Back to Journals » International Journal of Nanomedicine » Volume 15
Targeted Prodrug-Based Self-Assembled Nanoparticles for Cancer Therapy
Authors Wang W, Fan J, Zhu G, Wang J, Qian Y, Li H, Ju J ![]() , Shan L
, Shan L ![]()
Received 29 January 2020
Accepted for publication 7 April 2020
Published 24 April 2020 Volume 2020:15 Pages 2921—2933
DOI https://doi.org/10.2147/IJN.S247443
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yan Shen
Weiwei Wang,1 Junting Fan,2 Guang Zhu,1 Jing Wang,1 Yumei Qian,1 Hongxia Li,1 Jianming Ju,3 Lingling Shan1
1Institute of Pharmaceutical Biotechnology, School of Biology and Food Engineering, Key Laboratory of Spin Electron and Nanomaterials of Anhui Higher Education Institutes, Suzhou University, Suzhou 234000, People’s Republic of China; 2Department of Pharmaceutical Analysis, School of Pharmacy, Nanjing Medical University, Nanjing 211166, People’s Republic of China; 3Jiangsu Province Academy of Traditional Chinese Medicine, Nanjing, People’s Republic of China
Correspondence: Lingling Shan; Junting Fan Email [email protected]; [email protected]
Background: Targeted prodrug has various applications as drug formulation for tumor therapy. Therefore, amphoteric small-molecule prodrug combined with nanoscale characteristics for the self-assembly of the nano-drug delivery system (DDS) is a highly interesting research topic.
Methods and Results: In this study, we developed a prodrug self-assembled nanoplatform, 2-glucosamine-fluorescein-5(6)-isothiocyanate-glutamic acid-paclitaxel (2DA-FITC-PTX NPs) by integration of targeted small molecule and nano-DDS with regular structure and perfect targeting ability. 2-glucosamine (DA) and paclitaxel were conjugated as the targeted ligand and anti-tumor chemotherapy drug by amino acid group. 2-DA molecular structure can enhance the targeting ability of prodrug-based 2DA-FITC-PTX NPs and prolong retention time, thereby reducing the toxicity of normal cell/tissue. The fluorescent dye FITC or near-infrared fluorescent dye ICG in prodrug-based DDS was attractive for in vivo optical imaging to study the behavior of 2DA-FITC-PTX NPs. In vitro and in vivo results proved that 2DA-FITC-PTX NPs exhibited excellent targeting ability, anticancer activity, and weak side effects.
Conclusion: This work demonstrates a new combination of nanomaterials for chemotherapy and may promote prodrug-based DDS clinical applications in the future.
Keywords: targeted prodrug, nanoplatform, NIR imaging, chemotherapy
Introduction
Prodrugs represent a leading method in the current control release of drugs by chemical or enzymatic degradation to parent active agents.1,2 A prodrug skill can overcome certain limitations related to hydrophobic drugs, such as paclitaxel with high toxicity and low bioavailability. The targeted prodrug strategy is used to address poor water solubility, thus increasing targeting and membrane permeation. The prodrug structure is usually conducted with soluble small-molecule ligand, linker, and hydrophobic drugs to improve physicochemical characteristics and obtain good pharmacokinetic features.3,4 However, prodrugs, as small-molecule drugs, still encounter problems such as short half-life and rapid clearance.5 Similarly, a traditional nanoparticle drug delivery system (nano-DDS) also involves difficulties, such as low drug-loading efficiency, significant drug leakage, and poor biocompatibility.6,7
A prodrug-based nano-DDS can be considered to overcome certain disadvantages and has shown enormous potential applications in clinical cancer therapy.8,9 In particular, amphiphilic small-molecule-based nano-DDS can be self-assembled to nanoparticles for cancer treatment. These nanostructures are developed into the most promising nanoparticle DDS for anticancer therapy with high drug-loading efficiency and good biocompatibility.10,11 The metabolic pathway of these targeting amphiphilic small-molecule-based nano-DDS was dispersed into each targeted small molecule to perform the anticancer function; this process not only addressed insolubility issues but also enhanced the therapeutic effect.12,13 Therefore, amphiphilic small-molecule prodrug-based nano-DDS has attracted attention for the following characteristics: (i) self-assembled nanoarchitectures by amphiphilic small molecular prodrugs that can protect drugs from leakage and realize selective tumor accumulation through an enhanced permeation and retention effect,14,15 (ii) potentially achieving high drug-loading capacities without the incorporation of carriers,16 (iii) reducing the synthetic complexity of carriers17 and (iv) preventing the possible adverse effects induced by carriers.18
Malignant tumor cells undergo vigorous growth, leading to an aerobic capacity barrier. The important feature of their metabolism is anaerobic fermentation with glucose as the sole substrate to obtain energy. Cancer cells acquire energy only through this way, which is known as the “Warburg effect”.19 Glucose transporter1 (GLUT1) is the main carrier of glucose uptake in cells. Compared with normal cells, cancer cells show a significant increase in the expression and activity of GLUT1.20 Owing to the substitution of hydroxyl groups on the second carbon atom of glucose by markers in the metabolic pathway, GLUT1 cannot be isomerized by enzymes, thereby preventing the metabolism from proceeding normally, resulting in the accumulation of locally processed labeled glucose in cells to obtain relevant cancer information.21,22 Most importantly, glucosamine (DA) as a “targeting group” has a good targeting effect for cancer cells/tissues.23–25
Here, we report the research and development of a self-assembled DDS by targeting amphiphilic small molecules. The DDS nanoplatform conjugated the 2-glucosamine (2-DA), FITC/ICG, and PTX to an amphiphilic molecule and self-assembled these compounds in a nano-DDS. The DDS-based compound targeting the amphiphilic small molecule could maintain targeting ability, accumulate in tumor sites, and further enhance antitumor activity.
Materials and Methods
Materials
2-DA was prepared in our laboratory. Paclitaxel (PTX, MW 853.9) was purchased from Jiang Su Research Institute (Wuxi, China). Fmoc-Glu(OtBu)-OH (Glu, MW 425.49), Fmoc-Arg(Pbf)-OH (Arg, MW 648.77), fluorescent dye FITC (MW 389.38), succinic anhydride (SUC, MW 100.07), and ICG-Der-02 (ICG02, MW 995) were obtained from the Biomedical Engineering Laboratory of China Pharmaceutical University. All reagents used in this study were of certified analytical reagent grade.
Detection instruments of NMR, mass spectrometry LC-MS, and TEM were provided by China Pharmaceutical University. The following equipment were used: ZS-ultraviolet analytical photography instrument 180 (China Yuanming), ultraviolet spectrophotometer lambda 35 (Analytical Instrument Factory, Shanghai, China), Zetasizer 3000HSA laser particle sizing instrument (ZS90, Malvern Instruments, United Kingdom), inverted fluorescence microscope (IX71, Olympus, Japan), cell flow meter (Accuri C6, BD Biosciences, United States), fast frozen slicer (550, Meikang, Germany), and near-infrared imaging instrument by (Jinan Micro Technology Co. Ltd., China).
Synthesis of Glutamic Acid-Paclitaxel (PTX-SUC)
Exactly 100 mg (0.117 mmol) PTX was dissolved in 15 mL of DCM while adding 23.4 mg, 0.234 mmol, 2 equiv of succinic anhydride (SUC), and 14.29 mg of DMAP (0.117 mmol). Then, the mixture was stirred at room temperature for 40 h and traced by TLC. After the reaction was completed, the mixture was purified by silica gel column. Finally, 85% of crystal product PTX-SUC was obtained as white solid, and the structure was identified by LC-MS.26,27
Preparation of 2-Glucosamine-Glutamic Acid-Paclitaxel (2DA-FITC-NH2)
Exactly 0.2 mmol glucosamine was dissolved in 5 mL of DMF, added with 0.1 mmol Fmoc-Glu(OtBu)-NHS, and stirred overnight at room temperature. Then, the solvent was handled by CH3OH-HCl (OtBu removed), and the product was extracted by a freezer dryer. EDC and NHS (molar ratio 1:1.2:2) were added to activate the carboxyl group of 0.1 mmol Fmoc-Glu (COOH)-DA in DMF. After 4 h of stirring at room temperature, 0.2 mmol DA was added, and the mixture was stirred overnight at room temperature. The reaction solution was purified using a silica gel column, and 2-amino-glutamic acid (2DA-Fmoc-Glu) was obtained. 2DA-Fmoc-Glu was dissolved in DCM system and added with 2 mL of piperidine (20% of the total volume of piperidine). The 2DA-NH2-Glu prodrug was extracted with ether and traced by TLC for the next step, and the structure was identified by LC-MS.
