, but none have provided the long anticipated breakthrough direly needed for these patients. For now, therapy is still mostly based on surgery, radiation therapy, and traditional chemotherapy approaches. Especially in our pediatric and AYA population, even when cure can be achieved, long-term side effects are significant. Obstacles to improvements in the field are low patient numbers (and therefore slow accrual to clinical trials) and tremendous differences in tumor biology among histologic subtypes (grouping of tumors in clinical trials with vastly different responses to therapy). Our knowledge of the biology of these tumors has increased tremendously over the last two decades, and there is hope that further advances in our understanding will continue to produce targeted agents that can be tailored to the molecular drivers in various histologic subtypes of STSs.
Disclosure
The authors report no conflicts of interest in this work.
References
Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F. World Health Organization Classification of Tumours of Soft Tissue and Bone. Vol. 5. 4th ed. Lyon: International Agency for Research on Cancer (IARC); 2013:468. | ||
Ries LA, Smith MA, Gurney JG, et al. Cancer Incidence and Survival among Children and Adolescents: United States SEER Program 1975-1995. Bethesda, MD: National Cancer Institute; 1999. [NIH Pub. No. 99-4649]. | ||
Bleyer A, O’Leary M, Barr R, Ries LAG. Cancer Epidemiology in Older Adolescents and Young Adults 15 to 29 Years of Age, Including SEER Incidence and Survival: 1975-2000. National Cancer Institute, NIH Pub. No. 06-5767. Besthesda, MD, 2006. | ||
Bleyer A, Viny A, Barr R. Cancer in 15- to 29-year-olds by primary site. Oncologist. 2006;11(6):590–601. | ||
Fletcher CDM, Unni KK, Mertens F. World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Soft Tissue and Bone. Lyon: IARC Press; 2002. | ||
Davicioni E, Wai DH, Anderson MJ. Diagnostic and prognostic sarcoma signatures. Mol Diagn Ther. 2008;12(6):359–374. | ||
Hingorani P, Missiaglia E, Shipley J, et al. Clinical application of prognostic gene expression signature in fusion gene-negative rhabdomyosarcoma: a report from the Children’s Oncology Group. Clin Cancer Res. 2015;21(20):4733–4739. | ||
Skapek SX, Anderson J, Barr FG, et al. PAX-FOXO1 fusion status drives unfavorable outcome for children with rhabdomyosarcoma: a children’s oncology group report. Pediatr Blood Cancer. 2013;60(9):1411–1417. | ||
Williamson D, Missiaglia E, de Reynies A, et al. Fusion gene-negative alveolar rhabdomyosarcoma is clinically and molecularly indistinguishable from embryonal rhabdomyosarcoma. J Clin Oncol. 2010;28(13):2151–2158. | ||
Edge S, Byrd DR, Compton CC, Fritz AG, Greene FL, Totti A. AJCC Cancer Staging Manual. 7th ed. New York, NY: Springer-Verlag; 2010. | ||
Sobin LH, Gospodarowicz MK, Wittekind C. TNM Classificatin of Malignant Tumours. 7th ed. Hoboken, NJ: Wiley-Blackwell; 2009. | ||
Grufferman S, Schwartz AG, Ruymann FB, Maurer HM. Parents’ use of cocaine and marijuana and increased risk of rhabdomyosarcoma in their children. Cancer Causes Control. 1993;4(3):217–224. | ||
Bleyer A, Budd T, Montello M. Adolescents and young adults with cancer: the scope of the problem and criticality of clinical trials. Cancer. 2006;107(7 suppl):1645–1655. | ||
Bleyer A, Morgan S, Barr R. Proceedings of a workshop: bridging the gap in care and addressing participation in clinical trials. Cancer. 2006;107(7 suppl):1656–1658. | ||
O’Donnell PW, Griffin AM, Eward WC, et al. The effect of the setting of a positive surgical margin in soft tissue sarcoma. Cancer. 2014;120(18):2866–2875. | ||
Ferrari A, Miceli R, Rey A, et al. Non-metastatic unresected paediatric non-rhabdomyosarcoma soft tissue sarcomas: results of a pooled analysis from United States and European groups. Eur J Cancer. 2011;47(5):724–731. | ||
Pisters PW, Harrison LB, Leung DH, Woodruff JM, Casper ES, Brennan MF. Long-term results of a prospective randomized trial of adjuvant brachytherapy in soft tissue sarcoma. J Clin Oncol. 1996;14(3):859–868. | ||
Yang JC, Chang AE, Baker AR, et al. Randomized prospective study of the benefit of adjuvant radiation therapy in the treatment of soft tissue sarcomas of the extremity. J Clin Oncol. 1998;16(1):197–203. | ||
Haas RL, Miah AB, LePechoux C, et al. Preoperative radiotherapy for extremity soft tissue sarcoma; past, present and future perspectives on dose fractionation regimens and combined modality strategies. Radiother Oncol. 2016;119(1):14–21. | ||
Canter RJ. Surgical approach for soft tissue sarcoma: standard of care and future approaches. Curr Opin Oncol. 2015;27(4):343–348. | ||
