Back to Journals » International Journal of Nanomedicine » Volume 18
Targeted siRNA Delivery by Bioinspired Cancer Cell Membrane-Coated Nanoparticles with Enhanced Anti-Cancer Immunity
Authors Li J ![]() , Zhang J, Gao Y
, Zhang J, Gao Y ![]() , Lei S
, Lei S ![]() , Wu J, Chen X
, Wu J, Chen X ![]() , Wang K
, Wang K ![]() , Duan X, Men K
, Duan X, Men K ![]()
Received 5 July 2023
Accepted for publication 7 October 2023
Published 24 October 2023 Volume 2023:18 Pages 5961—5982
DOI https://doi.org/10.2147/IJN.S429036
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Yan Shen
Jingmei Li,1,* Jin Zhang,1,* Yan Gao,1 Sibei Lei,1 Jieping Wu,1 Xiaohua Chen,1 Kaiyu Wang,1 Xingmei Duan,2 Ke Men1
1Department of Biotherapy, Cancer Center and State Key Laboratory of Biotherapy, West China Hospital, Sichuan University, Chengdu, 610041, People’s Republic of China; 2Department of Pharmacy, Personalized Drug Therapy Key Laboratory of Sichuan Province Sichuan Academy of Medical Sciences & Sichuan Provincial People’s Hospital, School of Medicine, University of Electronic Science and Technology of China, Chengdu, 610072, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Ke Men, State Key Laboratory of Biotherapy and Cancer Center, National Clinical Research Center for Geriatrics, West China Hospital of Sichuan University, Chengdu, 610041, People’s Republic of China, Email [email protected] Xingmei Duan, Department of Pharmacy, Personalized Drug Therapy Key Laboratory of Sichuan Province, Sichuan Academy of Medical Sciences & Sichuan Provincial People’s Hospital, School of Medicine, University of Electronic Science and Technology of China, Chengdu, 610072, People’s Republic of China, Email [email protected]
Introduction: Cell-membrane nanocarriers are usually constructed by modifying the nanoparticle surface with cell membrane extracts, which has a direct benefit in endowing targeting capacity to nanocarriers based on their original cell types. However, delivering nucleic acid cargos by cell membrane–based nanoparticles is difficult owing to the strong negative charge of the cell membrane fraction. In this study, we developed a cancer cell membrane–based drug delivery system, the cMDS, for efficient siRNA delivery. Meanwhile, the cancer-specific immune response stimulated by the gene vector itself could offer synergistic anti-cancer ability.
Methods: The cMDS was prepared by ultrasound, and its transfection efficiency and anti-cancer ability were examined using cultures of CT26 cells. MTT and red blood cell hemolysis tests were performed to assess the safety of cMDS, while its targeted gene delivery and strong immune stimulation were investigated in a subcutaneous tumor model. Moreover, the detailed anti-cancer immune stimulation mechanisms of cMDS are uncovered by protein chip analysis.
Results: The cMDS was spherical core-shell structure. It showed high transfection efficiency and anti-cancer ability in vitro. In animal experiments, intravenously administered cMDS/siStat3 complex efficiently suppress the growth of colon cancer. Moreover, the result of protein chip analysis suggested that cMDS affect the migration and chemotaxis of immune cells.
Conclusion: The cMDS shows obvious tumor tissue-specific accumulation properties and strong immune stimulation ability. It is an advanced targeted gene delivery system with potent immunotherapeutic properties.
Keywords: bioinspired material, cancer cell membrane, targeted siRNA delivery, immunotherapeutic, colon cancer
Introduction
Bioinspired materials are advanced materials that are produced to provide biological functions similar to natural materials or body tissues, and replicate one or more attributes of a material produced by a living organism.1 Recently, bioinspired materials have attracted increasing attention owing to their potential applications in clinical therapy2 and tissue regeneration.3,4 For drug delivery, a bioinspired drug delivery system (BDDS) has been developed with the aim to achieve better delivery efficiency and biocompatibility.5 Current existing strategies include modifying the carrier with natural components6,7 or directly using cell vesicles as the carriers,8,9 thus making the delivery system less visible and heterogeneous in vivo. Several bioinspired drug delivery systems have made ideal progress in clinical applications.10
Recently, many new strategies have been developed to further expand the potential of bioinspired drug carriers, among which cell membrane–based nanocarriers have emerged as a novel drug delivery system with biofunction potential. The cell membrane comprises a lipid bilayer that is rich in cholesterol and glycoproteins, which acts as a border to protect the cytoplasm contents and function of intracellular components.11 The cell membrane also possesses important functions, including cell conjunction, uptake, and transmembrane signal transduction. Furthermore, even following isolation, cell membrane substrates still retain partial biological properties and identities; these remnants have a flexible lipid bilayer structure and are considered ideal resources for building bioinspired drug delivery systems. Cell-membrane nanocarriers are usually constructed by modifying the nanoparticle surface with cell membrane extracts, which has a direct benefit in endowing targeting capacity to nanocarriers based on their original cell types. For example, most immune membrane–based nanoparticles exhibit tissue wound site targeting properties.14 Meanwhile, cell membrane fractions reduce the in vivo clearance of nanocarriers, thereby improving biocompatibility and possibly increasing the safety of nanoformulation by endowing cell-like surface properties.15–17 However, delivering nucleic acid cargos by cell membrane–based nanoparticles is difficult owing to the strong negative charge of the cell membrane fraction, which contain high levels of phosphate. Additionally, negatively charged cell membrane fractions may change the delivery ability and internalization behavior of cationic nanocarriers. Therefore, although cell membrane–coated nanocarriers have great application potential, they are difficult to apply to the field of gene therapy. Moreover, there is another point to be considered when developing cell membrane delivery systems that has so far been rarely investigated: although the biological identity retained on the cell membrane extract can improve biocompatibility, it also provides biomarkers with immune properties, which may cause the carriers to induce antigenic effects.18,19 Although most cellular components (such as proteins) of their own origin are usually not recognized as foreign substances by the immune system, the situation of tumor cells is different. Tumor cells overexpress biomarkers and neoantigens during their development and treatment, which increases their potency in immune stimulation.11 In previous reports, mesenchymal stem cells, endothelial cells, and cancer cells-derived bioinspired materials have been used to deliver siRNA. But few reports have highlighted that the immune stimulation of the vector skeleton itself may play an auxiliary role in tumor immunotherapy, which provides an additional therapeutic function to the whole formulation and thus expands its application as a tumor vaccine.20,21 This is particularly important for tumor-targeted therapy if tumor immunotherapy is performed in parallel.
Hyperactivation of the transcription factor Stat3 is found in most tumors, and Stat3 can play a key role in the occurrence and development of tumors through various pathways. Previous studies have shown that hyperactivation of Stat3 was found in CT26 cells and silencing Stat3 in CT26 cells can cause strong anti-tumor effects. RNAi-based gene therapy is an effective method for cancer treatment.22,23 Cell membrane–coated nanocarriers with tumor targeting properties can safely deliver therapeutic siRNA to tumor tissues. Meanwhile, the cancer-specific immune response stimulated by the gene vector itself could offer synergistic anti-cancer ability, suggesting a novel application for bioinspired materials. However, to realize this, the gene delivery function of cell membrane–based nanoparticles should first be thoroughly investigated. In this study, we developed a cancer cell membrane–based drug delivery system, the cMDS, for efficient siRNA delivery (Scheme 1). Because the surface of the cMDS is coated with cancer cell membrane fractions, we attempted to combine this novel colon cancer-targeted gene therapy with anti-tumor immunotherapy and evaluated its therapeutic potential in detail.
 |
Scheme 1 Schematic illustration of cMDS for tumor prevention. (a) Preparation of cMDS. (b) Anti-tumor immunotherapy of cMDS. (c) Cancer-targeted gene therapy of cMDS. |
Materials and Methods
Materials
The 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and branched polyethyleneimine (PEI25K) were purchased from Sigma-Aldrich (US); RNase A and GoldView II nuclear staining dye were purchased from Solarbio; Dil (10 mg/mL) and LysoTracker red were purchased from Beyotime; DOTAP and DSPE-PEG-cy5.5 were purchased from xi’an ruixi Biological Technology Co., Ltd. (xi’an, China); and all other chemicals were purchased from Sigma-Aldrich unless otherwise mentioned. The Mus musculus colon carcinoma cell line, CT26; the human embryonic kidney cell line, HEK 293T; and the male mouse macrophage line, RAW264.7, were all purchased from the American Type Culture Collection (ATCC) and cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS; Gibco, US) at 37°C under a humidified atmosphere of 5% CO2. BABL/c mice were obtained from Beijing HFK Biotechnology Co., Ltd. (Beijing, China) and maintained under specific pathogen-free conditions. All mouse experiments were conducted under the guidance of the Animal Ethics Committee of the General Administration of Health Research of Sichuan University and the Guidelines and Standard Operating Procedure (SOP) for Laboratory Animals of Sichuan University. The primers (Stat3: forward 5′-TTC TCG TCC ACC AAG, reverse 3′-GAT ATT GTC TAG CCA GAC CC and β-actin: forward 5′-CCC AGG CAT TGC TGA CAG G, reverse 5′-TGG AAG GTG GAC AGT GAG GC) were synthesized and purified by TSINGKE Biological Technology (Chengdu, China). The siRNAs against Stat3 (sense: 5′-UUAGCCCAUGUGAUCUGACACCCUGAA, antisense: 5′ CAGGGUGUCAGAUCACAUGGGCUAA) and GFP (sense: 5′-AACUUCAGGGUCAGCUUGCdTdT, antisense: 5′ GCAAGCUGACCCUGAAGUUdTdT) were synthesized by Shanghai GenePharma Co., Ltd. (China).