Preparation of Amphoteric Small Molecular Monomer 2DA-FITC-PTX
After 0.15 mmol 2DA-NH2-Glu prodrug was dissolved with 7 mL of DMF, 0.1 mmol fluorescent dye FITC-Arg/ICG-Arg and 0.1 mmol PTX-SUC were added. The mixture was stirred overnight at room temperature, and then the reaction solution was purified by G15. Finally, the purified small molecular monomer of 2-glucosamine-fluorescent dye of paclitaxel prodrug (2DA-FITC-PTX/2DA-ICG-PTX) was obtained and confirmed by LC-MS and then stored at −20°C.
Self-Assembled DDS NPs (2DA-FITC-PTX NPs)
The targeted DDS of nanoparticles was prepared by the nano-precipitation method. Exactly 0.1 mmol 2DA-PTX-FITC amphoteric small-molecule monomer was dissolved in THF and stirred for 12 h at room temperature, and THF was removed via dialysis (8000 MWCO) overnight. The concentration of PTX was assayed by high-performance liquid chromatography (HPLC), and 2DA-FITC-PTX NPs was further employed for in vitro and in vivo experiments.
General Characterization
The chemical structures of amphiprotic targeted small-molecule intermediates and monomers were identified by NMR 1H and LC-MS.
The particle size (diameter nm), zeta potential (mV), and distribution of 2DA-FITC-PTX NPs can be measured using a Malvern particle size analyzer. Moreover, the morphology and appearance of the targeted nanoparticle DDS could be characterized by TEM.
Drug Loading Efficiency, Capacity, and Stability of 2DA-FITC-PTX NPs
The drug loading efficiency, capacity, and stability of 2DA-FITC-PTX NPs were measured as follows: nanoprecipitation from THF to water, and THF was removed by overnight evaporation in hood centrifuge at 3000 RPM for 3 min to remove any potential precipitate. Various concentrations of 2DA-FITC-PTX NP water solutions were placed at room temperature (RT) or 4°C for 2, 4, and 8 weeks, and the particle size of 2DA-FITC-PTX NPs was tested by a Malvern particle size analyzer. PTX content was measured by HPLC, and the others were measured by UV-Vis. The drug loading efficiency of 2DA-FITC-PTX NPs and PTX loading capacity were calculated.28
PTX Release from 2DA-FITC-PTX NPs
The PTX release feature of self-assembled targeting 2-DA-FITC-PTX NPs was determined by HPLC. Two dialysis cassettes were incubated in 50 mL of pH 7.429 and 5.5 buffer solutions30 at 300 rpm stirring speed. At pre-planned time points of 0, 0.05, 0.10, 0.5, 1, 2, 4, 8, 12, 24, 48, and 72 h, 100 μL of solution was removed to evaluate the released PTX amount by reversed-phase HPLC, and 100 μL of fresh buffer solution was simultaneously re-added into the cassettes.31
Cell Culture
Human breast cancer (MDA-MB-231) and human renal epithelial (293T) cell lines were purchased from ATCC. The cell line was cultured at 37°C in a humidified atmosphere containing 5% CO2 in DMEM medium supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin.
In vitro Targeting Ability Assessment of 2DA-FITC-PTX NPs
MDA-MD-231 and 293T cells were incubated in media with 90% MEM and 10% FBS at 37°C in 5% CO2 environment. The expression levels of GLUT1 in MDA-MB-231 and 293T cell lines were assessed by reverse transcriptase PCR. A four-well confocal microscopy plate was added into 104/well MDA-MB-231cells for 24 h. The same PTX concentrations (6.4 µmol/L) of 2DA-FITC-PTX and 2DA-FITC-PTX NPs were planted into the four wells. 2DA-FITC-PTX and 2DA-FITC-PTX NPs were cultured with cells for 1, 4, and 8 h. Then, PBS was used in all cells to remove free 2DA-FITC-PTX or 2DA-FITC-PTX NPs thrice, and fixed by Z-Fix solution at 37°C for 15 min. All cells were stained with DAPI for 30 min and then observed with a fluorescence microscope (Zeiss LSM 780).
In vitro Toxicity of 2DA-FITC-PTX NPs
293T and MDA-MB-231 cells were cultured with 2DA-FITC-PTX and 2DA-FITC-PTX NPs, respectively. After 48 h of incubation, the cells were handed with PBS thrice, and 180 μL of fresh medium in the wells was re-added and supplied with 20 μL MTT solution (5 mg/mL). After continuous culture for 4 h, each well was added to 150 L of DMSO and stirred gently at room temperature. Finally, each well with solution was evaluated using a microplate reader at absorbance 595 nm.
Cell Apoptosis Study of 2DA-PTX NPs
Cell apoptosis was qualitatively analyzed using a flow cytometer. Each well of MDA-MB-231 cells at 105 density was added into a six-well plate and then incubated for 24 h at 37°C. Cell media containing the same concentrations of PTX (0.64 μM) of 2DA-PTX or 2DA-PTX NPs were added and co-incubated for 24 or 48 h. After incubation, the cells were handed with PBS twice, and then 500 μL of binding buffer was seeded into each well. SYTOX green (5 μL) and Annexin V-PE (1 μL) from an apoptosis kit were added separately, and the results of quantitative apoptosis were graphed using flow cytometry. The cell morphology during apoptosis was determined through confocal microscopy.32
Tumor Model
BALB/c athymic nude mice (female) were purchased from Charles River Laboratories (Shanghai, China). All animals were maintained based on the Animal Management Rules of the Ministry of Health of the People’s Republic of China (Document No. 55, 2001) and the guidelines for the Care and Use of Laboratory Animals of Suzhou University. The animal experiment protocol was authorized by the Animal Experimental Ethics Committee of Suzhou University (No. 2016–0180).
When the tumor volumes reached 80–150 mm3, the nude mice were used to evaluate the targeting ability and therapeutic properties of 2DA-FITC-PTX or 2DA-FITC-PTX NPs.
Acute Toxicity Study
The in vivo acute toxicity of PTX, 2DA-FITC-PTX, and 2DA-FITC-PTX NPs on BALB/c mice was evaluated. A total of 20 BALB/c mice (5 weeks, 20 g) were randomly divided into three groups, which were treated with PBS, PTX, 2DA-FITC-PTX, and 2DA-FITC-PTX NPs. Exactly 200 µL each of 6 mg/mL PTX, 2DA-FITC-PTX, and 2DA-FITC-PTX NPs (equal to PTX: 6 mg/kg) was intravenously injected into the mice. Control group mice were injected with 200 µL of PBS. Post-injection blood at 1, 3, and 7 days was collected from the eye socket to assess the biochemical parameters, including aminotransferase (AST) and alanine aminotransferase (ALT). All mice were sacrificed 7 days after injection, and liver tissues were sectioned into slices for hematoxylin and eosin (H&E) analysis.