Spunt SL, Poquette CA, Hurt YS, et al. Prognostic factors for children and adolescents with surgically resected nonrhabdomyosarcoma soft tissue sarcoma: an analysis of 121 patients treated at St Jude Children’s Research Hospital. J Clin Oncol. 1999;17(12):3697–3705. | ||
Kepka L, DeLaney TF, Suit HD, Goldberg SI. Results of radiation therapy for unresected soft-tissue sarcomas. Int J Radiat Oncol Biol Phys. 2005;63(3):852–859. | ||
Slater JD, McNeese MD, Peters LJ. Radiation therapy for unresectable soft tissue sarcomas. Int J Radiat Oncol Biol Phys. 1986;12(10):1729–1734. | ||
Tepper JE, Suit HD. Radiation therapy alone for sarcoma of soft tissue. Cancer. 1985;56(3):475–479. | ||
Pratt CB, Pappo AS, Gieser P, et al. Role of adjuvant chemotherapy in the treatment of surgically resected pediatric nonrhabdomyosarcomatous soft tissue sarcomas: a Pediatric Oncology Group Study. J Clin Oncol. 1999;17(4):1219. | ||
Spunt SL, Hill DA, Motosue AM, et al. Clinical features and outcome of initially unresected nonmetastatic pediatric nonrhabdomyosarcoma soft tissue sarcoma. J Clin Oncol. 2002;20(15):3225–3235. | ||
Judson I, Verweij J, Gelderblom H, et al; European Organisation and Treatment of Cancer Soft Tissue and Bone Sarcoma Group. Doxorubicin alone versus intensified doxorubicin plus ifosfamide for first-line treatment of advanced or metastatic soft-tissue sarcoma: a randomised controlled phase 3 trial. Lancet Oncol. 2014;15(4):415–423. | ||
Edmonson JH, Ryan LM, Blum RH, et al. Randomized comparison of doxorubicin alone versus ifosfamide plus doxorubicin or mitomycin, doxorubicin, and cisplatin against advanced soft tissue sarcomas. J Clin Oncol. 1993;11(7):1269–1275. | ||
Demetri GD, Elias AD. Results of single-agent and combination chemotherapy for advanced soft tissue sarcomas. Implications for decision making in the clinic. Hematol Oncol Clin North Am. 1995;9(4):765–785. | ||
Patel SR, Vadhan-Raj S, Burgess MA, et al. Results of two consecutive trials of dose-intensive chemotherapy with doxorubicin and ifosfamide in patients with sarcomas. Am J Clin Oncol. 1998;21(3):317–321. | ||
Patel SR, Vadhan-Raj S, Papadopolous N, et al. High-dose ifosfamide in bone and soft tissue sarcomas: results of phase II and pilot studies – dose-response and schedule dependence. J Clin Oncol. 1997;15(6):2378–2384. | ||
Le Cesne A, Judson I, Crowther D, et al. Randomized phase III study comparing conventional-dose doxorubicin plus ifosfamide versus high-dose doxorubicin plus ifosfamide plus recombinant human granulocyte-macrophage colony-stimulating factor in advanced soft tissue sarcomas: a trial of the European Organization for Research and Treatment of Cancer/Soft Tissue and Bone Sarcoma Group. J Clin Oncol. 2000;18(14):2676–2684. | ||
Verweij J, Lee SM, Ruka W, et al. Randomized phase II study of docetaxel versus doxorubicin in first- and second-line chemotherapy for locally advanced or metastatic soft tissue sarcomas in adults: a study of the european organization for research and treatment of cancer soft tissue and bone sarcoma group. J Clin Oncol. 2000;18(10):2081–2086. | ||
Amodio A, Carpano S, Paoletti G, et al. [Phase II study of docetaxel in patients with advanced stage soft tissue sarcoma]. Clin Ter. 1998;149(2):121–125. Italian. | ||
Verschraegen CF, Arias-Pulido H, Lee SJ, et al. Phase IB study of the combination of docetaxel, gemcitabine, and bevacizumab in patients with advanced or recurrent soft tissue sarcoma: the Axtell regimen. Ann Oncol. 2012;23(3):785–790. | ||
Bay JO, Ray-Coquard I, Fayette J, et al; Groupe Sarcome Français. Docetaxel and gemcitabine combination in 133 advanced soft-tissue sarcomas: a retrospective analysis. Int J Cancer. 2006;119(3):706–711. | ||
Seddon B, Scurr M, Jones RL, et al. A phase II trial to assess the activity of gemcitabine and docetaxel as first line chemotherapy treatment in patients with unresectable leiomyosarcoma. Clin Sarcoma Res. 2015;5:13. | ||
Maki RG, Wathen JK, Patel SR, et al. Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. J Clin Oncol. 2007;25(19):2755–2763. | ||
Rapkin L, Qayed M, Brill P, et al. Gemcitabine and docetaxel (GEMDOX) for the treatment of relapsed and refractory pediatric sarcomas. Pediatr Blood Cancer. 2012;59(5):854–858. | ||
Mulder RL, Paulides M, Langer T, Kremer LC, van Dalen EC. Cyclophosphamide versus ifosfamide for paediatric and young adult bone and soft tissue sarcoma patients. Cochrane Database Syst Rev. 2012;12:CD006300. | ||
Lee SH, Chang MH, Baek KK, et al. High-dose ifosfamide as second- or third-line chemotherapy in refractory bone and soft tissue sarcoma patients. Oncology. 2011;80(3–4):257–261. | ||
Noujaim J, Constantinidou A, Messiou C, et al. Successful ifosfamide rechallenge in soft-tissue sarcoma. Am J Clin Oncol. Epub 2015 Oct 29. | ||