Nanoparticle Preparation
To prepare the cationic lipid nanoparticle (CLP), DOTAP, 1.2-dioleoyl-3-trimethylammonium propane and cholesterol (13:7, w/w) were dissolved in chloroform. After rotary evaporation for 45 min, the film was rehydrated with a 5% glucose solution. The resulting solution was redispersed by ultrasound to form a CLP solution and stored at 4°C. To prepare CLP-Cy5.5, DOTAP, cholesterol, and DSPE-PEG-Cy5.5 (13:7:0.5, W/W) were dissolved in chloroform. After rotary evaporation for 45 min, the film was rehydrated with a 5% glucose solution.
To prepare the cancer cell membrane fractions (CM), CT26 cells were cultured in DMEM with 10% FBS, before being collected and washed three times with phosphate buffered saline (PBS). Next, a hypotonic lysis buffer (10 mM Tris-HCl and 2 mM MgCl2 at pH 7.5) was added and the cells were homogenized using a Dounce homogenizer. The suspension was centrifuged at 3200 × g for 5 min at 4°C to collect the supernatant, and the pellet was re-homogenized, centrifuged again, and the supernatant containing the crude cell membrane extract was collected. After centrifuging the supernatant at 100,000 × g for 1 h, the precipitation was suspended in washing solution (10 mM Tris-HCl and 1 mM EDTA at pH 7.5) and centrifuged again at 100,000 × g for 1 h. Finally, the precipitate was resuspended by PBS and the CM was obtained by extruding the solution through a 400 nm polycarbonate membrane (LiposoFast, Avestin, Canada), and the protein concentration was detected.
To obtain the cancer membrane delivery system (cMDS), the CM and CLP were mixed (1:3, W/W) and incubated for 5 min. Then, the mixture was sonicated in a water bath type sonicator (MCS-2, Japan) at a frequency of 40 kHz for 3 min to form the cMDS nanoparticles.
Size and Zeta-Potential
We measured the size and zeta potential of the CLP, CM, and cMDS by dynamic light scattering (Nano ZS, Malvern), with a detection temperature of 25°C and a balancing time of 2 minutes.
Transmission Electron Microscopy (TEM)
The morphologies of the CLP and cMDS nanoparticles were detected and photographed by a transmission electron microscope (Tecnai G2 F20 S-Twin, FEI, USA).
Cytotoxicity Assays
To detect the cytotoxicity of CLP, HEK 293T cells were seeded in 96-well plates with DMEM (add 10% FBS) at a density of 5×103 cells/well. After 24 h, cells were incubated with CLP at the concentration gradient of 5, 10, 15, 20, 30, 40, 60, or 80 μg/mL, with PEI25K as the control. After incubation for 48 h in an incubator (37°C, 5% CO2), the reaction was terminated by adding 20 μL MTT (5 mg/mL) solution to each well. After incubation at 37°C for 4 h, the medium was replaced with 150 μL DMSO and shaken for 15 min at room temperature, before measuring the absorbance at 570 nm with a Spectramax M5 Microtiter Plate Luminometer (Molecular Devices, USA).
Biocompatibility of the cMDS
The blood of untreated female BABL/c mice was collected in a collection tube and centrifuged at 1500 rpm for 10 min to obtain the erythrocytes at the bottom of the tube. Subsequently, 0.9% NaCl solution was added, and the supernatant was discarded after centrifugation at 800 × g for 10 minutes. The erythrocytes were resuspended with 0.9% NaCl solution and a 2% red blood cell suspension was prepared. The 2% red blood cell suspension was treated with CLP and cMDS (final concentrations of 6, 12, 24, 48, or 60 μg/mL), with 0.9% normal saline as the negative control and distilled water as the positive control, and incubated at 37°C for 30 minutes. Following incubation, the suspension was collected and centrifuged at 1700 × g for 10 min and the absorbance of the supernatant was measured by the Spectramax M5 Microtiter Plate Luminometer at 545 nm, with the hemolysis rate calculated using the following formula: Hemolysis rate = (sample-negative control)/(positive control-negative control) × 100%.
Gel Retarding Assay
siRNA and CLP were mixed at ratios of 1:2, 1:4, and 1:6 (w/w). After incubation for 15 minutes at room temperature, electrophoresis was performed on a 1% (w/v) agarose gel at 120 V for 15 minutes, and then stained with 10,000× GoldViewII nuclear staining dyes. The bands on the gel were observed by an E-gel imager (Bio-Rad, ChemiDox XRS, USA).
Encapsulation Efficiency and Loading Content
After the preparation of the CLP/siRNA complex, free siRNA was quantified by Ribogreen (Invitrogen), and the total siRNA was similarly measured by Ribogreen after adding 5% Triton-X 100. The encapsulation efficiency and loading content were calculated using the following formulas:
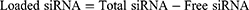
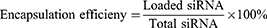
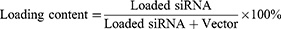
RNase Protection Assay
To study the ability of the cMDS to protect siRNA from nuclease degradation, RNase A (250 μg/mL final concentration) was added to the cMDS/siRNA complex. Free siRNA was treated with the same concentration of RNase A as a control. After 0, 2, 4, 8, and 24 h of incubation at 37°C, sodium dodecyl sulfate (SDS, 250 μg/mL final concentration) was added and incubated at 70°C for 10 minutes. The degradation was then observed by 15 minutes electrophoresis at 120 V on a 1% (w/v) agarose gel.
Vitro Intracellular Localization
First, 3×104 CT26 cells were seeded in chamber slides (Millicell, 4-well-glass) overnight before treating with the cMDS/siRNA complex (the cell membrane of cMDS was labeled with Dil). After 24 h, the nuclei were stained with DAPI and photographed with a confocal microscope (Nikon, Japan).
Endosomal Escape
To observe endosomal escape, CT26 cells were seeded in chamber slides at a density of 3×104 cells/well. The prepared CLP/siRNA complex and cMDS/siRNA complex were added to the cells (1 μg FAM-labeled siRNA) and allowed to culture at 37°C for 24 h before washing with PBS. Finally, the nuclei and lysosomes were stained with DAPI and LysoTracker red, respectively, and photographed using a confocal microscope.
In vitro Transfection
To detect the transfection efficiency of CLP and cMDS, CT26 cells were seeded into a 24-well plate, with 3×104 cells and 500 μL of medium per well (DMEM containing 10% FBS). After 24 h, the prepared cMDS/siRNA complex and CLP/siRNA complex were added to the cells (1 μg Cy3-labeled siRNA each well), and 1 μg Cy3-labeled siRNA mixed with PEI25K was used as a control. The plate was incubated at 37°C for 24 h, following which the cells were photographed using a fluorescence microscope (ZEN880, Bamboo Living) and the transfection efficiency was detected by flow cytometry (NovoCyte Flow Cytometer, ACEA Biosciences, USA).
To observe the transfection at different time points, CT26 cells were seeded in chamber slides at the same density and treated with the CLP/siRNA complex and cMDS/siRNA complex for various times (1 μg FAM-labeled siRNA each well). The cells were observed and photographed using a confocal microscope in different time points.
In vitro Interaction with Macrophages
To observe nanoparticle uptake by macrophages, RAW264.7 cells were seeded in chamber slides at a density of 1×105 cells/well, and incubated with the CLP/siRNA complex and cMDS/siRNA complex (1 μg FAM-labeled siRNA each well) at 37°C for 24 h. Following incubation, the nuclei were stained with DAPI and imaged using a fluorescence microscope. To evaluate nanoparticle uptake by macrophages, cells were collected and the transfection efficiency was detected by flow cytometry.
In vitro GFP Knockdown
The GFP-transduced CT26 cells were seeded in a 24-well plate overnight (5 × 103 cells/well), and then co-cultured with the CLP/siGFP complex and cMDS/siGFP complex for 48 h (2 μg siRNA each well). The gene knockout efficiency was measured by flow cytometry, with wild-type CT26 cells used to establish the baseline signal.
Real-Time PCR
To assess the level of Stat3 mRNA in cells after cMDS/siStat3 transfection, 48 h after transfection, total RNA was extracted from cells using the FastPure Cell/Tissue Total RNA Isolation Kit (Vazyme). RNA was used as the template, and the cDNA was synthesized by SuperScript II (Sigma-Aldrich). Subsequently, SYBR qPCR Master Mix (Vazyme, China) was used to perform real-time PCR, with β-actin used as the reference.
Western Blotting
To prove that the nanoparticles have been successfully coated with the cancer cell membrane, the protein expression of N cadherin, Sodium potassium ATPase, EPCAM, COXIV, Nucleoporin p62/NUP62, and β-actin in the cMDS nanoparticles was detected by Western blotting. Briefly, cMDS was mixed with loading buffer, while the cell lysate and cell membrane were used as controls. Proteins were separated by polyacrylamide gel electrophoresis (Beyotime), and then transferred to a polyvinylidene difluoride (PVDF) membrane (Millipore). The PVDF membrane was blocked with 5% skim milk (Servicebio Technology, Wuhan) for 90 min and incubated overnight with primary antibodies against N cadherin, Sodium potassium ATPase, EPCAM, COXIV, Nucleoporin p62/NUP62, and β-actin at 4°C. Following incubation, the PVDF membrane was washed and incubated with the secondary antibody (HRP-labeled Goat Anti-Rabbit and diluted with skim milk) for 90 min. The target protein was detected using a chemiluminescence system (Millipore, MA).
To evaluate the protein expression of Stat3, CT26 cells were transfected with the cMDS/siStat3 complex in 6-well plates. After 48 h, the cells were collected and lysed with RIPA Lysis Buffer (Beyotime, China) to extract the total protein. The protein concentration was determined by Coomassie brilliant blue G-250 (Beyotime) and the protein loading was adjusted to be consistent before mixing with loading buffer. Proteins were separated by polyacrylamide gel electrophoresis, and then transferred to a PVDF membrane. The PVDF membranes were blocked with 5% skim milk for 90 min, before incubating with primary antibodies (Stat3, β-actin) at 4°C overnight. Following incubation, the PVDF membranes were washed by PBST buffer 3 times, and then incubated with secondary antibody at room temperature for 90 minutes. The target protein was detected by a chemiluminescence system and quantitative analysis was conducted using ImageJ (National Institutes of Health, US).