Antitumor Activity
MDA-MB-231 tumor-bearing mice were randomly assigned to four groups (n = 5 per group). Each group was injected with PTX prodrugs by tail vein injection of PBS buffer (pH 7.4, control), pure PTX solution (6 mg/kg), 2DA-FITC-PTX (equal to PTX: 6 mg/kg), and 2DA-FITC-PTX NPs (equal to PTX: 6 mg/kg). MDA-MB-231 tumor-bearing mice underwent three intravenous injections every other day. The therapeutic efficacies of PTX nanoparticles and toxicities on the MDA-MB-231 tumor-bearing mice were estimated by measuring the tumor volume and body weight of each mouse every other day. To further evaluate the anti-tumor effect of PTX NPs, we excised the liver, kidney, and tumor tissues for pathology and measurement of p53 protein levels.33
Targeting Ability of 2DA-FITC-PTX NPs
The targeting ability of 2DA-FITC-PTX and 2DA-FITC-PTX NPs was evaluated by bearing MDA-MB-231 tumors (n = 4 per group). 2DA-FITC-PTX (0.2 mL, equal to PTX 6 mg/kg) and 2DA-FITC-PTX NPs (0.2 mL, equal to PTX 6 mg/kg) were administered to the mice by tail vein injection. At 48 h and 72 h after injection, the tumor tissues of the sacrificed mice were directly visualized by fluorescence microscopy.
In vivo Near-Infrared (NIR) Fluorescent Imaging
For NIR fluorescent imaging, the ICG-labelled 2DA-ICG-PTX or 2DA-ICG-PTX NPs were injected into the MDA-MB-231 xenograft tumor mice intravenously. The images were obtained using a Maestro II imaging system (PerkinElmer) with excitation filter of 661 nm and emission filter of 700 nm longpass at 1, 4, 24, 48, and 72 h post-injection.
To quantify the targeting ability of these drugs in MDA-MB-231 tumor, we determined the average fluorescence intensity in tumor regions by selecting the region of interest (ROI) and comparing them with that in normal muscle tissue. Tumor-to-normal tissue contrast ratios (T/N) were calculated using the ROI functions of Living Image software.34
Statistical Analysis
Statistical analysis was performed using Student’s t-test with statistical significance assigned at *P < 0.05 (significant), **P < 0.01 (moderately significant), and ***P < 0.001 (highly significant).
Results
Characterization of Amphoteric Small-Molecule-Based Self-Assembled Nanoplatform
The 2DA-FITC-PTX was synthesized by following the procedures described in Figure S1. Fmoc-Glu(OtBu)-COOH was used as linker to conjugate 2DA (Figure 1A), FITC-Arg (Figure 1B), and PTX-SUC (Figure 1C) to form the targeted amphoteric small-molecule 2DA-FITC-PTX (Figure 1D), and then further self-assembled to DDS (Figure 2A). In this NP delivery system, on the one hand, the high-cytotoxicity PTX functions as a core of 2DA-FITC-PTX NPs, leading to the low toxicity to normal cells or tissues. On the other hand, 2DA-FITC-PTX NPs could disperse to single-targeting amphoteric small molecule, and the release of PTX could efficiently inhibit the activity of tumor cells (Figure 2D). Most importantly, malignant tumor cells are characterized by the “Warburg effect”. Therefore, 2DA was used as the targeted group to enhance the tumor accumulation and retention time of 2DA-FITC-PTX NPs, which are beneficial for cancer treatment.
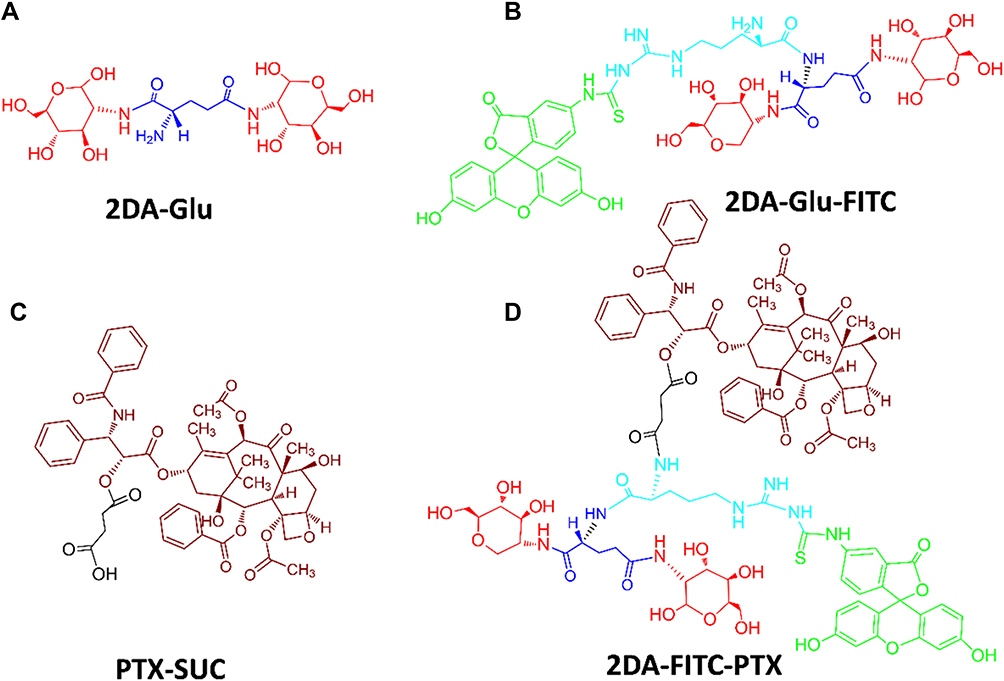 |
Figure 1 Chemical structures of 2DA-Glu (A), 2DA-Glu-FITC (B), PTX-SUC (C) and 2DA-FITC-PTX (D). Abbreviations: 2DA, 2-glucosamine, red; FITC, fluorescein-5(6)-isothiocyanate, green; PTX, paclitaxel, crimson; Glu, glutamic acid, linker, blue; Arg, arginine, linker, light blue; SUC, succinic anhydride, linker, black. |
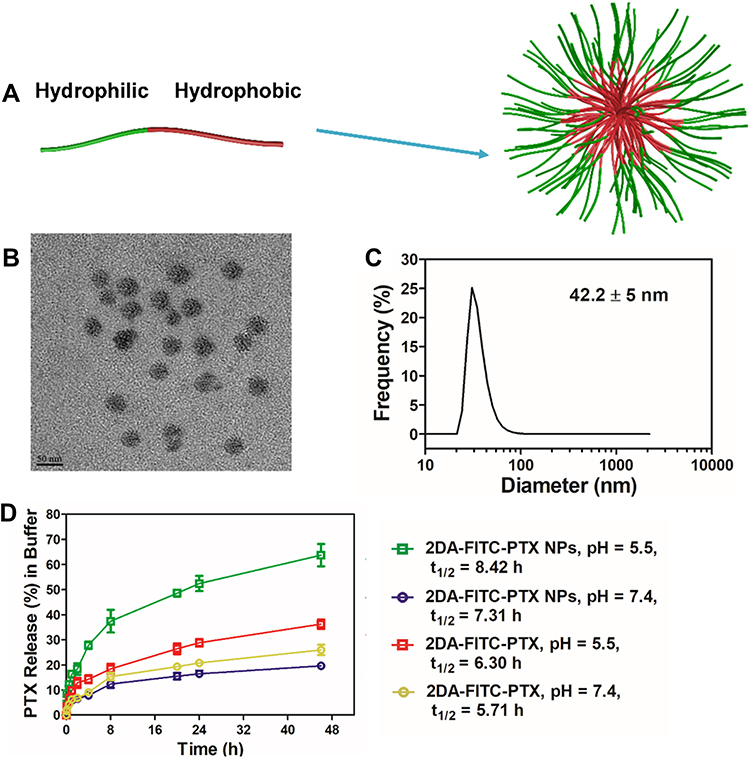 |
Figure 2 Formulation and characterization of prodrug-based nanoparticles. (A) Schematic illustration of the procedure for self-assembly of 2DA-FITC-PTX NPs. (B) TEM images of 2DA-FITC-PTX NPs. (C) DLS of 2DA-FITC-PTX NPs. (D) In vitro release of PTX in different buffer. PTX concentration was measured by HPLC. The release was expressed as mean ± standard deviation based on triplicate experiments. Abbreviations: 2DA-FITC-PTX NPs, 2-glucosamine-fluorescein-5(6)-isothiocyanate-glutamic acid-paclitaxel nanoparticles; TEM, transmission electron microscopy; DLS, dynamic light scattering; PTX, paclitaxel; HPLC, high-performance liquid chromatography. |
The purified 2DA-Glu, PTX-SUC, and 2DA-Glu-FITC samples were evaluated by LC-MS; the results were 468.4 ([M+H]+, Figure S2), 952.9 ([M+H]+, Figure S3) and 998.01 ([M+H]+, Figure S4). These values were identical to those of the theoretical molecule, as shown in Figures S1, S2, and S3. The structure of 2DA-FITC-PTX prodrug monomer was identified by NMR 1H (Figure S5). Based on the hydrogen spectrum shown in Figure S4, the covalent coupling 2-OH’ position between the hydrophilic end and hydrophobic end is between 4.77 ppm and 5.44 ppm, indicating the synthesis of this small-molecule monomer. The particle size of 2DA-FITC-PTX NPs was measured by Malvern particle size analyzer, as shown in Figure 2C. The results showed that the particle size (n = 4) of the DDS of targeting nanoparticles was 42.2 ± 5 nm, and the TEM images of 2DA-FITC-PTX NPs are provided in Figure 2B. The zeta potential (mV) was at ~16Mv positively charged (Figure S6).