D’Incalci M, Badri N, Galmarini CM, Allavena P. Trabectedin, a drug acting on both cancer cells and the tumour microenvironment. Br J Cancer. 2014;111(4):646–650. | ||
D’Incalci M, Galmarini CM. A review of trabectedin (ET-743): a unique mechanism of action. Mol Cancer Ther. 2010;9(8):2157–2163. | ||
Bui-Nguyen B, Butrynski JE, Penel N, et al; European Organisation for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group (EORTC/STBSG) and the Sarcoma Alliance for Research through Collaboration (SARC). A phase IIb multicentre study comparing the efficacy of trabectedin to doxorubicin in patients with advanced or metastatic untreated soft tissue sarcoma: the TRUSTS trial. Eur J Cancer. 2015;51(10):1312–1320. | ||
Cesne AL, Judson I, Maki R, et al. Trabectedin is a feasible treatment for soft tissue sarcoma patients regardless of patient age: a retrospective pooled analysis of five phase II trials. Br J Cancer. 2013;109(7):1717–1724. | ||
Samuels BL, Chawla S, Patel S, et al. Clinical outcomes and safety with trabectedin therapy in patients with advanced soft tissue sarcomas following failure of prior chemotherapy: results of a worldwide expanded access program study. Ann Oncol. 2013;24(6):1703–1709. | ||
Sanfilippo R, Dileo P, Blay JY, et al. Trabectedin in advanced synovial sarcomas: a multicenter retrospective study from four European institutions and the Italian Rare Cancer Network. Anticancer Drugs. 2015;26(6):678–681. | ||
Demetri GD, von Mehren M, Jones RL, et al. Efficacy and safety of trabectedin or dacarbazine for metastatic liposarcoma or leiomyosarcoma after failure of conventional chemotherapy: results of a phase iii randomized multicenter clinical trial. J Clin Oncol. 2016;34(8):786–793. | ||
Demetri GD, Chawla SP, Ray-Coquard I, et al. Results of an international randomized phase III trial of the mammalian target of rapamycin inhibitor ridaforolimus versus placebo to control metastatic sarcomas in patients after benefit from prior chemotherapy. J Clin Oncol. 2013;31(19):2485–2492. | ||
Cancer.gov [homepage on the Internet]. Bethesda: National Cancer Institute; 2016 [updated December 16, 2015]. Available from: http://www.cancer.gov/about-cancer/treatment/types/targeted-therapies/targeted-therapies-fact-sheet. Accessed August 10, 2016. | ||
Bukowinski AJ, Burns KC, Parsons K, Perentesis JP, O’Brien MM. Toxicity of cancer therapy in adolescents and young adults (AYAs). Semin Oncol Nurs. 2015;31(3):216–226. | ||
Record EO, Meacham LR. Survivor care for pediatric cancer survivors: a continuously evolving discipline. Curr Opin Oncol. 2015;27(4):291–296. | ||
Landier W, Armenian S, Bhatia S. Late effects of childhood cancer and its treatment. Pediatr Clin North Am. 2015;62(1):275–300. | ||
Metzger ML, Meacham LR, Patterson B, et al. Female reproductive health after childhood, adolescent, and young adult cancers: guidelines for the assessment and management of female reproductive complications. J Clin Oncol. 2013;31(9):1239–1247. | ||
Green DM, Liu W, Kutteh WH, et al. Cumulative alkylating agent exposure and semen parameters in adult survivors of childhood cancer: a report from the St Jude Lifetime Cohort Study. Lancet Oncol. 2014;15(11):1215–1223. | ||
Curino A, Mitola DJ, Aaronson H, et al. Plasminogen promotes sarcoma growth and suppresses the accumulation of tumor-infiltrating macrophages. Oncogene. 2002;21(57):8830–8842. | ||
Lee CH, Espinosa I, Vrijaldenhoven S, et al. Prognostic significance of macrophage infiltration in leiomyosarcomas. Clin Cancer Res. 2008;14(5):1423–1430. | ||
Mills CD, Lenz LL, Harris RA. A breakthrough: macrophage-directed cancer immunotherapy. Cancer Res. 2016;76(3):513–516. | ||
Torroella-Kouri M, Rodriguez D, Caso R. Alterations in macrophages and monocytes from tumor-bearing mice: evidence of local and systemic immune impairment. Immunol Res. 2013;57(1–3):86–98. | ||
Komohara Y, Fujiwara Y, Ohnishi K, Takeya M. Tumor-associated macrophages: potential therapeutic targets for anti-cancer therapy. Adv Drug Deliv Rev. 2016;99(pt B):180–185. | ||
Meyers PA, Schwartz CL, Krailo M, et al. Osteosarcoma: a randomized, prospective trial of the addition of ifosfamide and/or muramyl tripeptide to cisplatin, doxorubicin, and high-dose methotrexate. J Clin Oncol. 2005;23(9):2004–2011. | ||
Meyers PA, Schwartz CL, Krailo MD, et al; Children’s Oncology Group. Osteosarcoma: the addition of muramyl tripeptide to chemotherapy improves overall survival – a report from the Children’s Oncology Group. J Clin Oncol. 2008;26(4):633–638. | ||
Chou AJ, Kleinerman ES, Krailo MD, et al; Children’s Oncology Group. Addition of muramyl tripeptide to chemotherapy for patients with newly diagnosed metastatic osteosarcoma: a report from the Children’s Oncology Group. Cancer. 2009;115(22):5339–5348. | ||