Anti-Proliferation Assay in vitro
To evaluate the inhibitory effect of the CLP/siStat3 and cMDS/siStat3 complexes on the growth of tumor cells, CT26 cells were plated into 96-well plates (1 × 103 cells/well) and incubated overnight at 37°C. Subsequently, the cells were transfected with CLP, CLP/siStat3, or cMDS/siStat3, and 24 h later, the anti-proliferative effects were measured using an MTT assay.
Clonogenic Assay
To detect the cell proliferation capacity, the CLP, CLP/siStat3 complex, and cMDS/siStat3 complex were added to a 6-well plate containing 1×103 CT26 cells/well and incubated at 37°C for 7 days. Following incubation, the supernatant was discarded and the cells were washed twice with PBS. Colonies were fixed with paraformaldehyde for 15 minutes and then stained with crystal violet (0.1%) for at least 30 minutes at room temperature. The colonies were counted the inhibition rate was calculated.
In vitro Apoptosis Assay
To verify the anti-tumor mechanism of the cMDS/siStat3, CT26 cells were plated in 24-well plates (3 × 104 cells per well) and transfected with CLP, CLP/siStat3 (2 μg siRNA each well), or cMDS/siStat3 (2 μg siRNA each well) for 48 h later. Subsequently, the cells of each group were collected and centrifuged. The CT26 cells were then resuspended in Annexin V-fluorescein isothiocyanate and propidium iodide staining solution (Genechem, Shanghai), and the rate of apoptosis was detected by flow cytometry.
Uptake of the cMDS by Dendritic Cells (DCs)
To examine whether the cMDS could be taken up by DCs, cMDS was prepared by CLP and Dil-labeled cancer cell membranes, then added to DCs and incubated for 24 h. Following incubation, the nuclei were stained with DAPI and photographed using a confocal microscope.
Immune Responses to cMDS
In Vitro To verify that T cells could be indirectly activated by cMDS, bone marrow-derived dendritic cells (BMDCs) were flushed from the marrow cavities of femurs and tibias of mice and cultured in 1640 medium containing 10% FBS and GMCSF (20 ng/mL). Following a 6-day culture in incubation (37°C, 5%CO2, the BMDCs were collected, seeded in a 6-well plate (1 × 106 cells/well), and incubated with CM, CLP, and cMDS at 37°C for 24 h. Following incubation, the cells were stained with anti-CD80 (FITC) and anti-CD86 (PerCP) antibodies for 20 minutes at room temperature. Subsequently, the cells were washed twice with PBS and then subjected to flow cytometry to assess the maturation of DCs. Similarly, anti-MHC-II (FITC) and anti-CD11C (APC) antibodies were used to evaluate the expression of MHC II. Furthermore, T cells were added to the pretreated BMDCs for 24-h co-culture (1:5). Following incubation, the cells were incubated with anti-CD3 (FITC) and anti-CD4 (PE) antibodies for 20 minutes at room temperature. After washing with PBS, the fluorescence was detected by flow cytometry to assess the level of T cell activation.
To investigate whether cMDS can directly activate NK cells, CM, CLP, and cMDS were added to splenic lymphocytes. The cells were collected after 24 h and then stained with anti-CD3 (FITC) and anti-NK1.1 (PE) antibodies to assess the changes in CD3−NK1.1+ cells.
In Vivo To further assess the immune response in vivo, BABL/c mice were grouped and injected with normal saline, CLP, or cMDS. After 24 h, the spleens were isolated and triturated to obtain a single cell suspension, which was then filtered with a 70-µm cell strainer and separated with lymphocyte separation medium. Lastly, the erythrocytes were removed using red blood cell lysis buffer. After incubating the splenic lymphocytes with antibodies, the CD80+CD86+ cells, MHCII+CD11C+ cells, CD3+ CD4+ cells, and CD3−NK1.1+ cells were detected by flow cytometry. The lymph nodes were also processed as above after harvesting.
In vivo Fluorescence Imaging
To evaluate the distribution of the cMDS after intravenous administration, 1×107 CT26 cells were subcutaneously inoculated into the right side of female BABL/c mice (5-weeks-old). One week later, mice were intravenously injected with Cy5.5-labeled CLP or Cy5.5-labeled cMDS. An in vivo imaging system (IVIS) (PerkinElmer, USA) was used to image at 2 h, 4 h, 6 h, 24 h, 144 h and 264 h post administration, and 264 h after administration, the mice were euthanized and the major organs (hearts, livers, spleens, lungs, and kidneys) were imaged using the IVIS. The fluorescence images, with a region of intensity (ROI) for each organ, were analyzed using Living Image 3.2 software.
In vivo Tumor Inhibition Assay
To evaluate the therapeutic effect of the cMDS/siStat3 complex in a subcutaneous tumor model, female BABL/c mice were subcutaneously injected with 1.5×106 CT26 cells (Day 0). On Day 3, the mice were randomly divided into five groups (n=5): normal saline, CLP/siNC, cMDS/siNC, CLP/siStat3, and cMDS/siStat3 (10 μg siRNA per mouse), in which each treatment was intravenously administered every day for 12 treatments. The tumor volume was measured every 2 days, and was calculated as 0.5 × length × width2. All mice were sacrificed on day 12, and the tumor tissue and organs (heart, liver, spleen, lungs, and kidneys) were collected immediately. After the tumor was weighed, all tumors and organs were stored in 4% paraformaldehyde until further use.
Immunohistochemistry Analysis
Tumor tissues and organs obtained from in vivo tumor suppression experiments were fixed with paraformaldehyde and embedded in paraffin blocks, which were then made into 3–4-μm-thick tissue sections. Paraffin sections were dewaxed, rehydrated, and stained with Mayer’s hematoxylin and eosin (HE). To detect apoptosis, we used TUNEL kit (Promega, Madison, WI) to stain the paraffin sections. For immunohistochemistry, the antigen recovery was performed before the sections were incubating with IFN-γ, TNF-α, CD4, CD31, and Stat3 antibodies (Abcam) overnight at 4°C. Subsequently, the sections were incubated with horseradish peroxidase (HRP)-conjugated secondary antibody for 20 minutes at 37°C. The sections were observed and photographed by a microscope.
Protein Chip Technology
The tumors that were harvested from the subcutaneous tumor model were lysed with lysate including protease inhibitors according to the instructions of the Quantibody R Mouse Cytokine Antibody Array 4000 (Guangzhou RayBiotech Biotechnology Co., Ltd) to quantitatively analyze 200 cytokines in the tumor lysate. The fluorescence signal was measured by a laser scanner (InnoScan 300 Microarray Scanner, French), with an excitation wavelength of 532 nm. The experimental data were analyzed by GSM-CAA-4000 data analysis software, which including normalizing the original data, then performing significance analysis, drawing heat maps of differential genes, and performing gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses on differential genes.
Blood Test
Nine female BABL/c mice were divided into three groups: One group was intravenously injected with CLP/siRNA complex (10 μg siRNA each mouse), one group was intravenously injected with cMDS/siRNA complex (10 μg siRNA each mouse), and one group was untreated. After 24 h, the blood of the mice in all groups was collected and subjected to the following analyses: mean corpuscular volume (MVC), mean platelet volume (MPV), hemoglobin concentration (HGB), mean corpuscular hemoglobin concentration (MCHC), red blood cell (RBC) count, mean corpuscular hemoglobin (MCH), platelet distribution width (PDW), lymphocyte (Lymph), monocytes (Mon), neutrophil (Gran), red blood cell specific volume (HCT), red blood cell distribution width (RDW), platelet distributing width (PCT), white blood cell (WBC) count, and platelet (PLT) count.
Statistical Analysis
Data analysis was performed using t-test and one-way analysis of variance (ANOVA) by GraphPad Prism 8.0.2 statistical software. All data were defined as the mean ± standard deviation (SD). A p-value of < 0.05 was considered as statistically significant; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Results
Synthesis and Characterization of the cMDS
The cancer cell membrane was coated on the surface of cationic lipid nanoparticle (CLP) to form the cMDS (Figure 1a). As shown in Figure 1b and c, the size of the CLP was 60.9 ± 17.8 nm (Polymer dispersity index [PDI]: 0.572) and the zeta potential was 36.7 ± 6.1 mV. The cytotoxicity of CLP was tested on HEK 293T cells.
 |
Figure 1 Preparation and characterization of the cMDS. (a) Schematic of the preparation of the cMDS. (b) Size of the CLP, CM and cMDS. (c) Zeta potential of the CLP, CM, and cMDS. (d) Cell viability assay of CLP and PEI25k on HEK 293T cells. (e) TEM images of CLP (scale bar: 200 nm) and cMDS (scale bar: 50 nm). (f) Western blots for plasma membrane specific markers (Sodium potassium ATPase, EPCAM, and N Cadherin), mitochondrial inner membrane (COX IV), and nuclear protein markers (β-actin and Nucleoporin p62/NUP62) in the cell lysate, CM and cMDS. (g) In vitro hemolysis test of the CLP and cMDS. (h) The hemolysis rate of the CLP and cMDS. (i) Gel regarding assay of the CLP/siRNA complex. (j) The siRNA loading ability and efficiency of the cMDS. (k) RNase protection assay. *p < 0.05, ***p < 0.001, ****p < 0.0001. Abbreviations: cMDS, cancer membrane delivery system; CLP, cationic lipid nanoparticle; CM, cancer cell membrane fractions. |
The results of the MTT assay showed that the IC50 value of the gold standard transfection reagent PEI25k was < 15 μg/mL, while the IC50 value of CLP was > 80 μg/mL, indicating that CLP is safer than PEI25k (Figure 1d). To prepare cMDS, we then isolated the CM from the CT26 murine colon cancer cell line. During this process, the intracellular components of cultured CT26 cells were removed by differential centrifugation. As shown in Figure 1c, the size and zeta potential of CM was 218.9 ± 34.8 nm and −29.1 ± 5.6 mV, respectively. We then coated CLP with the cancer cell membrane by sonication. As shown in Figure 1b, the size of the cMDS was 79.24 ± 8.22 nm, which was slightly larger than that of the CLP, which may be due to the extra layer of cell membrane on the surface of the cMDS. Furthermore, compared with the zeta potential of CLP (36.7 ± 6.1 mV), the zeta potential of the cMDS decreased to −24.6 ± 3.4 mV, which was close to that of the CM, suggesting that the negatively charged cancer cell membrane fractions had coated the positively charged CLP (Figure 1c). We further investigated the morphology of the cMDS. As shown in Figure 1e, compared with the CLP, the cMDS had an additional membrane–like layer on the outer surface.