The drug loading efficiency, capacity, and stability of 2DA-FITC-PTX NP water solution at different concentrations were measured at RT or 4°C for 2, 4, and 8 weeks. The drug loading efficiency shows the percentage (%) PTX drug in 2DA-FITC-PTX that is successfully entrapped in the 2DA-FITC-PTX NPs. Loading capacity helps to calculate the PTX drug content in 2DA-FITC-PTX NPs after their separation from the medium.28 The corresponding calculation formula is shown in Table S1. Results in Table S1 show that the drug loading efficiency was more than 95% with different concentrations of 2DA-FITC-PTX NPs solution. The 2DA-FITC-PTX NPs exhibited high drug loading capacity at 66.2% with the concentration of 2DA-FITC-PTX 50 μg/mL. Whether at room temperature (RT) or 4°C, all 2DA-FITC-PTX NP water solutions displayed good stability and their particle size did not change significantly (Table S1).
To study the drug release characteristics of 2DA-FITC-PTX NPs, we detected paclitaxel by HPLC after incubation in PBS buffer at different pH values. In the simulated physiological environment PBS buffer at pH = 7.4, the release rate of 2DA-FITC-PTX NPs or 2DA-FITC-PTX was slow, and sustained release was observed regardless of the presence of duration in PBS buffer with pH 7.4, as shown in Figure 2D. Moreover, the release rate was 17.2% or 20.2% (at 24 h) and 18.6% or 24.5% (at 46 h) with half-lives of 7.31 h (2DA-FITC-PTX NPs) and 5.71 h (2DA-FITC-PTX), respectively. However, the 2DA-FITC-PTX NPs or 2DA-FITC-PTX had a faster release rate in acidic PBS buffer (pH = 5.5); the release rates were 52.8% and 31.2% at 24 h, and the half-lives were 8.42 h and 6.3 h. This finding indicates that the acidic conditions and enzyme-containing serum favor the cleavage of the ester bond and contribute to the release of the paclitaxel drug.
Cell Uptake
To study prodrug-based NP DDS cellular uptake of 2DA-FITC-PTX and 2DA-FITC-PTX NPs, we evaluated the GLUT1 expression levels on different cells. The results displayed that the GLUT1 expression levels were higher in MDA-MB-231 tumor cells than in 293T normal cell lines, as shown in Figure S7.
The 2DA-FITC-PTX NPs and 2DA-FITC-PTX cell uptakes were measured via confocal microscopy and flow cytometry. 2DA-FITC-PTX NPs and 2DA-FITC-PTX were incubated in MDA-MB-231 cells at 1, 4, and 8 h. As shown in Figure 3A and B, the efficient images were observed from cell uptakes in 2DA-FITC-PTX NPs and 2DA-FITC-PTX by confocal microscopy. The data of flow cytometry proved that cell uptake increased sharply with time (Figure 3C and D). When the treatment time was delayed from 1 h to 8 h, fluorescence of FITC was observed around the blue cell nucleus (Figure 3A and B), explaining the release of PTX from the 2DA-FITC-PTX NPs or 2DA-FITC-PTX in the cytoplasm.
 |
Figure 3 Evaluation of cell uptake of FITC-labeled 2DA-PTX or 2DA-PTX NPs after incubation with MDA-MB-231 cells for 1, 4, and 8 h via confocal microscopy and flow cytometry analysis. The confocal microscopy images of MDA-MB-231 cell uptake from 2DA-FITC-PTX NPs (A) and 2DA-FITC-PTX (B) following various incubation times. Both the 2DA-FITC-PTX NPs and 2DA-FITC-PTX were labeled with FITC (green). The blue fluorescence was from the cell nucleus (DAPI, blue). Scale bars are 21 μm. The flow cytometric analyses of cell uptake from 2DA-FITC-PTX NPs (C) and 2DA-FITC-PTX (D). Abbreviations: 2DA-FITC-PTX NPs, 2-glucosamine-fluorescein-5(6)-isothiocyanate-glutamic acid-paclitaxel nanoparticles; 2DA-FITC-PTX, 2-glucosamine-fluorescein-5(6)-isothiocyanate-glutamic acid-paclitaxel; FITC, fluorescein isothiocyanate; DAPI, 4,6-diamino-2-phenyl indole. |
Thereafter, quantitative determination of cell uptake was measured by flow cytometry. Results showed that the fluorescence intensity of 2DA-FITC-PTX NPs uptake in MDA-MB-231 cancer cells was significantly shifted, as shown in Figure 3C and D. The cell uptake of 2DA-FITC-PTX NPs in MDA-MB-231 tumor cells was greater than that of 2DA-FITC-PTX. Thus, 2DA-FITC-PTX NPs could target tumor cells accurately as mediated by the high expression of GLUT1 transport receptor on the surface of the tumor cells.
Studies on Cytotoxicity and Cell Apoptosis
MDA-MB-231 and 293T two cell lines were used to measure the in vitro cytotoxic effect of 2DA-FITC-PTX. Based on Figure 4A and B, PTX, 2DA-FITC-PTX, and 2DA-FITC-PTX NPs displayed dose-dependent toxicity from tumor and normal cell lines. In particular, in the tumor cells of MDA-MB-231, 2DA-FITC-PTX NPs showed inhibition ratios above 90%. The maximum inhibition ratio of PTX and 2DA-FITC-PTX was 65% and 70% of MDA-MB-231 cells, respectively. Low toxicity was observed for 293T cell from both 2DA-FITC-PTX and 2DA-FITC-PTX NPs. The half-maximal inhibitory concentration (IC50) of free PTX was higher than that of 2DA-FITC-PTX or 2DA-FITC-PTX NPs, indicating the targeted tumor effect of 2DA-FITC-PTX or 2DA-FITC-PTX NPs (Figure 4C). Interestingly, 2DA-FITC-PTX or 2DA-FITC-PTX NPs showed relatively lower inhibition ratio to 293T normal cells compared with MDA-MB-231 tumor cells with the same drug doses; this condition can be attributed to the relatively low expression of GLUT1 in 293T normal cells.