Verweij J, Judson I, Steward W, et al. Phase II study of liposomal muramyl tripeptide phosphatidylethanolamine (MTP/PE) in advanced soft tissue sarcomas of the adult. An EORTC Soft Tissue and Bone Sarcoma Group study. Eur J Cancer. 1994;30A(6):842–843. | ||
Gonzalez-Fernandez Y, Imbuluzqueta E, Patino-Garcia A, Blanco-Prieto MJ. Antitumoral-lipid-based nanoparticles: a platform for future application in osteosarcoma therapy. Curr Pharm Des. 2015;21(42):6104–6124. | ||
O’Brien SG, Guilhot F, Larson RA, et al; IRIS Investigators. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2003;348(11):994–1004. | ||
Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347(7):472–480. | ||
Verweij J, van Oosterom A, Blay JY, et al. Imatinib mesylate (STI-571 Glivec, Gleevec) is an active agent for gastrointestinal stromal tumours, but does not yield responses in other soft-tissue sarcomas that are unselected for a molecular target. Results from an EORTC Soft Tissue and Bone Sarcoma Group phase II study. Eur J Cancer. 2003;39(14):2006–2011. | ||
Chugh R, Wathen JK, Maki RG, et al. Phase II multicenter trial of imatinib in 10 histologic subtypes of sarcoma using a bayesian hierarchical statistical model. J Clin Oncol. 2009;27(19):3148–3153. | ||
Schuetze SM, Wathen JK, Lucas DR, et al. SARC009: phase 2 study of dasatinib in patients with previously treated, high-grade, advanced sarcoma. Cancer. 2016;122(6):868–874. | ||
Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438(7070):932–936. | ||
Potti A, Ganti AK, Tendulkar K, et al. Determination of vascular endothelial growth factor (VEGF) overexpression in soft tissue sarcomas and the role of overexpression in leiomyosarcoma. J Cancer Res Clin Oncol. 2004;130(1):52–56. | ||
Zhang L, Hannay JA, Liu J, et al. Vascular endothelial growth factor overexpression by soft tissue sarcoma cells: implications for tumor growth, metastasis, and chemoresistance. Cancer Res. 2006;66(17):8770–8778. | ||
Heymach JV, Desai J, Manola J, et al. Phase II study of the antiangiogenic agent SU5416 in patients with advanced soft tissue sarcomas. Clin Cancer Res. 2004;10(17):5732–5740. | ||
van der Graaf WT, Blay JY, Chawla SP, et al; EORTC Soft Tissue and Bone Sarcoma Group; PALETTE Study Group. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2012;379(9829):1879–1886. | ||
Wilky BA, Meyer CF, Trent JC. Pazopanib in sarcomas: expanding the PALETTE. Curr Opin Oncol. 2013;25(4):373–378. | ||
Glade Bender JL, Lee A, Reid JM, et al. Phase I pharmacokinetic and pharmacodynamic study of pazopanib in children with soft tissue sarcoma and other refractory solid tumors: a children’s oncology group phase I consortium report. J Clin Oncol. 2013;31(24):3034–3043. | ||
Tan AR, Johannes H, Rastogi P, et al. Weekly paclitaxel and concurrent pazopanib following doxorubicin and cyclophosphamide as neoadjuvant therapy for HER-negative locally advanced breast cancer: NSABP Foundation FB-6, a phase II study. Breast Cancer Res Treat. 2015;149(1):163–169. | ||
Eichbaum M, Mayer C, Eickhoff R, et al. The PACOVAR-trial: a phase I/II study of pazopanib (GW786034) and cyclophosphamide in patients with platinum-resistant recurrent, pre-treated ovarian cancer. BMC Cancer. 2011;11:453. | ||
Munhoz RR, D’Angelo SP, Gounder MM, et al. A Phase Ib/II study of gemcitabine and docetaxel in combination with pazopanib for the neoadjuvant treatment of soft tissue sarcomas. Oncologist. 2015;20(11):1245–1246. | ||
Sleijfer S, Gorlia T, Lamers C, et al. Cytokine and angiogenic factors associated with efficacy and toxicity of pazopanib in advanced soft-tissue sarcoma: an EORTC-STBSG study. Br J Cancer. 2012;107(4):639–645. | ||
Brodowicz T, Liegl-Atzwager B, Tresch E, et al. Study protocol of REGOSARC trial: activity and safety of regorafenib in advanced soft tissue sarcoma: a multinational, randomized, placebo-controlled, phase II trial. BMC Cancer. 2015;15:127. | ||
Demetri GD, Reichardt P, Kang YK, et al; GRID Study Investigators. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):295–302. | ||
Grothey A, Van Cutsem E, Sobrero A, et al; CORRECT Study Group. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):303–312. | ||
Olivier M, Thomas B, Jennifer W, et al. Activity of regorafenib (RE) in leiomyosarcomas (LMS) and other types of soft-tissue sarcomas (OTS): results of a double-blind, randomized placebo (PL) controlled phase II trial. J Clin Oncol. 2015;33(15 suppl):10504. | ||
Chow LQ, Eckhardt SG. Sunitinib: from rational design to clinical efficacy. J Clin Oncol. 2007;25(7):884–896. | ||
Mendel DB, Laird AD, Xin X, et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2003;9(1):327–337. | ||
O’Farrell AM, Abrams TJ, Yuen HA, et al. SU11248 is a novel FLT3 tyrosine kinase inhibitor with potent activity in vitro and in vivo. Blood. 2003;101(9):3597–3605. | ||