Meanwhile, the protein ingredients of the obtained CM and cMDS were analyzed by Western blot. As shown in Figure 1f, the plasma membrane–specific markers (Sodium potassium ATPase, EPCAM, and N Cadherin) and the mitochondrial inner membrane (COX IV) could be detected in both the CM and cMDS. Conversely, the typical cytoplasm and nuclear protein markers (β-actin and Nucleoporin p62/NUP62) were rarely detected in the CM and cMDS. These results implied that the CM was successfully coated onto the CLP surface.
Next, the biocompatibility of the cMDS was investigated by red blood cell hemolysis test. As shown in Figure 1g, obvious hemolysis was evident in the CLP group, but no obvious hemolysis was observed in the cMDS group. Additionally, the cell supernatants of all groups were collected and the absorbance was measured. The results showed that the hemolysis rate of the CLP group was significantly higher than that of the cMDS group, especially at concentrations of 24, 48, and 60 μg/mL, suggesting that the cMDS has acquired additional biocompatibility as a result of the CM coating (Figure 1h).
The siRNA delivery ability of the cMDS was evaluated using a siRNA retarding assay. As shown in Figure 1i, the nucleic acid bands were almost invisible when the mass ratio was 6:1 (CLP: siRNA), suggesting that siRNA was completely blocked by the CLP. The encapsulation efficiency and loading content of CLP/siRNA complex was 99.7% and 12.5%, respectively, and as a result, this mass ratio was selected for the follow-up study. The CLP/siRNA complex was then coated with the cancer cell membrane to form the cMDS/siRNA complex. As shown in Figure 1j, siRNA bands with the same brightness could be observed in the both the CLP/siRNA and cMDS/siRNA groups after extraction, suggesting that the cancer cell membranes did not affect the siRNA loading ability and efficiency of the CLP. We then studied ability of the cMDS to protect siRNA against RNase. As shown in Figure 1k, the cMDS could effectively protect siRNA from RNase degradation for up to 24 h, while naked siRNA was completely degraded within 15 min.
In vitro Transfection Study
To study the ability of the cMDS to deliver nucleic acids, CT26 cells were incubated with the cMDS/siRNA complexes (FAM-siRNA and Dil-labeled CM). As shown in Figure 2a, the fluorescence signals of FAM (green) were observed in the cytoplasm of CT26 cells, suggesting that siRNA could be delivered into tumor cells by the cMDS. Meanwhile, the presence of Dil (red) signal also indicated the successful coating of the CM and the internalization of the cMDS by CT26 cancer cells. Due to the complexity of the cMDS/siRNA complex composition, we then evaluated whether it could escape from lysosomes after cellular uptake. As shown in Figure 2b, FAM-labeled siRNA (green) and lysosomes (red) had no obvious co-localization, suggesting that the cMDS/siRNA complex could efficiently escape from lysosomes. Subsequently, the siRNA transfection efficiency of the cMDS/siRNA complex was studied on CT26 cells. As shown in Figure 2c, the siRNA signal in the cMDS group increased compared with that in the CLP group at the timepoint of 5 min, 30 min, 1 h, 2h, and 8h post transfection. Meanwhile, as shown in Figure 2d, 24 h post transfection, the efficiency of the cMDS was 99.55% ± 0.12%, which was slightly higher than that of CLP (97.93% ± 0.08%) (p < 0.001). These results suggest that the siRNA delivery ability of the CLP backbone was fully retained by the cMDS. Moreover, the coating of CLP with CM could slightly enhance the delivery ability in vitro. As nanoparticle/gene complexes are considered to be cleared by macrophages when being delivered in vivo, we then studied whether this clearance effect could be reduced by CM coating. As shown in Figure 2e, after incubating the cMDS/siRNA complex with RAW264.7 cells, fewer FAM-labeled siRNA (in green) was detected in the plasma of RAW264.7 cells. Moreover, the uptake rate of the cMDS/siRNA complex was 64.98% ± 2.6%, which was lower than that of the CLP group (70.85% ± 1.35%) (p < 0.05), suggesting that coating CM on the surface of the CLP/siRNA complex could improve the biocompatibility and avoid macrophage clearance to a certain extent. To confirm that the gene silencing function of the cMDS-delivered siRNA could be fully retained, we then verified the interference ability of GFP-against siRNA on GFP-transduced CT26 cells. As shown in Figure 2f, both the CLP/siGFP complex and cMDS/siGFP complex significantly inhibited the expression of GFP protein in GFP-transduced CT26 cells. The silencing efficiency of the CLP/siGFP complex and cMDS/siGFP showed no significant difference. Taken together, these results suggest that the cMDS can efficiently deliver siRNA into CT26 cells and that the delivery ability of CLP could be fully retained after CM coating.
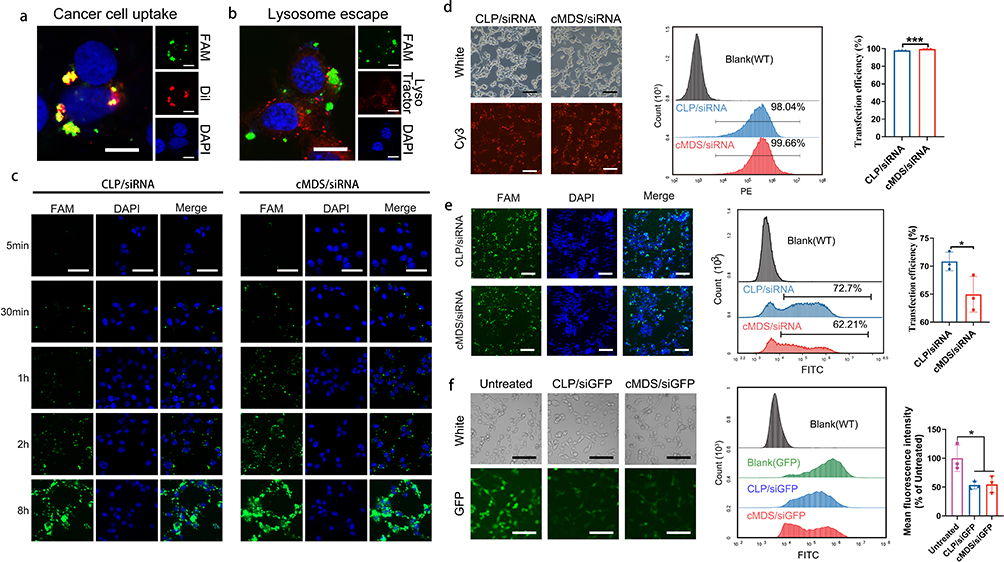 |
Figure 2 Uptake of cMDS/siStat3 by cells in vitro. (a) In vitro intracellular localization (green: FAM-siRNA; red: Dil-labeled membrane; blue: nucleus; scale bar: 10 μm). (b) cMDS/siRNA complexes escape from the lysosome (green: FAM-siRNA; red: lysosome; blue: nucleus; scale bar: 10 μm). (c) Fluorescence images of CT26 cells after incubation with CLP/siRNA and cMDS/siRNA complexes for 5 min, 30 min, 1 h, 2 h, and 8 h (green: FAM-siRNA; blue: nucleus; scale bar: 50 μm). (d) Efficiency of CLP and cMDS to deliver siRNA into CT26 cells (red: Cy3-siRNA; scale bar: 100 μm). (e) Uptake rate of CLP/siRNA and cMDS/siRNA complexes in macrophages (green: FAM-siRNA; blue: nucleus; scale bar: 100 μm). (f) Gene knockdown in GFP-transduced CT26 cells after incubation with CLP/siRNA and cMDS/siRNA complexes (green: GFP; scale bar: 100 μm). *p < 0.05, ***p < 0.001. |
cMDS/siStat3 Inhibits Tumor Growth in vitro
Stat3-targeting siRNA was next selected to form a cMDS-based therapeutic complex. As shown in Figure 3a, compared with the untreated group, the level of Stat3 mRNA in the CLP/siStat3 group and cMDS/siStat3 group decreased by 34.3% ± 8.1% and 23.8% ± 3.1% respectively (p < 0.01, versus untreated group). Furthermore, as shown in Figure 3b, the expression of Stat3 protein in CT26 cells was obviously downregulated after treatment with CLP/siStat3 or cMDS/siStat3 complex. As shown Figure 3c, compared with the untreated group, the expression level of Stat3 protein in the CLP/siStat3 group and cMDS/siStat3 group decreased by 46.4% ± 17.8% and 33.6% ± 13.3%, respectively (p < 0.05, versus untreated group), suggesting that the Stat3 gene was efficiently silenced by the cMDS/siStat3 complex.
 |
Figure 3 cMDS/siStat3 inhibits tumor growth in vitro. (a) Quantitative RT-PCR analysis of Stat3 mRNA levels in CT26 cells after transfection for 48 hours. (b) Western blot analysis of Stat3 protein levels in CT26 cells after transfection for 48 hours. (c) Quantitative analysis of Stat3 protein levels in CT26 cells. (d) Clonogenic assay. (e) Mean clones per well. (f) Viability of CT26 cells after transfection with CLP/siStat3 and cMDS/siStat3 complexes for 24 hours. (g) Apoptosis of CT26 cells after transfection with CLP/siStat3 and cMDS/siStat3 complexes for 48 hours. (h) Histogram analysis of the apoptosis rate. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. |
Next, the anti-proliferation effect of the cMDS/siStat3 complex was studied using a clonogenic assay. As shown in Figure 3d, the clonogenesis of the CLP/siStat3 and cMDS/siStat3 groups significantly decreased compared with those of the untreated and CLP groups. The number of clones in the cMDS/siStat3 group and CLP/siStat3 group was 125 ± 7 and 122 ± 4, respectively (p < 0.001 versus untreated and CLP groups) (Figure 3e). The anti-proliferation ability of the cMDS/siRNA complex was also evaluated using an MTT assay. As shown in Figure 3f, the cell viability of the CLP/siStat3 and cMDS/siStat3 groups decreased to 49.6% ± 10.6% and 43.9% ± 4.8%, respectively, compared with that of the untreated group (p < 0.0001). As it has been reported that the suppression of Stat3 activation can induce apoptosis, we next examined whether the anti-proliferation effect of the cMDS/siStat3 complex was a result of the induction of apoptosis in CT26 cells.24 As shown in Figure 3g and h, the CLP/siStat3 complex and cMDS/siStat3 complex increased apoptosis, with a mean apoptosis rate of 32.9% ± 4% and 38.9% ± 9%, respectively. Taken together, these results suggest that cMDS could efficiently deliver Stat3 siRNA into CT26 cells and induce anti-proliferative effects through the induction of apoptosis.