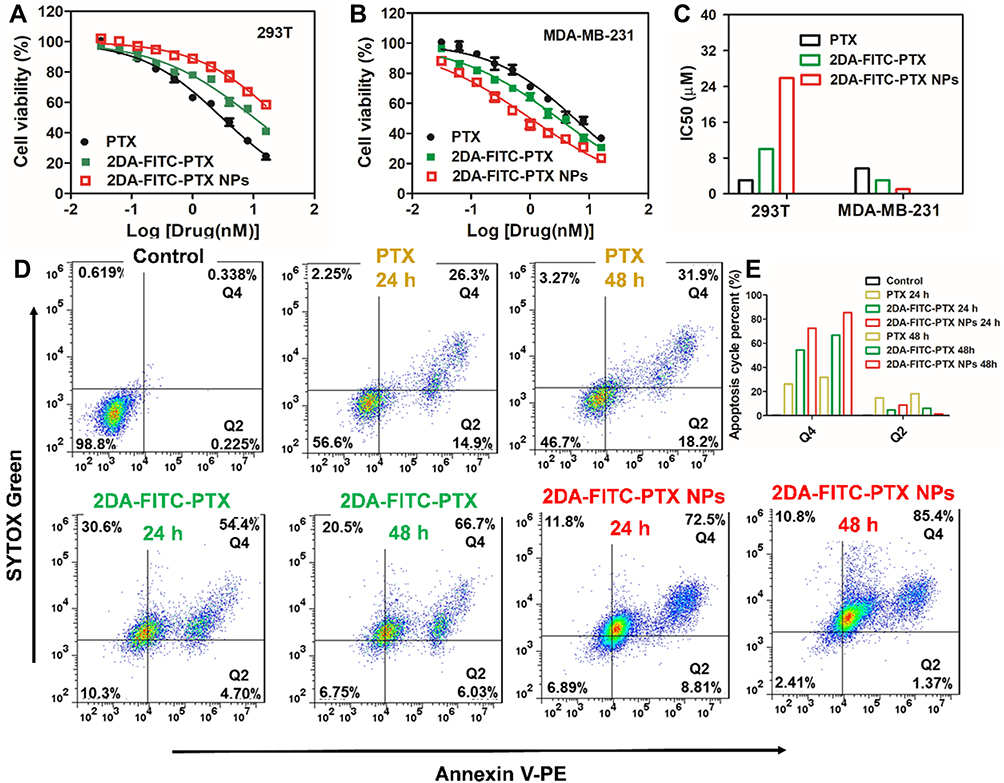 |
Figure 4 Cell viability and apoptosis analysis after treatment with 2DA-FITC-PTX and 2DA-FITC-PTX NPs. Cell viability of 293T cells (A) and MDA-MB-321 cells (B) following incubation with free PTX drug, 2DA-FITC-PTX, and 2DA-FITC-PTX NPs for 48 h. The IC50 of free PTX drug, 2DA-FITC-PTX, and 2DA-FITC-PTX NPs for 293T cells and MDA-MB-231 (C). The cell apoptosis of MDA-MB-231 cells following incubation with free PTX drug, 2DA-FITC-PTX, and 2DA-FITC-PTX NPs for 24 and 48 h (D), and the apoptosis cycle percentage in Q2 and Q4 zones (E). Abbreviations: PTX, paclitaxel; 2DA-FITC-PTX, 2-glucosamine-fluorescein-5(6)-isothiocyanate-glutamic acid-paclitaxel; 2DA-FITC-PTX NPs, 2-glucosamine-fluorescein-5(6)-isothiocyanate-glutamic acid-paclitaxel nanoparticles, and the PE Annexin V/Dead Cell Apoptosis Kit with SYTOX® green dye for flow cytometry. |
Cell apoptosis study of MDA-MB-231 was conducted using the dead cell apoptosis kit. Based on Figure 4D and E, the quantitative flow cytometry results proved that the cells underwent early apoptosis (Q2) to late apoptosis (Q4). The percentage of apoptotic cells increased with incubation time, and the highest percentage of apoptosis of 2DA-FITC-PTX NPs was observed at 48 h post-incubation. All results showed that the 2DA-FITC-PTX NPs or 2DA-FITC-PTX displayed relatively low cytotoxicity to normal cells and increased cytotoxicity to tumor cells with the overexpression of GLUT1 transporter.35,36
In vivo Toxicity and Targeting Ability Studies
The mice blood from the aforementioned groups was analyzed at 1, 3, and 7 days post-injection. As shown in Figure 5A and B, serious hepatotoxicity was observed from the free PTX treatment group with significantly increased levels of AST and ALT. However, the AST and ALT of mice from 2DA-FITC-PTX NPs or 2DA-FITC-PTX were within the normal range. The results support that 2DA-FITC-PTX NPs or 2DA-FITC-PTX could significantly reduce the toxicity of PTX to blood cells. The hepatotoxicity was further evaluated by H&E staining of liver tissue after euthanasia of the mice at 7 days post-injection (Figure S8). The free PTX caused slight damage to the liver. No obvious histological signs were observed in the 2DA-FITC-PTX or 2DA-FITC-PTX group of liver damage.
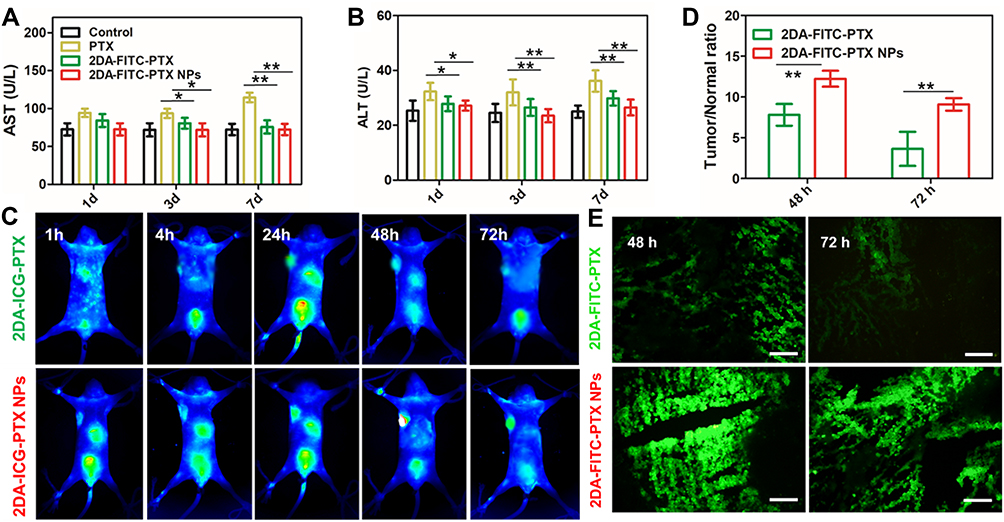 |
Figure 5 Acute toxicity and in vivo NIR and optical imaging analysis. (A) Blood biochemistry test of AST (A) and ALT (B) after various treatments (n = 4). (C) In vivo NIR imaging in MDA-MB-231 xenograft tumor mice (n = 4). (D) Time courses of tumor-to-normal tissue (T/N) ratio in MDA-MB-231 tumor for 2DA-ICG-PTX and 2DA-ICG-PTX NPs. Statistical analysis indicates significant difference of T/N ratio in the MDA-MB-231tumors between the two drugs (n = 4). (E) Ex vivo fluorescence images of tumor tissues after 48, 72 h post-injection of FITC-labelled 2DA-FITC-PTX and 2DA-FITC-PTX NPs intravenously (n = 4). *P < 0.05; **P < 0.01. Scale bars are 100 μm. Abbreviations: AST, aminotransferase; ALT, alanine aminotransferase; NIR, near-infrared; 2DA-ICG-PTX, 2-glucosamine-fluorescein-5(6)-isothiocyanate-glutamic acid-paclitaxel, 2DA-ICG-PTX NPs, 2-glucosamine-fluorescein-5(6)-isothiocyanate-glutamic acid-paclitaxel nanoparticles. |
In vivo targeting ability studies were further measured in detail. First, the NIR fluorescence imaging was collected by both the ICG-labelled 2DA-PTX to form the structure of 2DA-ICG-PTX and 2DA-ICG-PTX NPs. The MDA-MB-231 tumor-bearing mice were injected into the 2DA-ICG-PTX NPs and 2DA-ICG-PTX through the tail vein (n = 4 per group). The mice were observed at various post-injection time points. The quantitative NIR imaging results are shown in Figure 5C, indicating whether 2DA-ICG-PTX NPs or 2DA-ICG-PTX could stay in the tumor tissues for 72 h. The targeting ability of 2DA-ICG-PTX NPs or 2DA-ICG-PTX was quantified using ROI to analyze the fluorescence intensity in tumor tissues. The dynamics of 2DA-ICG-PTX NPs or 2DA-ICG-PTX signals in tumor tissues are described in Figure 5C and D. The NIR image from 2DA-ICG-PTX or 2DA-ICG-PTX NPs reached the maximum peak at 7.81 ± 1.34 (T/N, fluorescence intensity ratio between tumor and normal tissue) and 12.23 ± 0.98, respectively, at 48 h post-injection. 2DA-ICG-PTX slowly decreased to 3.36 ± 0.82 by 72 h, and that of 2DA-ICG-PTX NPs gradually declined to 9.09 ± 0.76 until 72 h. Compared with 2DA-ICG-PTX, the 2DA-ICG-PTX NPs showed good tumor tissue accumulation due to the nanoplatform.37,38
According to the above in vivo NIR imaging studies, after the injection of 2DA-FITC-PTX and 2DA-FITC-PTX NPs at 48 h, mice bearing MDA-MB-231 tumor xenografts were sacrificed, and the excised tumor tissues were sectioned. According to 2DA-PTX or 2DA-PTX NPs labeled FITC, these tissue sections were observed by fluorescence microscopy. In Figure 5E, compared with 2DA-FITC-PTX, the highest fluorescence signal was observed in tumor tissues for 2DA-FITC-PTX NPs, suggesting the higher uptake of 2DA-FITC-PTX NPs than 2DA-FITC-PTX. Thus, 2DA-FITC-PTX NPs could enhance the drug accumulation and retention time.