Motzer RJ, Hutson TE, Tomczak P, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356(2):115–124. | ||
Reichardt P, Kang YK, Rutkowski P, et al. Clinical outcomes of patients with advanced gastrointestinal stromal tumors: safety and efficacy in a worldwide treatment-use trial of sunitinib. Cancer. 2015;121(9):1405–1413. | ||
Stacchiotti S, Tamborini E, Marrari A, et al. Response to sunitinib malate in advanced alveolar soft part sarcoma. Clin Cancer Res. 2009;15(3):1096–1104. | ||
Orbach D, Brennan B, Casanova M, et al. Paediatric and adolescent alveolar soft part sarcoma: a joint series from European cooperative groups. Pediatr Blood Cancer. 2013;60(11):1826–1832. | ||
Lewin J, Khamly KK, Young RJ, et al. A phase Ib/II translational study of sunitinib with neoadjuvant radiotherapy in soft-tissue sarcoma. Br J Cancer. 2014;111(12):2254–2261. | ||
Yoon SS, Stangenberg L, Lee YJ, et al. Efficacy of sunitinib and radiotherapy in genetically engineered mouse model of soft-tissue sarcoma. Int J Radiat Oncol Biol Phys. 2009;74(4):1207–1216. | ||
Judson I, Scurr M, Gardner K, et al. Phase II study of cediranib in patients with advanced gastrointestinal stromal tumors or soft-tissue sarcoma. Clin Cancer Res. 2014;20(13):3603–3612. | ||
Kummar S, Allen D, Monks A, et al. Cediranib for metastatic alveolar soft part sarcoma. J Clin Oncol. 2013;31(18):2296–2302. | ||
Fox E, Aplenc R, Bagatell R, et al. A phase 1 trial and pharmacokinetic study of cediranib, an orally bioavailable pan-vascular endothelial growth factor receptor inhibitor, in children and adolescents with refractory solid tumors. J Clin Oncol. 2010;28(35):5174–5181. | ||
Morton CL, Maris JM, Keir ST, et al. Combination testing of cediranib (AZD2171) against childhood cancer models by the pediatric preclinical testing program. Pediatr Blood Cancer. 2012;58(4):566–571. | ||
Ren W, Korchin B, Lahat G, et al. Combined vascular endothelial growth factor receptor/epidermal growth factor receptor blockade with chemotherapy for treatment of local, uterine, and metastatic soft tissue sarcoma. Clin Cancer Res. 2008;14(17):5466–5475. | ||
Daw NC, Furman WL, Stewart CF, et al; Children’s Oncology Group. Phase I and pharmacokinetic study of gefitinib in children with refractory solid tumors: a Children’s Oncology Group Study. J Clin Oncol. 2005;23(25):6172–6180. | ||
Furman WL, Navid F, Daw NC, et al. Tyrosine kinase inhibitor enhances the bioavailability of oral irinotecan in pediatric patients with refractory solid tumors. J Clin Oncol. 2009;27(27):4599–4604. | ||
Ren W, Korchin B, Zhu QS, et al. Epidermal growth factor receptor blockade in combination with conventional chemotherapy inhibits soft tissue sarcoma cell growth in vitro and in vivo. Clin Cancer Res. 2008;14(9):2785–2795. | ||
Jakacki RI, Hamilton M, Gilbertson RJ, et al. Pediatric phase I and pharmacokinetic study of erlotinib followed by the combination of erlotinib and temozolomide: a Children’s Oncology Group Phase I Consortium Study. J Clin Oncol. 2008;26(30):4921–4927. | ||
Kim A, Widemann BC, Krailo M, et al. Phase 2 trial of sorafenib in children and young adults with refractory solid tumors: a report from the Children’s Oncology Group. Pediatr Blood Cancer. 2015;62(9):1562–1566. | ||
Agulnik M, Costa RL, Milhem M. A phase II study of tivozanib in patients with metastatic and non-resectable soft tissue sarcomas. J Clin Oncol. 2015;33(suppl abstr):10515. | ||
Voss SD, Glade-Bender J, Spunt SL, et al. Growth plate abnormalities in pediatric cancer patients undergoing phase 1 anti-angiogenic therapy: a report from the Children’s Oncology Group Phase I Consortium. Pediatr Blood Cancer. 2015;62(1):45–51. | ||
Bagatell R, Norris R, Ingle AM, et al. Phase 1 trial of temsirolimus in combination with irinotecan and temozolomide in children, adolescents and young adults with relapsed or refractory solid tumors: a Children’s Oncology Group Study. Pediatr Blood Cancer. 2014;61(5):833–839. | ||
Thornton KA, Chen AR, Trucco MM, et al. A dose-finding study of temsirolimus and liposomal doxorubicin for patients with recurrent and refractory bone and soft tissue sarcoma. Int J Cancer. 2013;133(4):997–1005. | ||
Schuetze SM, Zhao L, Chugh R, et al. Results of a phase II study of sirolimus and cyclophosphamide in patients with advanced sarcoma. Eur J Cancer. 2012;48(9):1347–1353. | ||
Bryce AH, Rao R, Sarkaria J, et al. Phase I study of temsirolimus in combination with EKB-569 in patients with advanced solid tumors. Invest New Drugs. 2012;30(5):1934–1941. | ||
Schwartz GK, Tap WD, Qin LX, et al. Cixutumumab and temsirolimus for patients with bone and soft-tissue sarcoma: a multicentre, open-label, phase 2 trial. Lancet Oncol. 2013;14(4):371–382. | ||