Distribution of the cMDS in vivo
To verify that the cancer cell membrane coating could provide additional tumor-targeting properties to the original backbone, we next characterized the in vivo distribution of the cMDS. For this purpose, Cy5.5-labeled CLP and cMDS were intravenously injected into mice bearing CT26 tumors. As shown in Figure 4a, the cMDS group showed stronger fluorescence signals in the tumor tissues than the CLP group did. This difference could still be observed 264 h after administration, suggesting clear tumor accumulation ability of the cMDS in vivo. As calculated from the quantitative fluorescence analysis, the distribution behavior of the cMDS in the tumor is also shown in Figure 4b. The fluorescence intensity of the cMDS in tumor tissues gradually increased following administration, with a peak radiant efficiency value of 1.49 ± 0.28×108 [p/s/cm²/sr] / [µW/cm²] 144 h post injection. However, the tumor site fluorescence signal in the CLP group decreased from 24 h post injection, with peak radiant efficiency value of only 1.09 ± 0.64×108 [p/s/cm²/sr] / [µW/cm²]. In addition, compared with the CLP group, the radiant efficiency fluorescence intensity in the cMDS group was quite high 264 h after injection (p < 0.05), which also indicated the specific accumulation property of the cMDS in tumor tissues. Additionally, the fluorescence signals of the main organs and tumor tissues of the mice in each group were analyzed 264 h after administration. As shown in Figure 4c, obvious tumor tissue-specific accumulation of fluorescence signals could be detected in the cMDS group, but CLP showed less significant tumor specific accumulation of fluorescence signals. The fluorescence signals in each tissue were also analyzed by quantification. As shown in Figure 4d, in the cMDS group, the fluorescence in tumor tissue accounted for 68.47% of all tissues, with an average radiant efficiency of 1.38 ± 0.23×108 [p/s/cm²/sr] / [µW/cm²]; this was notably higher than that of the CLP group (44.36% of all tissues, with 1.386 ± 2.52×107 [p/s/cm²/sr] / [µW/cm²], p < 0.05). These results suggest that cMDS could efficiently accumulate in tumor tissues upon systemic administration for a comparatively long period. Coating the CLP backbone with CM successfully introduced cancer targeting properties to cMDS, thus providing the basics for targeted siRNA delivery in vivo.
 |
Figure 4 Biodistribution of the cMDS in vivo. (a) In vivo imaging of Cy5.5-labeled CLP and cMDS over an 11-day period after intravenous injection (n=3). (b) Quantification of the radiant efficiency in the tumor site at each time point. (c) Imaging of major organs and tumors after intravenous injection of CLP and cMDS. (d) Quantification of the radiant efficiency in major organs and tumors. *p < 0.05. |
Immunostimulatory Effect of cMDS
In addition to having tumor targeting characteristics, the cMDS may cause tumor related immune stimulation. Therefore, we next investigated the immune stimulation ability of the cMDS. To this end, we first studied whether cMDS could be taken up by DCs, which play a primary role in antigen processing.25 For this purpose, bone marrow-derived dendritic cells (BMDCs) were incubated with Dil-labeled cMDS for 24 h. As shown in Figure 5a, the Dil signal was clearly observed in the cytoplasm of DCs, indicating that cMDS could be easily taken up by DCs. We then studied whether cMDS could induce the DC maturation and promote immune stimulatory activities. To this end, an in vitro study was designed as shown in Figure 5b. We first verified the activation ability of the cMDS against DCs. The percentage of mature DCs (CD80+ CD86+) in the CM, CLP, and cMDS groups was evaluated via flow cytometry.26 As shown in Figure 5c, the percentage of mature DCs in the CM, CLP, and cMDS groups was 16.42% ± 1.05%, 5.04% ± 0.9%, and 32.27% ± 0.35% respectively, which was notably higher than that of the untreated group (3.63% ± 0.06%) (p < 0.0001), among which the cMDS demonstrated the strongest ability to induce DC maturation. Moreover, as MHC molecules expressed on the surface of DCs are key components mediating antigen presentation and recognition of T cells,27 we next measured the proportion of MHC-II+ DCs (CD11C+ MHC-II+) following incubation with CM, CLP, and cMDS. As shown in Figure 5d, the percentage of MHC-II+ DCs in the CM and cMDS groups was 45.52% ± 3% (p < 0.01, versus untreated group) and 59.37% ± 2.14% (p < 0.0001, versus untreated group) respectively, which was obviously higher than that of the untreated group (32.55% ± 0.96%), among which the cMDS showed the greatest ability to induce MHC-II molecule expression. These results suggest that compared with CM and CLP, the cMDS could more effectively induce the maturation and activation of DCs in vitro. We then studied whether cMDS-stimulated DCs could further promote T cell activation.28 To this end, we incubated spleen-derived primary lymphocytes with DCs subjected to different treatments. As shown in Figure 5e, the number of activated T cells (CD3+ CD4+) after treatment with cMDS (4.83% ± 0.38%, p < 0.01, versus untreated group) and CLP (3.87% ± 0.3%, p < 0.05, versus untreated group) was notably increased. Meanwhile, the cMDS exhibited stronger T cell activation ability than CM. We also evaluated whether the cMDS could directly promote NK cell activation.29 To achieve this, the cDMS was incubated with spleen-derived primary lymphocytes for 24 h. As shown in Figure 5f, a notable increase in the number of activated NK cells (CD3− NK1.1+) was observed in the cMDS group (2.32% ± 0.06%, p < 0.001, versus untreated group) and the CLP group (0.71% ± 0.09%, p < 0.01, versus untreated group), which was in contrast to the results of the untreated group (0.26% ± 0.01%) and the CM group (0.2% ± 0.09%). These results suggest that compared with CM and CLP, the cMDS has an increased capacity to activate T and NK cells in vitro.
 |
Figure 5 Immune cell activation by cMDS in vivo and in vitro. (a) Uptake of the cMDS by DC (red: Dil-labeled membrane; blue: nucleus; scale bar: 10 μm). (b) diagram of in vitro immunostimulation experiments. (c) Flow cytometric analyses of the percentage of mature DCs (CD80 and CD86) after incubation with CM, CLP, and cMDS. (d) Flow cytometric analyses of the percentage of CD11C+ and MHC-II+ cells after incubation of DCs with CM, CLP, and cMDS. (e) Flow cytometric analyses of the percentage of activated T cells (CD3+ and CD4+) after incubation of T cells with the above-pretreated DCs. (f) Flow cytometric analyses of the percentage of activated NK cells (CD3− and NK1.1+) after incubation of splenic lymphocytes with CM, CLP, and cMDS. (g) Analysis of DCs, T cells, and NK cell activation in the spleens of mice after cMDS injection. (h) Analysis of DC and T cell activation in the lymph nodes of mice after cMDS injection. *p < 0.05, **p < 0.01, ****p < 0.0001. |
To further verify the immunostimulatory effect of the cMDS in vivo, mice were injected with normal saline (NS), CLP, or cMDS, and after 24 h, the lymph nodes were isolated from each group of mice. The proportions of various immune cells were measured via flow cytometry. As shown in Figures 5g and S1, compared with the NS group, cMDS administration notably increased the number of CD11C+ cells (66.02%), mature DCs (CD80+ CD86+, 21.57%), MHC-II+ DCs (CD11C+ MHC-II+, 36.91%), total T cells (CD3+, 48.68%), CD4+ T cells (CD3+ CD4+, 31.68%), and activated NK cells (CD3− NK1.1+, 6.46%) in the spleen. The stimulation effects of the cMDS on the above cells were much stronger than those of the CLP group. As shown in Figures 5h and S1, similar stimulation effects were also detected in lymph nodes. Administration of the cMDS obviously increased the number of CD11C+ cells (33.5%), mature DCs (36.53%), MHC-II+ DCs (31.01%), total T cells (92.51%), and CD4+ T cells (72.31%) in lymph nodes. Similar to the effects observed in the spleen, the cMDS also demonstrated stronger stimulation effects than CLP in the above cell populations. These results suggest that the cMDS could efficiently stimulate the maturation and activation of multiple immune cell types both in vitro and in vivo. The cancer cell membrane fractions coated on the surface of the nanobackbone successfully introduced some immunological properties of cancer cells to the vector, highlighting the immunotherapeutic potential of the cMDS.