Antitumor Activity of 2DA-FITC-PTX NPs
The tumor inhibition was further evaluated by injecting 2DA-FITC-PTX NPs and 2DA-FITC-PTX to MDA-MB-231 tumor-bearing mice. All mice samples were randomly divided into three groups and administered with PTX, 2DA-FITC-PTX, and 2DA-FITC-PTX NPs intravenously. The tumor size and body weight were measured every other day. As shown in Figure 6A and B, compared with the free drug group, 2DA-FITC-PTX NPs and 2DA-FITC-PTX could inhibit tumor growth significantly due to their targeting ability and high tumor accumulation. The 2DA-FITC-PTX NPs exhibited the highest tumor growth inhibition rates (75.64%), followed by 2DA-FITC-PTX (59.41%) and PTX (50.82%), supporting the significantly enhanced tumor therapy (Figure 5D). Moreover, the 2DA-FITC-PTX NPs group could remarkably extend the mice survival time (Figure 6C). Interestingly, no obvious body weight loss was observed from the 2DA-FITC-PTX NPs group (Figure 6B). By contrast, the mice body weight decreased with free PTX administration due to the severe side effects of PTX. The representative tumor tissue sections from various treatment groups are provided in Figure 6E. Compared with the other free drug administration group, the tumor tissues from the 2DA-FITC-PTX NPs groups exhibited high populations of dead cells, intercellular blank spots, and necrosis, indicating the effective tumor therapy. All results could be attributed to the high tumor aggregation of 2DA-FITC-PTX and 2DA-FITC-PTX NPs, which can target to release PTX. Meanwhile, the p53 protein expression levels of the 2DA-FITC-PTX and 2DA-FITC-PTX NPs groups were enhanced, supporting the critical damage in tumor cells (Figure 6F). The liver and kidney tissues of mice from all the treatment groups were excised and evaluated through histopathological examination after the treatment (Figure S9). No significant pathologic changes were observed in the liver and kidney of the mice administered with PTX, 2DA-FITC-PTX, and 2DA-FITC-ICG NPs.
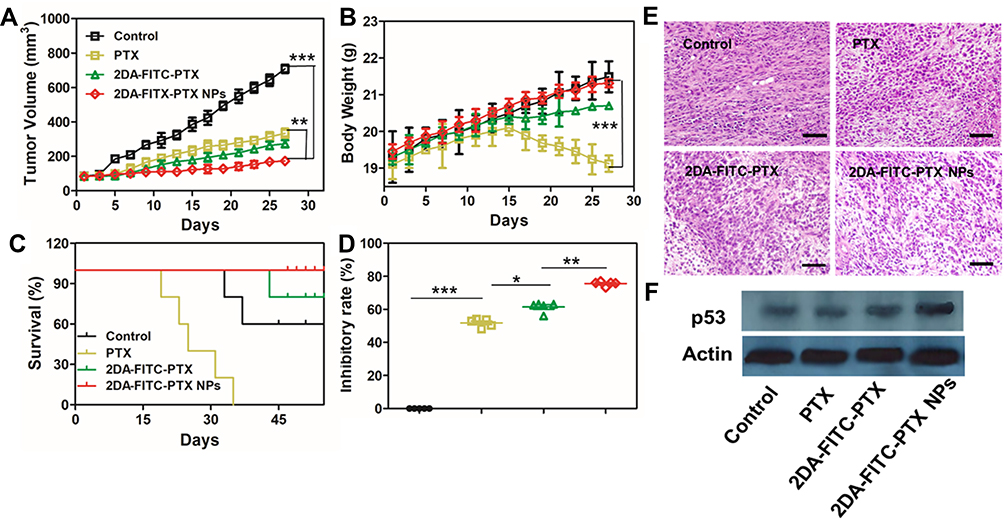 |
Figure 6 In vivo antitumor evaluation. (A) MDA-MB-231 tumor growth curves, (B) body weight with various treatments, (C) survival curve, and (D) tumor growth inhibition rates (n = 5). *P < 0.05 and **P < 0.01. (E) Tumor tissue images under various treatments. Scale bars are 100 μm. (F) Tumor tissue Western blotting results of p53 expression under various treatments: *P < 0.05, **P < 0.01, and ***P < 0.001. Abbreviations: PTX, paclitaxel; 2DA-ICG-PTX, 2-glucosamine-fluorescein-5(6)-isothiocyanate-glutamic acid-paclitaxel; 2DA-ICG-PTX NPs, 2-glucosamine-fluorescein-5(6)-isothiocyanate-glutamic acid-paclitaxel nanoparticles. |
Discussion
Targeted drug delivery is an important issue that can promote enhanced drug accumulation in tumors through the selectivity of drugs to tumor sites and decreased toxicity for normal cells. Prodrug-based nanoparticles can induce drug release in target cells and guarantee the enhanced drug accumulation in the tumor by enhanced permeability and retention (EPR) effect.39,40 In this study, we developed a new class of nano-DDS based on targeted prodrug, which utilized hydrophilic–hydrophobic conjugated amphiphilic nature to form the nano-DDS.18 The delivery drug system for 2DA-FITC-PTX NPs displayed a strong self-assembly, which significantly enhanced the aqueous solubility of PTX, drug release half-life, and tumor uptake. The FITC fluorescence signal of 2DA-FITC-PTX or 2DA-FITC-PTX NPs was used to study the dynamic distribution of the prodrug-based DDS at the cellular and tissue levels.
The design of prodrug-based DDS aims to enhance the PTX drug release half-life and tumor site retention time and reduce the side effect toxicity of free PTX to normal tissue. Characterized by high stability and high loading efficiency, small-sized 2DA-FITC-PTX NPs may preferentially serve as an advanced DDS because it accumulates in the tumor for a long time. In vivo pharmacokinetic and tissue distribution were studied, thus strongly supporting the enhanced drug delivery in tumor sites.41,42 Notably, the ICG/FITC fluorescence intensity of 2DA-ICG/FITC-PTX in the tumor section was lower than that of 2DA-ICG/FITC-PTX NPs at 48 h post-injection (Figure 5C and E). Similar results were observed in in vitro studies. The cellular uptake of prodrug-based 2DA-FITC-PTX NPs was qualitatively and quantitatively analyzed (Figure 2). The percentage of cell uptake of 2DA-FITC-PTX NPs (23.2%, 45.4%, and 75.1%) was far higher than that of 2DA-FITC-PTX (11.5%, 21.2%, and 35.8%). Prodrug-based DDS significantly enhanced the targeting ability of tumors via receptor-mediated uptake and EPR effect as evidenced by fluorescence microscopy. These results prove that prodrug-based DDS enhances the targeting ability of PTX delivery to tumor sites. Overall, this functional prodrug delivery system can combine the prodrug and nanoparticle advantage to enhance the drug retention time and concentration in tumor tissue.