Wagner LM, Fouladi M, Ahmed A, et al. Phase II study of cixutumumab in combination with temsirolimus in pediatric patients and young adults with recurrent or refractory sarcoma: a report from the Children’s Oncology Group. Pediatr Blood Cancer. 2015;62(3):440–444. | ||
Quek R, Wang Q, Morgan JA, et al. Combination mTOR and IGF-1R inhibition: phase I trial of everolimus and figitumumab in patients with advanced sarcomas and other solid tumors. Clin Cancer Res. 2011;17(4):871–879. | ||
Yoo C, Lee J, Rha SY, et al. Multicenter phase II study of everolimus in patients with metastatic or recurrent bone and soft-tissue sarcomas after failure of anthracycline and ifosfamide. Invest New Drugs. 2013;31(6):1602–1608. | ||
Ho AL, Vasudeva SD, Lae M, et al. PDGF receptor alpha is an alternative mediator of rapamycin-induced Akt activation: implications for combination targeted therapy of synovial sarcoma. Cancer Res. 2012;72(17):4515–4525. | ||
Keohan ML, Tap WD, Dickson MA, et al. A phase Ib/II study of imatinab and everolimus in patients with PDGFRA+ synovial sarcoma. J Clin Oncol. 2013;31(suppl abstract):10558. | ||
Katz D, Azraq Y, Eleyan F, Gill S, Peretz T, Merimsky O. Pazolimus: pazopanib plus sirolimus following progression on pazopanib, a retrospective case series analysis. BMC Cancer. 2016;16:616. | ||
Chawla SP, Staddon AP, Baker LH, et al. Phase II study of the mammalian target of rapamycin inhibitor ridaforolimus in patients with advanced bone and soft tissue sarcomas. J Clin Oncol. 2012;30(1):78–84. | ||
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546–1558. | ||
Bennani-Baiti IM. Epigenetic and epigenomic mechanisms shape sarcoma and other mesenchymal tumor pathogenesis. Epigenomics. 2011;3(6):715–732. | ||
Chu QS, Nielsen TO, Alcindor T, et al. A phase II study of SB939, a novel pan-histone deacetylase inhibitor, in patients with translocation-associated recurrent/metastatic sarcomas-NCIC-CTG IND 200 dagger. Ann Oncol. 2015;26(5):973–981. | ||
Zorzi AP, Bernstein M, Samson Y, et al. A phase I study of histone deacetylase inhibitor, pracinostat (SB939), in pediatric patients with refractory solid tumors: IND203 a trial of the NCIC IND program/C17 pediatric phase I consortium. Pediatr Blood Cancer. 2013;60(11):1868–1874. | ||
Choy E, Flamand Y, Balasubramanian S, et al. Phase 1 study of oral abexinostat, a histone deacetylase inhibitor, in combination with doxorubicin in patients with metastatic sarcoma. Cancer. 2015;121(8):1223–1230. | ||
Schelman WR, Traynor AM, Holen KD, et al. A phase I study of vorinostat in combination with bortezomib in patients with advanced malignancies. Invest New Drugs. 2013;31(6):1539–1546. | ||
Cassier PA, Lefranc A, Amela EY, et al. A phase II trial of panobinostat in patients with advanced pretreated soft tissue sarcoma. A study from the French Sarcoma Group. Br J Cancer. 2013;109(4):909–914. | ||
Witt O, Milde T, Deubzer HE, et al. Phase I/II intra-patient dose escalation study of vorinostat in children with relapsed solid tumor, lymphoma or leukemia. Klin Padiatr. 2012;224(6):398–403. | ||
Lopez G, Song Y, Lam R, et al. HDAC inhibition for the treatment of epithelioid sarcoma: novel cross talk between epigenetic components. Mol Cancer Res. 2016;14(1):35–43. | ||
Lopez G, Bill KL, Bid HK, et al. HDAC8, a potential therapeutic target for the treatment of malignant peripheral nerve sheath tumors (MPNST). PLoS One. 2015;10(7):e0133302. | ||
Nathan DF, Vos MH, Lindquist S. In vivo functions of the Saccharomyces cerevisiae Hsp90 chaperone. Proc Natl Acad Sci U S A. 1997;94(24):12949–12956. | ||
Ellis RJ. Molecular chaperones: assisting assembly in addition to folding. Trends Biochem Sci. 2006;31(7):395–401. | ||
Workman P, Burrows F, Neckers L, Rosen N. Drugging the cancer chaperone HSP90: combinatorial therapeutic exploitation of oncogene addiction and tumor stress. Ann N Y Acad Sci. 2007;1113:202–216. | ||
Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5(10):761–772. | ||
Neckers L, Workman P. Hsp90 molecular chaperone inhibitors: are we there yet? Clin Cancer Res. 2012;18(1):64–76. | ||
Wagner AJ, Chugh R, Rosen LS, et al. A phase I study of the HSP90 inhibitor retaspimycin hydrochloride (IPI-504) in patients with gastrointestinal stromal tumors or soft-tissue sarcomas. Clin Cancer Res. 2013;19(21):6020–6029. | ||
Demetri GD, Le Cesne A, Von Mehren M, et al. Final results from a phase III study of IPI-504 (retaspimycin hydrochloride) versus placebo in patients (pts) with gastrointestinal stromal tumors (GIST) following failure of kinase inhibitor therapies. Paper presented at: 2010 Gastrointestinal Cancers Symposium 2010. | ||