Antitumor Effect of the cMDS/siStat3 Complex in vivo
The antitumor effect of the cMDS/siStat3 complex has been proved in vitro, and cMDS can target tumors and stimulate tumor related immune response in vivo. Therefore, we next evaluated the anti-cancer effect of the cMDS/siStat3 complex on the CT26 subcutaneous xenografted model through intravenous injection (Figure 6a). As shown in the tumor growth curve, the tumor growth of the NS, CLP/siNC and cMDS/siNC group were the fastest, followed by that of the CLP/siStat3 group and cMDS/siStat3 group (Figure 6b). The mice were euthanized and the tumors were photographed. As shown in Figure 6c, the tumors of the cMDS/siStat3 group were the smallest. The tumor volume was measured and statistically analyzed at the treatment endpoint. As shown in Figure 6b, the mean tumor volume of the cMDS/siStat3 group was 426 ± 122 mm3, which was much smaller than that of the NS group (1059 ± 175 mm3, p < 0.01), CLP/siNC (1048 ± 256 mm3, p < 0.01), cMDS/siNC (997 ± 256 mm3, p < 0.01) and the CLP/siStat3 group (659 ± 125 mm3, p < 0.05). Then, the tumors were weighed and analyzed statistically. As shown in Figure 6d, the weight of the cMDS/siStat3 group (0.27 ± 0.06 g) was lower that of the NS group (0.87 ± 0.17 g, p < 0.001), CLP/siNC (0.76 ± 0.13 g, p < 0.001) group, cMDS/siNC group (0.74 ± 0.13 g, p < 0.001), and CLP/siStat3 (0.53 ± 0.15 g, p < 0.05) suggesting that the cMDS/siStat3 complex had antitumor effects in vivo. Daily intravenous administration of CLP/siStat3 or cMDS/siStat3 nanoparticles had little effect on the body weight of the mice compared with the NS group (Figure 6e). To study the immune cell infiltration of tumors, we performed flow cytometry. As shown in Figure 6f, the number of mature DC, CD4+ T cells and NK cells in cMDS/siStat3 was higher than in other groups.
 |
Figure 6 cMDS/siStat3 inhibits subcutaneous xenograft growth in vivo. (a) A schematic view of the experimental design. (b) Tumor growth curves of each group (n=5). (c) Tumor tissues collected from each group. (d) Mean tumor weight of each group. (e) body weight of the mice. (f) Flow cytometric quantification of immune cells in tumor microenvironment. *p < 0.05, ***p < 0.001. |
Next, to study the therapeutic mechanism of the cMDS/siStat3 complex, immunohistochemistry and TUNEL assays were performed. As shown in Figure 7a, the expression of Stat3 in the cMDS/siStat3 group was lower than that in other groups, indicating that the cMDS/siStat3 complex could silence the Stat3 gene in vivo. Compared with the other groups, significantly increased cell apoptosis was induced after treatment with the cMDS/siStat3 complex, suggesting that the cMDS/siStat3 complex can promote apoptosis. Moreover, less CD31 was expressed in the cMDS/siStat3 group compared with that in other groups, suggesting that cMDS/siStat3 has anti-angiogenesis effects. Additionally, enhanced TNF-α and IFN-γ secretion and CD4+ T cells was found in the cMDS/siStat3 group, indicating that the cMDS/siStat3 complex can also achieve anti-cancer effects via immune stimulation. In conclusion, the cMDS/siStat3 complex could effectively inhibit tumor growth through gene silencing, anti-angiogenesis, apoptosis induction, and immune stimulation. To study the therapeutic safety of the cMDS/siStat3 complex, the mice were euthanized and major organs were collected for HE staining. As shown in Figure 7b, no obvious organ damage was observed after treatment with the CLP/siStat3 complex and the cMDS/siStat3 complex. Additionally, routine blood analysis was performed after intravenous injection of the CLP/siStat3 complex and cMDS/siStat3 complex. As shown in Figure 7c, there was no significant difference between the three groups. These results show that cMDS/siStat3 can safely and effectively treat tumors in vivo.
 |
Figure 7 Anti-tumor mechanism and safety research. (a) Immunohistochemistry and immunofluorescence analysis of tumor tissues (scale bars: 100 μm). (b) HE analysis of the main organs for all groups (scale bars: 100 μm). (c) Routine blood analysis after intravenous administration of CLP/siStat3 and cMDS/siStat3 complexes (n=3). |
Mechanism of the cMDS Enhancing Immune Response
As a good therapeutic effect was observed in the mice treated with the cMDS/siStat3 complex, we hypothesized that CM in the surface of the cMDS played a considerable role in further uncovering the immune response mechanism of cancer cell membrane fractions. We next prepared protein lysates from mouse tumors in the NS, CLP/siStat3, and cMDS/siStat3 groups, and then analyzed 200 cytokines by protein chip (Figure S2), CLP/siStat3 and cMDS/siStat3 groups were mainly analyzed. As shown in the fluorescence image of the protein chip, the cytokines showed many differences between the CLP/siStat3 and cMDS/siStat3 groups (Figure 8a). Then, the fluorescence of the protein chip was quantified and analyzed. As shown in Figure 8b, 21 factors were upregulated, and 35 factors were downregulated in the cMDS/siStat3 group compared with the CLP/siStat3 group (fold change ≥ 1.2 and p < 0.05). Then, the top 20 significantly up- and downregulated genes were statistically analyzed. As shown in Figure 8c, the cytolytic Granzyme B was secreted by cytotoxic T and NK cells more frequently in the cMDS/siStat3 group than that in the CLP/siStat3 group. Additionally, chemokines such as MIP-2, MIG, MIP-1g, TECK, RANTES, and TARC in the cMDS/siStat3 group were also significantly higher than those in the CLP/siStat3 group, suggesting that the chemotactic response of cells in the cMDS/siStat3 group was stronger, and that the toxic cells secreted more cytolytic granules to promote tumor killing. However, VEGF B protein was significantly downregulated in the cMDS/siStat3 group, which may be related to the inhibition of tumor angiogenesis. Next, the differentially expressed genes (DEGs) were analyzed via GO enrichment analysis. As shown in Figure 8d, from the perspective of molecular function, the differential genes between the CLP/siStat3 and cMDS/siStat3 groups were enriched in “chemokine activity” and “chemokine receptor binding”. The activity and binding of chemokines are closely related to the immune response, indicating that the CM on the surface of the cMDS played a key role in enhancing immunity. From the perspective of the biological process, the differential genes between the CLP/siStat3 and cMDS/siStat3 groups were mainly enriched in the migration and chemotaxis of leukocytes such as monocytes and granulocytes, which are essential components of the immune response, further proving that cMDS/siStat3 complex enhances the immune response (Figure 8e). KEGG enrichment analysis was also performed. As shown in Figure 8f, cMDS/siStat3 caused immune activation through various pathways, including cytokine–cytokine receptor interaction, chemokine signaling pathway, TNF signaling pathway, and IL-17 signaling pathway. In conclusion, compared with the CLP/siStat3 group, the cMDS/siStat3 group showed additional obvious immune stimulation. These effects are mainly achieved by promoting the migration of immune cells, and the CM fractions are the main reason for enhanced immune effect. These findings indicate that the CM can enhance the anti-cancer immunity of nanoparticle/gene complexes while conferring tumor-targeting properties to achieve a synergistic therapeutic effect.
 |
Figure 8 Protein chip analysis of tumors. (a) Representative chip of the CLP/siStat3 group and cMDS/siStat3 group. (b) Heatmap of differential genes. (c) Histogram analysis of the top 20 upregulated and downregulated genes. (d) GO enrichment of differential genes (molecular function). (e) GO enrichment of differential genes (biological process). (f) KEGG enrichment analysis of differential genes. |
Discussion
In this study, a cMDS was established to deliver siRNA against Stat3 for tumor-targeted gene therapy and immunotherapy. Our results demonstrated that the prepared cMDS efficiently delivered siRNA into CT26 cells with tumor-targeting properties. The cMDS could also induce immune stimulation in vivo and in vitro. After systemic administration with the cMDS/siStat3 complex, the combination of tumor-targeted gene therapy and immunotherapy showed obvious anti-tumor effects. Our results demonstrated that the cMDS can deliver siRNA for tumor targeting and immunotherapy.
Gene therapy, as a new treatment method, has shown potential in the field of tumor therapy. Indeed, specially designed siRNAs can bind and induce silencing of target genes,30 although their application is limited due to their rapid degradation in vivo and poor tumor targeting.31 Current strategies for tumor-targeted therapy mainly include conjugating ligands32–34 and antibodies,35,36 or coating carriers with cell membranes.37–39 Indeed, various types of cell membrane–coated nanocarriers have been developed as novel tumor-targeted therapeutic strategies by taking advantage of the homozygous adhesion properties of cancer cells,40 the recognition of tumors by NK cells,8 macrophages,41 and leukocytes,42 the binding of neutrophils43 and platelets44 to circulating cancer cells, and the cancer cell homing properties of stem cells.45 For example, Wang et al used a fusion cell membrane (erythrocytes and cancer cells)–coated nanocarrier to deliver doxorubicin.46 Hu et al delivered TRAIL and DOX with platelet membrane–coated nanoparticles,47 while Ho et al used the fusion cell membrane (leukocytes and cancer cells) coated nanocarrier to deliver PTX.38 Moreover, Zhang et al delivered siRNA against TERT via macrophage membrane–coated nanoparticles.48 The above strategies promoted the tumor-targeting ability of the nanocarrier, improved anti-cancer efficacy, and achieved targeted therapy for tumors. However, many existing studies of cell membrane–coated nanoparticles only used nanocarriers as “supports” and mainly relied on the cell membrane to function.49,50 Other studies used nanocarriers to deliver chemical drugs, while the cell membranes coated on the surface of nanocarriers performed unique functions.38,45,51 Only a few studies have used membrane–coated nanoparticles to deliver nucleic acid drugs.48 Therefore, cell membrane–coated nanocarriers are rarely applied in the field of gene therapy, largely because both nucleic acids and the cell membrane are negatively charged, which makes them difficult to combine to form nanoparticles. In this study, after combining siRNA with cationic nanoparticles to prepare a complex, we coated the complex with a cancer cell membrane. TEM images showed a membrane-like layer on the surface of the CLP. Additionally, the size of the cMDS was larger than that of CLP, and the zeta potential of the cMDS was close to that of the CM. These results demonstrate that the cell membrane was successfully coated with CLP nanoparticles. In addition, the cMDS could also protect siRNA from RNase degradation within 24 h, and the transfection efficiency of the formed cMDS/siRNA complexes in tumor cells was as high as 99%. The uptake rate of the cMDS/siRNA complex in macrophages was 65%, which was less than that of the CLP/siRNA complex (71%), indicating that the cMDS coated with CM can reduce the uptake of macrophages. Importantly, the cancer cell membranes endow cMDS with tumor-targeting properties. After loading with Stat3-against siRNA, the cMDS/siStat3 complex can exert a good anti-tumor effect in vitro, causing 56% of cancer cells to die. Moreover, in animal experiments, the tumor volume was reduced by 62% after cMDS/siStat3 treatment. The designed cMDS can target siRNA delivery to the tumor site, and the membrane-coated drug delivery vector can be applied to the field of gene therapy. These results may be because CLP itself is an efficient siRNA delivery vector, while the efficiency of CLP transfecting siRNA in tumor cells is nearly 99%. The surface of CLP has a strong cationic charge (nearly 40 mV), which can simultaneously bind negatively charged siRNA and the cell membrane. Additionally, as the positively charged CLP could be firmly bound to siRNA, it also protects siRNA from being degraded in the presence of RNase, which is also the premise of siRNA for in vivo therapy. As a liposome material, CLP has a similar structure to the cell membrane, meaning that they can be easily combined, with CLP coated on the cell surface after simple sonication. When the CLP/siRNA complexes are coated with CM, both their accumulation in the tumor site and their therapeutic effects are greater.