Paclitaxel is among the most promising antitumor drugs as a microtubule stabilizer, and it plays a key role in the clinical treatment of breast cancer.43 Compared with free PTX, the IC50 value of prodrug-based DDS has a lower value of 1.01 nM, indicating the enhancement of cytotoxicity via nanoparticles. Interestingly, the IC50 values of 2DA-FITC-PTX NPs, as a prodrug-based DDS, were approximately 8-fold higher than free PTX, 25.8 nM in normal cells, thereby supporting the specificity of drug cytotoxicity (Figure 4C). Prodrug-based DDS is more effective than 2DA-FITC-PTX or free PTX in vitro. Therefore, prodrug-based NP DDS increased the intracellular delivery of 2DA-FITC-PTX prodrugs, thus increasing the cytotoxicity in tumor cells. The similar percentage of late apoptotic cells (PI+) in tumor groups was higher for the DDS formulations. The percentage of necrotic/dead cells (Q2 + Q4) was 86.7% for 2DA-FITC-PTX NPs compared with 50.1% or 70.2% in free PTX or 2DA-FITC-PTX prodrug treated cells (MDA-MB-231) at 48 h, respectively (Figure 4E).
The in vivo anticancer efficacy of 2DA-FITC-PTX NPs was confirmed with nude mice-bearing MDA-MB-231 tumor model by tail vein injection at an equivalent dose of 6 mg/kg PTX (Figure 6). Compared with the free PTX group, the 2DA-FITC-PTX NPs group had a remarkable inhibitory effect. Moreover, the inhibitory effect of tumor is related to apoptosis in tumor tissues (Figure 6E). Importantly, no obvious changes in mice body weight were displayed after treatment with 2DA-FITC-PTX or 2DA-FITC-PTX NPs. The tumor inhibition rate of 2DA-FITC-PTX NPs reached 75.64%, which was approximately 1.2 times higher than that of 2DA-FITC-PTX and 1.5 times higher than that of PTX. Thus, prodrug-based nano-DDS synergistically strengthened the antitumor effect of 2DA-FITC-PTX prodrugs.
The mouse weight of the free PTX treatment group dropped obviously, confirming that free PTX has serious side effects and toxicity. The average body weight of the PTX group was 18.67 ± 0.01 mg, accounting for a 2.8% loss of the original weight. By contrast, the increase in body weight of other groups treated with 2DA-FITC-PTX and 2DA-FITC-PTX NPs showed that the prodrug-based nano-DDS improved the pharmacokinetic properties and biodistribution of PTX, thereby causing low toxicity. The H&E staining section showed no histological change of toxicity in major organs for all groups. However, necrotic areas were clearly shown in the tumor tissues treated with 2DA-FITC-PTX NPs. The antitumor effects of prodrug-based nano-DDS were observed by p53 protein expression analysis (Figure 6F). The enhanced therapeutic effect of the 2DA-FITC-PTX NPs might be due to the synergistic function of targeting, prodrug, EPR effect, GLUT1-mediated targeted endocytosis, and the prodrug-based nano-DDS-enhanced tumor retention.44
Conclusions
Overall, we have developed the advantages of a prodrug-based nano-DDS as a multifunctional nano-DDS for active and passive targeted delivery of PTX, resulting in improved pharmacokinetics, high antitumor activity, and low toxicity. Furthermore, compared with free PTX or prodrug formulations, prodrug-based 2DA-FITC-PTX NP also exhibited high tumor retention and antitumor activity and reduced toxicity. Therefore, multifunctional targeting nano-DDS is expected to develop the clinical implications of prodrug-based nano-platforms.
Acknowledgments
We gratefully acknowledge support from the Program of Study for Outstanding Young Scholar sponsored in Universities by Education Department of Anhui Province (gxgnfx2019051), National Science Foundation Committee of Anhui Province (1808085MH256, 1908085MC100), Open fund project of national and local joint engineering laboratory for crop stress resistance breeding and disaster reduction (NELCOF20190108) and Suzhou science-technology plan projects (2019071, 2019087), and the Innovation Team of Pharmaceutical Biotechnology (2016KYTD01), Innovative Research Team of Anhui Provincial Education Department (2016SCXPTTD), Key Discipline of Material Science and Engineering of Suzhou University (2017XJZDXK3, 2017yzd11, 2019jb06).
Disclosure
The authors declare no competing financial interest.
References
1. Walther R, Rautio J, Zelikin AN. Prodrugs in medicinal chemistry and enzyme prodrug therapies. Adv Drug Deliv Rev. 2017;118:65–77. doi:10.1016/j.addr.2017.06.013
2. Ruparelia KC, Zeka K, Ijaz T, et al. The synthesis of chalcones as anticancer prodrugs and their bioactivation in cyp1 expressing breast cancer cells. Med Chem. 2018;14(4):322–332. doi:10.2174/1573406414666180112120134
3. Wang Y, Cheetham AG, Angacian G, et al. Peptide-drug conjugates as effective prodrug strategies for targeted delivery. Adv Drug Deliv Rev. 2017;110-111:112–126. doi:10.1016/j.addr.2016.06.015
4. Lee MH, Sharma A, Chang MJ, et al. Fluorogenic reaction-based prodrug conjugates as targeted cancer theranostics. Chem Soc Rev. 2018;47(1):28–52. doi:10.1039/c7cs00557a
5. Li H, Zhang P, Luo J, et al. Chondroitin sulfate-linked prodrug nanoparticles target the golgi apparatus for cancer metastasis treatment. ACS Nano. 2019;13(8):9386–9396. doi:10.1021/acsnano.9b04166
6. Barhoumi A, Wang W, Zurakowski D, et al. Photothermally targeted thermosensitive polymer-masked nanoparticles. Nano Lett. 2014;14(7):3697–3701. doi:10.1021/nl403733z
7. Kalam MA, Alshamsan A. Poly (d, l-lactide-co-glycolide) nanoparticles for sustained release of tacrolimus in rabbit eyes. Biomed Pharmacother. 2017;94:402–411. doi:10.1016/j.biopha.2017.07.110
8. Xiao H, Guo Y, Liu H, et al. Structure-based design of charge-conversional drug self-delivery systems for better targeted cancer therapy. Biomaterials. 2019;232:119701. doi:10.1016/j.biomaterials.2019.119701
9. Shi J, Kantoff PW, Wooster R, et al. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. 2017;17(1):20–37. doi:10.1038/nrc.2016.108
10. Jing F, Guo Q, Xu W, et al. Docetaxel prodrug self-assembled nanosystem: synthesis, formulation and cytotoxicity. Bioorg Med Chem Lett. 2018;28(4):826–830. doi:10.1016/j.bmcl.2017.07.041
11. Zhang H, Zhu Y, Sun C, et al. GSH responsive nanomedicines self-assembled from small molecule prodrug alleviate the toxicity of cardiac glycosides as potent cancer drugs. Int J Pharm. 2019;575:118980. doi:10.1016/j.ijpharm.2019.118980
12. Cui D, Huang J, Zhen X, et al. A semiconducting polymer nano-prodrug for hypoxia-activated photodynamic cancer therapy. Angew Chem Int Ed Engl. 2019;58(18):5920–5924. doi:10.1002/anie.201814730
13. Nishimura T, Sasaki Y, Akiyoshi K. Biotransporting self-assembled nanofactories using polymer vesicles with molecular permeability for enzyme prodrug cancer therapy. Adv Mater. 2017;29(36):24–43. doi:10.1002/adma.201702406