Bagatell R, Gore L, Egorin MJ, et al. Phase I pharmacokinetic and pharmacodynamic study of 17-N-allylamino-17-demethoxygeldanamycin in pediatric patients with recurrent or refractory solid tumors: a pediatric oncology experimental therapeutics investigators consortium study. Clin Cancer Res. 2007;13(6):1783–1788. | ||
Mahipal A, Malafa M. Importins and exportins as therapeutic targets in cancer. Pharmacol Ther. 2016;164:135–143. | ||
Nakayama R, Zhang YX, Czaplinski JT, et al. Preclinical activity of selinexor, an inhibitor of XPO1, in sarcoma. Oncotarget. 2016;7(13):16581–16592. | ||
Gounder MM, Zer A, Tap WD, et al. Phase IB study of selinexor, a first-in-class inhibitor of nuclear export, in patients with advanced refractory bone or soft tissue sarcoma. J Clin Oncol. 2016;34(26):3166–3174. | ||
Alexander TB, Lacayo NJ, Choi JK, Ribeiro RC, Pui CH, Rubnitz JE. Phase I study of selinexor, a selective inhibitor of nuclear export, in combination with fludarabine and cytarabine, in pediatric relapsed or refractory acute leukemia. J Clin Oncol. 2016;34(34):4094–4101. | ||
Rosenberg SA, Lotze MT, Yang JC, et al. Experience with the use of high-dose interleukin-2 in the treatment of 652 cancer patients. Ann Surg. 1989;210(4):474–484. | ||
Weigel B, Malempati S, Reid JM, et al. Phase 2 trial of cixutumumab in children, adolescents, and young adults with refractory solid tumors: a report from the Children’s Oncology Group. Pediatr Blood Cancer. 2014;61(3):452–456. | ||
Kolb EA, Gorlick R, Maris JM, et al. Combination testing (Stage 2) of the Anti-IGF-1 receptor antibody IMC-A12 with rapamycin by the pediatric preclinical testing program. Pediatr Blood Cancer. 2012;58(5):729–735. | ||
Shin DH, Min HY, El-Naggar AK, Lippman SM, Glisson B, Lee HY. Akt/mTOR counteract the antitumor activities of cixutumumab, an anti-insulin-like growth factor I receptor monoclonal antibody. Mol Cancer Ther. 2011;10(12):2437–2448. | ||
Kurzrock R, Patnaik A, Aisner J, et al. A phase I study of weekly R1507, a human monoclonal antibody insulin-like growth factor-I receptor antagonist, in patients with advanced solid tumors. Clin Cancer Res. 2010;16(8):2458–2465. | ||
Macaulay VM, Middleton MR, Protheroe AS, et al. Phase I study of humanized monoclonal antibody AVE1642 directed against the type 1 insulin-like growth factor receptor (IGF-1R), administered in combination with anticancer therapies to patients with advanced solid tumors. Ann Oncol. 2013;24(3):784–791. | ||
Reidy-Lagunes DL, Vakiani E, Segal MF, et al. A phase 2 study of the insulin-like growth factor-1 receptor inhibitor MK-0646 in patients with metastatic, well-differentiated neuroendocrine tumors. Cancer. 2012;118(19):4795–4800. | ||
Tabernero J, Chawla SP, Kindler H, et al. Anticancer activity of the type I insulin-like growth factor receptor antagonist, ganitumab, in combination with the death receptor 5 agonist, conatumumab. Target Oncol. 2015;10(1):65–76. | ||
Okada K, Yamasaki K, Tanaka C, Fujisaki H, Osugi Y, Hara J. Phase I study of bevacizumab plus irinotecan in pediatric patients with recurrent/refractory solid tumors. Jpn J Clin Oncol. 2013;43(11):1073–1079. | ||
Wagner L, Turpin B, Nagarajan R, Weiss B, Cripe T, Geller J. Pilot study of vincristine, oral irinotecan, and temozolomide (VOIT regimen) combined with bevacizumab in pediatric patients with recurrent solid tumors or brain tumors. Pediatr Blood Cancer. 2013;60(9):1447–1451. | ||
Olsen RJ, Lydiatt WM, Koepsell SA, et al. C-erb-B2 (HER2/neu) expression in synovial sarcoma of the head and neck. Head Neck. 2005;27(10):883–892. | ||
Ebb D, Meyers P, Grier H, et al. Phase II trial of trastuzumab in combination with cytotoxic chemotherapy for treatment of metastatic osteosarcoma with human epidermal growth factor receptor 2 overexpression: a report from the children’s oncology group. J Clin Oncol. 2012;30(20):2545–2551. | ||
Tap WD, Jones RL, Van Tine BA, et al. Olaratumab and doxorubicin versus doxorubicin alone for treatment of soft-tissue sarcoma: an open-label phase 1b and randomised phase 2 trial. Lancet. 2016;388(10043):488–497. | ||
Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. | ||
Maki RG, Jungbluth AA, Gnjatic S, et al. A Pilot Study of anti-CTLA4 antibody ipilimumab in patients with synovial sarcoma. Sarcoma. 2013;2013:168145. | ||
D’Angelo SP, Shoushtari AN, Agaram NP, et al. Prevalence of tumor-infiltrating lymphocytes and PD-L1 expression in the soft tissue sarcoma microenvironment. Hum Pathol. 2015;46(3):357–365. | ||
Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369(2):134–144. | ||
Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. | ||
Topalian SL, Sznol M, McDermott DF, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32(10):1020–1030. | ||