The CM not only provides tumor-targeting properties, but can also cause cancer-related immune stimulation in vivo, implying that it could be used as a vaccine to treat tumors.52 However, the cell membrane cannot be used directly because the cell membrane aggregates easily and has poor stability,53 and also because the immune stimulation of the cell membrane alone is insufficient to successfully kill tumors. Therefore, we propose that nanocarriers coated with CM have potential use as tumor vaccines. This strategy has two main advantages: First, nanocarriers themselves can have a certain immune stimulating effect, which could be combined with the cell membrane to enhance the stimulating effect; second, as a delivery vector, nanoparticles can promote the uptake of cancer cell membranes by DCs, thereby enhancing immune stimulation. Indeed, Jiang et al have coated PEG nanoparticles with tumor cell membranes expressing OVA and CD80 to achieve tumor prevention,54 while Liu et al coated PEG nanoparticles with cancer cells and DC fusion membranes as a tumor vaccine.19 However, in these studies, the preventive effect of the nanoparticles coated with the cancer cell membrane alone is not obvious, and they need to be fused with other cells or modified cancer cell membranes to exert a better curative effect. Additionally, they stimulated mice with cell membrane–coated nanoparticles before cancer cell inoculation, while in clinical applications, treatment measures are often required after tumor occurrence. Therefore, the immunostimulatory effect of cancer cell membranes alone have a poor anti-tumor effect. This may be because the antigens on the surface of the cell membrane are not sufficiently abundant. Moreover, it only relies on the immune effect of autoimmunity to fight tumors, and the mode of action is single. Therefore, in this study, to improve the therapeutic effect of cell membrane–coated nanoparticles, we employed them to deliver gene therapy drugs. The in vitro results showed that the cMDS caused a 30% increase in mature CD80+ CD86+ DCs, while CD11C+ MHC-II+ DCs increased by 27%; this indirectly stimulated a 2% increase in T cell activation. Additionally, the cMDS directly caused a 2% increase in NK cell activation. Moreover, after the mice were injected with cMDS, CD80+ CD86+ DCs, MHC-II-expressing DCs, activated T cells and NK cells in the spleen increased by 9%, 18.1%, 9.9%, and 4.6%, respectively. Moreover, mature DCs, MHC-II-expressing DCs, and activated T cells in lymph nodes increased by 31%, 17.4% and 5.5%, respectively. In the subcutaneous tumor model, the delivery of Stat3-targeting siRNA with cMDS achieved good therapeutic effects. Furthermore, through protein chip technology, we found significant differences in cytokine expression levels between the CLP/siStat3 and cMDS/Stat3 groups, which confirmed the results of in vivo and in vitro immune stimulation. Additionally, using GO enrichment, we further discovered that these differences in cytokines mainly affect the migration and chemotaxis of immune cells. Therefore, we have identified the detailed mechanism underlying the cancer cell membrane–based immune stimulation effect, providing an important basis for the subsequent application of cancer cell membrane bioinspired nanocarriers. However, in our study, the tumor targeting effect of the cancer cell membrane was not very good. We will subsequently screen for cancer cell membranes with better targeting effects and optimize the manufacturing process of cancer cell membrane-coated nanoparticle, and further study the targeting mechanism of nanoparticles. Meanwhile, cancer cell membrane-based nanoparticles also have potential safety concerns for their potential applications in clinic. After the surface of nanoparticles was coated with cancer cell membrane, the particle size and uniformity of nanoparticles may be affected, and there is a certain risk in intravenous injection. In addition, the components on the cancer cell membrane are complex, which may cause side effects such as uncontrolled immune response. Therefore, it is necessary to optimize the process, strengthen quality control, and try different routes of administration. Additionally, preclinical safety evaluation is required, including assays such as mutation, teratogenicity, and carcinogenicity tests.
Conclusion
Herein, we prepared a cancer membrane–based drug delivery system (cMDS) for efficient targeted siRNA delivery. The cMDS showed obvious tumor tissue-specific accumulation properties strong immune stimulation ability. The growth of colon cancer can be suppressed after intravenously administered cMDS/siStat3 complex suggesting that the cMDS is an advanced targeted gene delivery system.
Funding
This work is supported by Medico-Engineering Cooperation Funds from university of Electronic Science and Technology of China (ZYGX2021YGLH225), the Key Research Program of Science and Technology Department of Sichuan Province (2023YFS0165, 23NSFJQ0104) and the Science Foundation of Chengdu (2022-YF05-01793-SN).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Suresh Kumar N, Padma Suvarna R, Chandra Babu Naidu K, Banerjee P, Ratnamala A, Manjunatha H. A review on biological and biomimetic materials and their applications. Applied Physics A. 2020;126(6):445. doi:10.1007/s00339-020-03633-z
2. Kim K, Ryu JH, Koh M-Y, et al. Coagulopathy-independent, bioinspired hemostatic materials: a full research story from preclinical models to a human clinical trial. Sci Adva. 2021;7(13):eabc9992. doi:10.1126/sciadv.abc9992
3. Rajabi-Zeleti S, Jalili-Firoozinezhad S, Azarnia M, et al. The behavior of cardiac progenitor cells on macroporous pericardium-derived scaffolds. Biomaterials. 2014;35(3):970–982. doi:10.1016/j.biomaterials.2013.10.045
4. Fan L, Liu C, Chen X, et al. Directing induced pluripotent stem cell derived neural stem cell fate with a three-dimensional biomimetic hydrogel for spinal cord injury repair. ACS Appl Mater Interfaces. 2018;10(21):17742–17755. doi:10.1021/acsami.8b05293
5. Chen L, Hong W, Ren W, Xu T, Qian Z, He Z. Recent progress in targeted delivery vectors based on biomimetic nanoparticles. Signal Transduct Target Therap. 2021;6(1):225. doi:10.1038/s41392-021-00631-2
6. Chi Y, Yin X, Sun K, et al. Redox-sensitive and hyaluronic acid functionalized liposomes for cytoplasmic drug delivery to osteosarcoma in animal models. J Cont Rel. 2017;261:113–125. doi:10.1016/j.jconrel.2017.06.027
7. Li L, Zhang R, Gu W, Xu ZP. Mannose-conjugated layered double hydroxide nanocomposite for targeted siRNA delivery to enhance cancer therapy. Nanomedicine. 2018;14(7):2355–2364. doi:10.1016/j.nano.2017.06.006
8. Pitchaimani A, Nguyen TDT, Aryal S. Natural killer cell membrane infused biomimetic liposomes for targeted tumor therapy. Biomaterials. 2018;160:124–137. doi:10.1016/j.biomaterials.2018.01.018
9. Liu G, Zhao X, Zhang Y, et al. Engineering biomimetic platesomes for pH-responsive drug delivery and enhanced antitumor activity. Adva Mat. 2019;31(32):e1900795. doi:10.1002/adma.201900795
10. Cully M. Exosome-based candidates move into the clinic. Nat Rev Drug Discov. 2021;20(1):6–7. doi:10.1038/d41573-020-00220-y
11. Lombard J. Once upon a time the cell membranes: 175 years of cell boundary research. Biol Direct. 2014;9:32. doi:10.1186/s13062-014-0032-7
12. Papa S, Ferrari R, De Paola M, et al. Polymeric nanoparticle system to target activated microglia/macrophages in spinal cord injury. J Cont Rel. 2014;174:15–26. doi:10.1016/j.jconrel.2013.11.001
13. Tang W, Yang Y, Yang L, Tang M, Chen Y, Li C. Macrophage membrane-mediated targeted drug delivery for treatment of spinal cord injury regardless of the macrophage polarization states. Asian J Pharm Sci. 2021;16(4):459–470. doi:10.1016/j.ajps.2021.03.005
14. Yu Q, Jiang X, Liu X, et al. Glutathione-modified macrophage-derived cell membranes encapsulated metformin nanogels for the treatment of spinal cord injury. Biomat Adva. 2022;133:112668. doi:10.1016/j.msec.2022.112668
15. Pei Q, Hu X, Zheng X, et al. Light-activatable red blood cell membrane-camouflaged dimeric prodrug nanoparticles for synergistic photodynamic/chemotherapy. ACS Nano. 2018;12(2):1630–1641. doi:10.1021/acsnano.7b08219