14. Dai L, Si C. Recent advances on cellulose-based nano-drug delivery systems: design of prodrugs and nanoparticles. Curr Med Chem. 2019;26(14):2410–2429. doi:10.2174/0929867324666170711131353
15. Li H, Zhao Y, Jia Y, et al. Covalently assembled dopamine nanoparticle as an intrinsic photosensitizer and pH-responsive nanocarrier for potential application in anticancer therapy. Chem Commun. 2019;55(100):15057–15060. doi:10.1039/c9cc08294h
16. Wang Y, Huang P, Hu M, et al. Self-delivery nanoparticles of amphiphilic methotrexate-gemcitabine prodrug for synergistic combination chemotherapy via effect of deoxyribonucleotide pools. Bioconjug Chem. 2016;27(11):2722–2733. doi:10.1021/acs.bioconjchem.6b00503
17. Dong S, He J, Sun Y, et al. Efficient click synthesis of a protonized and reduction-sensitive amphiphilic small-molecule prodrug containing camptothecin and gemcitabine for a drug self-delivery system. Mol Pharm. 2019;16(9):3770–3779. doi:10.1021/acs.molpharmaceut.9b00349
18. Huang P, Wang DL, Su Y, et al. Combination of small molecule prodrug and nanodrug delivery: amphiphilic drug-drug conjugate for cancer therapy. J Am Chem Soc. 2014;136(33):11748–11756. doi:10.1021/ja505212y
19. Tekade RK, Sun X. The Warburg effect and glucose-derived cancer theranostics. Drug Discov Today. 2017;22(11):1637–1653. doi:10.1016/j.drudis.2017.08.003
20. Hamann I, Krys D, Glubrecht D, et al. Expression and function of hexose transporters GLUT1, GLUT2, and GLUT5 in breast cancer-effects of hypoxia. FASEB J. 2018;32(9):5104–5118. doi:10.1096/fj.201800360R
21. Rivenzon-Segal D, Boldin-Adamsky S, Seger D, et al. Glycolysis and glucose transporter 1 as markers of response to hormonal therapy in breast cancer. Int J Cancer. 2003;107(2):177–182. doi:10.1002/ijc.11387
22. Almahmoud S, Wang X, Vennerstrom JL, et al. Conformational studies of glucose transporter 1 (GLUT1) as an anticancer drug target. Molecules. 2019;24(11):E2159. doi:10.3390/molecules24112159
23. Pawar S, Vavia P. Glucosamine anchored cancer targeted nano-vesicular drug delivery system of doxorubicin. J Drug Target. 2016;24(1):68–79. doi:10.3109/1061186X.2015.1055572
24. Pawar S, Shevalkar G, Vavia P. Glucosamine-anchored doxorubicin-loaded targeted nano-niosomes: pharmacokinetic, toxicity and pharmacodynamic evaluation. J Drug Target. 2016;24(8):730–743. doi:10.3109/1061186X.2016.1154560
25. Pawar SK, Vavia P. Efficacy Interactions of PEG-DOX-N-acetyl glucosamine prodrug conjugate for anticancer therapy. Eur J Pharm Biopharm. 2015;97(2):454–463. doi:10.1016/j.ejpb.2015.07.019
26. Shan L, Cui S, Du C, et al. A paclitaxel-conjugated adenovirus vector for targeted drug delivery for tumor therapy. Biomaterials. 2012;33(1):146–162. doi:10.1016/j.biomaterials.2011.09.025
27. Shan L, Zhuo X, Zhang F, et al. A paclitaxel prodrug with bifunctional folate and albumin binding moieties for both passive and active targeted cancer therapy. Theranostics. 2018;8(7):2018–2030. doi:10.7150/thno.24382
28. Tang J, Yao J, Shi J, et al. Synthesis, characterization, drug-loading capacity and safety of novel pH-independent amphiphilic amino acid copolymer micelles. Pharmazie. 2012;67(9):756–764.
29. Klein S, Seeger N, Mehta R, et al. Robustness of barrier membrane coated metoprolol tartrate matrix tablets: drug release evaluation under physiologically relevant in vitro conditions. Int J Pharm. 2018;543(1–2):368–375. doi:10.1016/j.ijpharm.2018.04.005
30. Kanamala M, Wilson WR, Yang M, Palmer BD, Wu Z. Mechanisms and biomaterials in pH-responsive tumor targeted drug delivery: A review. Biomaterials. 2016;85:152–167. doi:10.1016/j.biomaterials
31. Shan L, Fan W, Wang W, et al. Organosilica-based hollow mesoporous bilirubin nanoparticles for antioxidation-activated self-protection and tumor-specific deoxygenation-driven synergistic therapy. ACS Nano. 2019;13(8):8903–8916. doi:10.1021/acsnano.9b02477
32. Feng J, Feng T, Yang C, et al. Feasibility study of stain-free classification of cell apoptosis based on diffraction imaging flow cytometry and supervised machine learning techniques. Apoptosis. 2018;23(5–6):290–298. doi:10.1007/s10495-018-1454-y
33. Blagih J, Zani F, Chakravarty P, et al. Cancer-specific loss of p53 leads to a modulation of myeloid and T cell responses. Cell Rep. 2020;30(2):481–496. doi:10.1016/j.celrep.2019.12.028
34. Shan L, Liu M, Wu C, et al. Multi-small molecule conjugations as new targeted delivery carriers for tumor therapy. Int J Nanomed. 2015;10:5571–5591. doi:10.2147/IJN.S85402
35. Kolesnik DL, Pyaskovskaya ON, Yakshibaeva YR, et al. Time-dependent cytotoxicity of dichloroacetate and metformin against Lewis lung carcinoma. Exp Oncol. 2019;41(1):14–19.
36. Wallberg F, Tenev T, Meier P. Time-lapse imaging of cell death. Cold Spring Harb Protoc. 2016;20(3):395–403. doi:10.1101/pdb.prot087395
37. Mishra AK, Mishra A, Pragya, et al. Screening of acute and sub-chronic dermal toxicity of Calendula officinalis L essential oil. Regul Toxicol Pharmacol. 2018;98:184–189. doi:10.1016/j.yrtph.2018.07.027
38. Gao J, Chen K, Xie R, et al. In vivo tumor-targeted fluorescence imaging using near-infrared non-cadmium quantum dots. Bioconjug Chem. 2010;21(4):604–609. doi:10.1021/bc900323v
39. Kalyane D, Raval N, Maheshwari R, et al. Employment of enhanced permeability and retention effect (EPR): nanoparticle-based precision tools for targeting of therapeutic and diagnostic agent in cancer. Mater Sci Eng C Mater Biol Appl. 2019;98:1252–1276. doi:10.1016/j.msec.2019.01.066
40. Maeda H, Nakamura H, Fang J. The EPR effect for macromolecular drug delivery to solid tumors: improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv Drug Deliv Rev. 2013;65(1):71–79. doi:10.1016/j.addr.2012.10.002
41. Sun B, Luo C, Cui W, et al. Chemotherapy agent-unsaturated fatty acid prodrugs and prodrug-nanoplatforms for cancer chemotherapy. J Control Release. 2017;264:145–159. doi:10.1016/j.jconrel.2017.08.034
42. Gao C, Bhattarai P, Chen M, et al. Amphiphilic drug conjugates as nanomedicines for combined cancer therapy. Bioconjug Chem. 2018;29(12):3967–3981. doi:10.1021/acs.bioconjchem.8b00692
43. Weaver BA. How Taxol/paclitaxel kills cancer cells. Mol Biol Cell. 2014;25(18):2677–2681. doi:10.1091/mbc.E14-04-0916
44. Ma P, Sun Y, Chen J, et al. Enhanced anti-hepatocarcinoma efficacy by GLUT1 targeting and cellular microenvironment-responsive PAMAM-camptothecin conjugate. Drug Deliv. 2018;25(1):153–165. doi:10.1080/10717544.2017.1419511
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.