Motzer RJ, Escudier B, McDermott DF, et al; CheckMate 025 Investigators. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373(19):1803–1813. | ||
Borghaei H, Paz-Ares L, Horn L, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015;373(17):1627–1639. | ||
Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373(1):23–34. | ||
Chan TA, Wolchok JD, Snyder A. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2015;373(20):1984. | ||
Sioud M, Nyakas M, Saeboe-Larssen S, Mobergslien A, Aamdal S, Kvalheim G. Diversification of antitumour immunity in a patient with metastatic melanoma treated with ipilimumab and an IDO-silenced dendritic cell vaccine. Case Rep Med. 2016;2016:9639585. | ||
Krishnadas DK, Shapiro T, Lucas K. Complete remission following decitabine/dendritic cell vaccine for relapsed neuroblastoma. Pediatrics. 2013;131(1):e336–e341. | ||
Sharma A, Bode B, Studer G, et al. Radiotherapy of human sarcoma promotes an intratumoral immune effector signature. Clin Cancer Res. 2013;19(17):4843–4853. | ||
Lai JP, Rosenberg AZ, Miettinen MM, Lee CC. NY-ESO-1 expression in sarcomas: a diagnostic marker and immunotherapy target. Oncoimmunology. 2012;1(8):1409–1410. | ||
Lai JP, Robbins PF, Raffeld M, et al. NY-ESO-1 expression in synovial sarcoma and other mesenchymal tumors: significance for NY-ESO-1-based targeted therapy and differential diagnosis. Mod Pathol. 2012;25(6):854–858. | ||
Kawaguchi S, Wada T, Ida K, et al. Phase I vaccination trial of SYT-SSX junction peptide in patients with disseminated synovial sarcoma. J Transl Med. 2005;3(1):1. | ||
Kawaguchi S, Tsukahara T, Ida K, et al. SYT-SSX breakpoint peptide vaccines in patients with synovial sarcoma: a study from the Japanese Musculoskeletal Oncology Group. Cancer Sci. 2012;103(9):1625–1630. | ||
Chang HR, Cordon-Cardo C, Houghton AN, Cheung NK, Brennan MF. Expression of disialogangliosides GD2 and GD3 on human soft tissue sarcomas. Cancer. 1992;70(3):633–638. | ||
Chapman PB, Morrisey D, Panageas KS, et al. Vaccination with a bivalent G(M2) and G(D2) ganglioside conjugate vaccine: a trial comparing doses of G(D2)-keyhole limpet hemocyanin. Clin Cancer Res. 2000;6(12):4658–4662. | ||
Carvajal RD, Agulnik M, Ryan CW et al. Trivalent ganglioside vaccine and immunologic adjuvant versus adjuvant alone in metastatic sarcoma patients rendered disease-free by surgery: a randomized phase 2 trial. Paper presented at: 2014 ASCO Annual Meeting 2014; Chicago, IL. | ||
Krishnadas DK, Shusterman S, Bai F, et al. A phase I trial combining decitabine/dendritic cell vaccine targeting MAGE-A1, MAGE-A3 and NY-ESO-1 for children with relapsed or therapy-refractory neuroblastoma and sarcoma. Cancer Immunol Immunother. 2015;64(10):1251–1260. | ||
Robbins PF, Morgan RA, Feldman SA, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29(7):917–924. | ||
Spear TT, Nagato K, Nishimura MI. Strategies to genetically engineer T cells for cancer immunotherapy. Cancer Immunol Immunother. 2016;65(6):631–649. | ||
Simon-Keller K, Paschen A, Eichmuller S, et al. [Adoptive T-cell therapy of rhabdomyosarcoma]. Pathologe. 2010;31(suppl 2):215–220. German. | ||
Simon-Keller K, Paschen A, Hombach AA, et al. Survivin blockade sensitizes rhabdomyosarcoma cells for lysis by fetal acetylcholine receptor-redirected T cells. Am J Pathol. 2013;182(6):2121–2131. | ||
Gattenlohner S, Marx A, Markfort B, et al. Rhabdomyosarcoma lysis by T cells expressing a human autoantibody-based chimeric receptor targeting the fetal acetylcholine receptor. Cancer Res. 2006;66(1):24–28. | ||
Lehner M, Gotz G, Proff J, et al. Redirecting T cells to Ewing’s sarcoma family of tumors by a chimeric NKG2D receptor expressed by lentiviral transduction or mRNA transfection. PLoS One. 2012;7(2):e31210. | ||
Rainusso N, Brawley VS, Ghazi A, et al. Immunotherapy targeting HER2 with genetically modified T cells eliminates tumor-initiating cells in osteosarcoma. Cancer Gene Ther. 2012;19(3):212–217. | ||
Ahmed N, Brawley VS, Hegde M, et al. Human epidermal growth factor receptor 2 (HER2) -specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol. 2015;33(15):1688–1696. | ||
Beavis PA, Slaney CY, Kershaw MH, Gyorki D, Neeson PJ, Darcy PK. Reprogramming the tumor microenvironment to enhance adoptive cellular therapy. Semin Immunol. 2016;28(1):64–72. | ||
Mirzaei HR, Mirzaei H, Yun Lee S, Hadjati J, Till BG. Prospects for chimeric antigen receptor (CAR) gammadelta T cells: a potential game changer for adoptive T cell cancer immunotherapy. Cancer Lett. 2016;380(2):413–423. | ||
Huang X, Yang Y. Driving an improved CAR for cancer immunotherapy. J Clin Invest. 2016;126(8):2795–2798. | ||
Cherkassky L, Morello A, Villena-Vargas J, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. 2016;126(8):3130–3144. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.