16. Sahoo K, Karumuri S, Hikkaduwa Koralege RS, et al. Molecular and biocompatibility characterization of red blood cell membrane targeted and cell-penetrating-peptide-modified polymeric nanoparticles. Mol Pharm. 2017;14(7):2224–2235. doi:10.1021/acs.molpharmaceut.7b00053
17. Wang B, Yang P, Ding Y, Qi H, Gao Q, Zhang C. Improvement of the biocompatibility and potential stability of chronically implanted electrodes incorporating coating cell membranes. ACS Appl Mater Interfaces. 2019;11(9):8807–8817. doi:10.1021/acsami.8b20542
18. Ke Y, Zhu J, Chu Y, et al. Bifunctional fusion membrane-based hydrogel enhances antitumor potency of autologous cancer vaccines by activating dendritic cells. Adv Funct Mater. 2022;2201306. doi:10.1002/adfm.202201306
19. Liu W-L, Zou M-Z, Liu T, et al. Cytomembrane nanovaccines show therapeutic effects by mimicking tumor cells and antigen presenting cells. Nat Commun. 2019;10(1):3199. doi:10.1038/s41467-019-11157-1
20. Liu R, Luo C, Pang Z, et al. Advances of nanoparticles as drug delivery systems for disease diagnosis and treatment. Chin Chem Letters. 2023;34(2):107518. doi:10.1016/j.cclet.2022.05.032
21. Kubiatowicz LJ, Mohapatra A, Krishnan N, Fang RH, Zhang L. mRNA nanomedicine: design and recent applications. Exploration. 2022;2(6):20210217. doi:10.1002/EXP.20210217
22. Huang Z, Liu S, Lu N, et al. Nucleus-specific RNAi nanoplatform for targeted regulation of nuclear lncRNA function and effective cancer therapy. Exploration. 2022;2(5):20220013. doi:10.1002/EXP.20220013
23. Xin Y, Huang M, Guo WW, Huang Q, Zhang LZ, Jiang G. Nano-based delivery of RNAi in cancer therapy. Mol Cancer. 2017;16(1):134. doi:10.1186/s12943-017-0683-y
24. Dasgupta A, Raychaudhuri B, Haqqi T, et al. Stat3 activation is required for the growth of U87 cell-derived tumours in mice. Eur J Cancer. 2009;45(4):677–684. doi:10.1016/j.ejca.2008.11.027
25. Wang X, Wang N, Yang Y, et al. Polydopamine nanoparticles carrying tumor cell lysate as a potential vaccine for colorectal cancer immunotherapy. Biomat Sci. 2019;7(7):3062–3075. doi:10.1039/C9BM00010K
26. Benencia F, Sprague L, McGinty J, Pate M, Muccioli M. Dendritic cells the tumor microenvironment and the challenges for an effective antitumor vaccination. J Biomed Biotechnol. 2012;2012:425476. doi:10.1155/2012/425476
27. Drozina G, Kohoutek J, Jabrane-Ferrat N, Peterlin BM. Expression of MHC II Genes. In: Singh H, Grosschedl R, editors. Molecular Analysis of B Lymphocyte Development and Activation. Berlin, Heidelberg: Springer Berlin Heidelberg; 2005:147–170.
28. Binnewies M, Mujal AM, Pollack JL, et al. Unleashing type-2 dendritic cells to drive protective antitumor CD4(+) T cell immunity. Cell. 2019;177(3):556–571.e16. doi:10.1016/j.cell.2019.02.005
29. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. 2008;9(5):503–510. doi:10.1038/ni1582
30. Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411(6836):494–498. doi:10.1038/35078107
31. Ozpolat B, Sood AK, Lopez-Berestein G. Liposomal siRNA nanocarriers for cancer therapy. Adv Drug Deliv Rev. 2014;66:110–116. doi:10.1016/j.addr.2013.12.008
32. Sun T, Wang Y, Wang Y, et al. Using SV119-gold nanocage conjugates to eradicate cancer stem cells through a combination of photothermal and chemo therapies. Adv Healthcare Mater. 2014;3(8):1283–1291. doi:10.1002/adhm.201400026
33. Soe ZC, Thapa RK, Ou W, et al. Folate receptor-mediated celastrol and irinotecan combination delivery using liposomes for effective chemotherapy. Colloids Surf B Biointerfaces. 2018;170:718–728. doi:10.1016/j.colsurfb.2018.07.013
34. Patil Y, Shmeeda H, Amitay Y, Ohana P, Kumar S, Gabizon A. Targeting of folate-conjugated liposomes with co-entrapped drugs to prostate cancer cells via prostate-specific membrane antigen (PSMA). Nanomedicine. 2018;14(4):1407–1416. doi:10.1016/j.nano.2018.04.011
35. Kim I-Y, Kang Y-S, Lee DS, et al. Antitumor activity of EGFR targeted pH-sensitive immunoliposomes encapsulating gemcitabine in A549 xenograft nude mice. J Cont Rel. 2009;140(1):55–60. doi:10.1016/j.jconrel.2009.07.005
36. Kirpotin DB, Drummond DC, Shao Y, et al. Antibody targeting of long-circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models. Cancer Res. 2006;66(13):6732–6740. doi:10.1158/0008-5472.CAN-05-4199
37. Pan W-L, Tan Y, Meng W, et al. Microenvironment-driven sequential ferroptosis, photodynamic therapy, and chemotherapy for targeted breast cancer therapy by a cancer-cell-membrane-coated nanoscale metal-organic framework. Biomaterials. 2022;283:121449. doi:10.1016/j.biomaterials.2022.121449
38. He H, Guo C, Wang J, et al. Leutusome: a biomimetic nanoplatform integrating plasma membrane components of leukocytes and tumor cells for remarkably enhanced solid tumor homing. Nano Lett. 2018;18(10):6164–6174. doi:10.1021/acs.nanolett.8b01892
39. Ye H, Wang K, Lu Q, et al. Nanosponges of circulating tumor-derived exosomes for breast cancer metastasis inhibition. Biomaterials. 2020;242:119932. doi:10.1016/j.biomaterials.2020.119932
40. Sun H, Su J, Meng Q, et al. Cancer-cell-biomimetic nanoparticles for targeted therapy of homotypic tumors. Adv Mater. 2016;28(43):9581–9588. doi:10.1002/adma.201602173
41. Xuan M, Shao J, Dai L, He Q, Li J. Macrophage cell membrane camouflaged mesoporous silica nanocapsules for in vivo cancer therapy. Adv Healthcare Mater. 2015;4(11):1645–1652. doi:10.1002/adhm.201500129
42. Parodi A, Quattrocchi N, van de Ven AL, et al. Synthetic nanoparticles functionalized with biomimetic leukocyte membranes possess cell-like functions. Nat Nanotechnol. 2013;8(1):61–68. doi:10.1038/nnano.2012.212
43. Kang T, Zhu Q, Wei D, et al. Nanoparticles coated with neutrophil membranes can effectively treat cancer metastasis. ACS Nano. 2017;11(2):1397–1411. doi:10.1021/acsnano.6b06477
44. Li J, Ai Y, Wang L, et al. Targeted drug delivery to circulating tumor cells via platelet membrane-functionalized particles. Biomaterials. 2016;76:52–65. doi:10.1016/j.biomaterials.2015.10.046
45. Gao C, Lin Z, Jurado‐Sánchez B, Lin X, Wu Z, He Q. Stem cell membrane‐coated nanogels for highly efficient in vivo tumor targeted drug delivery. Small. 2016;12(30):4056–4062. doi:10.1002/smll.201600624
46. Wang D, Dong H, Li M, et al. Erythrocyte–cancer hybrid membrane camouflaged hollow copper sulfide nanoparticles for prolonged circulation life and homotypic-targeting photothermal/chemotherapy of melanoma. ACS nano. 2018;12(6):5241–5252. doi:10.1021/acsnano.7b08355
47. Hu Q, Sun W, Qian C, Wang C, Bomba HN, Gu Z. Anticancer platelet‐mimicking nanovehicles. Adv Mater. 2015;27(44):7043–7050. doi:10.1002/adma.201503323
48. Zhang F, Zhao L, Wang S, et al. Construction of a biomimetic magnetosome and its application as a SiRNA Carrier for high‐performance anticancer therapy. Adv Funct Mater. 2018;28(1):1703326. doi:10.1002/adfm.201703326
49. Wang F, Gao W, Thamphiwatana S, et al. Hydrogel retaining toxin-absorbing nanosponges for local treatment of methicillin-resistant staphylococcus aureus infection. Adva Mat. 2015;27(22):3437–3443. doi:10.1002/adma.201501071
50. Zhang Q, Honko A, Zhou J, et al. Cellular nanosponges inhibit SARS-CoV-2 Infectivity. Nano Letter. 2020;20(7):5570–5574. doi:10.1021/acs.nanolett.0c02278
51. Chai Z, Hu X, Wei X, et al. A facile approach to functionalizing cell membrane-coated nanoparticles with neurotoxin-derived peptide for brain-targeted drug delivery. J Control Release. 2017;264:102–111. doi:10.1016/j.jconrel.2017.08.027
52. Wang D, Liu C, You S, et al. Bacterial vesicle-cancer cell hybrid membrane-coated nanoparticles for tumor specific immune activation and photothermal therapy. ACS Appl Mater Interfaces. 2020;12(37):41138–41147. doi:10.1021/acsami.0c13169
53. Frasch CE, van Alphen L, Holst J, Poolman JT, Rosenqvist E. Outer Membrane Protein Vesicle Vaccines for Meningococcal Disease. In: Pollard AJ, Maiden MCJ, editors. Meningococcal Vaccines: Methods and Protocols. Totowa, NJ: Humana Press; 2001:81–107.
54. Jiang Y, Krishnan N, Zhou J, et al. Engineered cell‐membrane‐coated nanoparticles directly present tumor antigens to promote anticancer immunity. Adv Mater. 2020;32(30):2001808. doi:10.1002/adma.202001808
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.