Back to Journals » Infection and Drug Resistance » Volume 19
Targeted Next-Generation Sequencing Analysis of BALF Microbiota and Clinical Characteristics in Severe versus Non-Severe Community-Acquired Pneumonia
Authors Fan Y ![]() , Ren Y
, Ren Y ![]() , An J, Wang X
, An J, Wang X
Received 13 December 2025
Accepted for publication 3 March 2026
Published 16 April 2026 Volume 2026:19 588781
DOI https://doi.org/10.2147/IDR.S588781
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Hazrat Bilal
Yafei Fan,1,2 Yingzheng Ren,3 Junjie An,4 Xia Wang1,2
1Department of Respiratory and Critical Care Medicine, Putuo District Central Hospital, Shanghai, 200062, People’s Republic of China; 2Department of Respiratory and Critical Care Medicine, Yuncheng Central Hospital Affiliated to Shanxi Medical University, Yuncheng, 044000, People’s Republic of China; 3Department of General Surgery, The Second Hospital of Dalian Medical University, Dalian, 116011, People’s Republic of China; 4Department of Respiratory and Critical Care Medicine, Shanxi Provincial People’s Hospital, Taiyuan, 030012, People’s Republic of China
Correspondence: Xia Wang, Department of Respiratory and Critical Care Medicine, Putuo District Central Hospital, Shanghai, 200062, People’s Republic of China, Email [email protected]
Background: Severe community-acquired pneumonia (SCAP) is associated with high mortality. However, data on the bronchoalveolar lavage fluid (BALF) microbiota in Chinese SCAP patients remain limited. This study aimed to characterize the clinical features and BALF microbiome composition in patients with SCAP compared to non-severe CAP using targeted next-generation sequencing (tNGS).
Methods: We conducted a retrospective study involving 224 CAP and 97 SCAP patients from two hospitals in Shanxi, China (January 2023–January 2025). Clinical characteristics and inflammatory cytokines were compared between groups. BALF samples were analyzed via tNGS to evaluate microbial alpha and beta diversity. Differentially abundant taxa were identified using Linear Discriminant Analysis Effect Size (LEfSe).
Results: Compared to the CAP group, SCAP patients were significantly older, had a higher prevalence of comorbidities (hypertension, coronary heart disease, diabetes), and exhibited elevated inflammatory indices (CRP, IL-6, PCT, ESR). SCAP patients also demonstrated a higher likelihood of mixed infections, and the number of detected pathogens showed a positive correlation with the length of hospital stay. tNGS analysis revealed significant differences in alpha diversity and distinct beta diversity clustering between the two groups. LEfSe analysis identified Pseudomonas as a potential biomarker enriched in SCAP, whereas Streptococcus was predominant in CAP.
Conclusion: In patients with SCAP, the BALF microbiota showed a significant increase in alpha diversity, which appears to be closely associated with inflammatory cytokine production and correlates with disease severity. There were pronounced differences between SCAP and CAP in both clinical characteristics and microbiome profiles, highlighting the necessity of integrated diagnostic approaches in pneumonia care. Future research should prioritize delineating the dynamic shifts of microbial communities and their influence on pneumonia severity, with the goal of refining and optimizing treatment strategies.
Keywords: community-acquired pneumonia, severe community-acquired pneumonia, clinical characteristics, targeted next-generation sequencing, microbiological profiles
Introduction
Community-acquired pneumonia (CAP) is an acute lower respiratory tract infection involving the lung parenchyma and represents a substantial global public health burden, with an incidence of approximately 250–400 cases per 100,000 population; it poses a particularly significant threat to older adults and individuals with comorbidities who require hospitalization.1,2 Previous studies indicate that more than half of hospitalized CAP patients progress to severe community-acquired pneumonia (SCAP), and about 27% die within one year.3,4 As China’s population continues to age and life expectancy rises, the number of older adults is steadily increasing; consequently, the incidence and mortality of CAP are expected to rise markedly, further exacerbating the public health and economic burden attributable to CAP.5 Fujikura et al reported that only approximately 55% of CAP cases have an identified etiologic pathogen, whereas 44% remain pathogen-negative even after completing a full panel of conventional tests, including sputum and blood cultures, serology, rapid antigen assays, and limited PCR.5 These limitations in etiological diagnosis may delay diagnosis and treatment, facilitate progression to SCAP, and adversely affect clinical outcomes and prognosis. Therefore, enabling clinicians to distinguish the pathogen spectrum between SCAP and CAP and to institute timely, targeted therapy is crucial to preventing disease deterioration.
The etiologic spectrum of CAP is complex. Common bacterial pathogens include Streptococcus pneumoniae and Haemophilus influenzae, whereas, prior to the COVID-19 pandemic, human rhinovirus and influenza virus were the most frequently detected viral pathogens.6 In an international, multicenter, prospective CAP cohort, Waldeck et al further characterized this distribution: bacterial CAP accounted for approximately 29.0% (n=161), viral CAP for 20.6% (n=114), and bacterial–viral coinfection for 13. 5% (n=76).2,7 Such coinfections are common and serious problem and are associated with high mortality, underscoring the urgency of precise etiologic diagnosis. Moreover, in the early stages of illness, clinical presentation and inflammatory biomarkers such as C-reactive protein (CRP), interleukin-6 (IL-6), procalcitonin (PCT), erythrocyte sedimentation rate (ESR) are not sufficient to differentiate between bacterial and viral pneumonia. Therefore, timely pathogen identification and elucidation of microbiological differences between CAP and SCAP are critical for effective treatment and management.
Conventional pathogen diagnostic methods include morphologic examination, microbiological culture, immunologic assays (antigen or antibody detection), and molecular techniques such as PCR. However, these approaches have inherent limitations with respect to detecting polymicrobial infections, analytical sensitivity, turnaround time, and the breadth of detectable pathogens. With rapid advances in next-generation sequencing over the past decade, targeted next-generation sequencing (tNGS) has emerged as an important tool in precision medicine, offering high sensitivity, rapid turnaround, streamlined sample processing, and a broad detection spectrum.8 Although metagenomic next-generation sequencing (mNGS) can provide unbiased and comprehensive detection, its clinical application is often limited by high host DNA contamination, complex bioinformatics analysis, and high costs. In contrast, tNGS significantly improves the detection sensitivity for low-abundance pathogens by performing targeted amplification of predefined pathogenic genes. At the same time, it effectively reduces the interference from host genetic material, shortens the detection turnaround time, and lowers the cost, making it more accessible and targeted in clinical diagnosis. To date, numerous studies have examined the utility of tNGS for pathogen identification and for shortening the duration of empiric therapy when definitive microbiologic evidence is lacking.9–12
Prior studies have identified several key factors associated with progression from CAP to SCAP, including the composition of the pulmonary microbiota, host immune activity, age, underlying diseases, and complications. Unlike a balanced lung microbial community, patients with SCAP exhibit dysbiosis of the pulmonary microbiome.13 Therefore, this study aimed to analyze tNGS derived microbial profiles from the lower respiratory tract to elucidate potential differences in microbial distributions between CAP and SCAP, and to compare their clinical characteristics, with the goal of identifying distinguishing clinical features and pathogen-associated markers that may indicate progression to severe disease.
Materials and Methods
Study Population
This study extracted complete clinical and microbiological data for 321 pneumonia patients admitted between January 2023 and January 2025 to Shanxi Provincial People’s Hospital and Yuncheng Central Hospital. Diagnostic criteria for CAP followed the Chinese Guidelines for the Diagnosis and Treatment of Adult CAP (See Table 1 and Table 2).
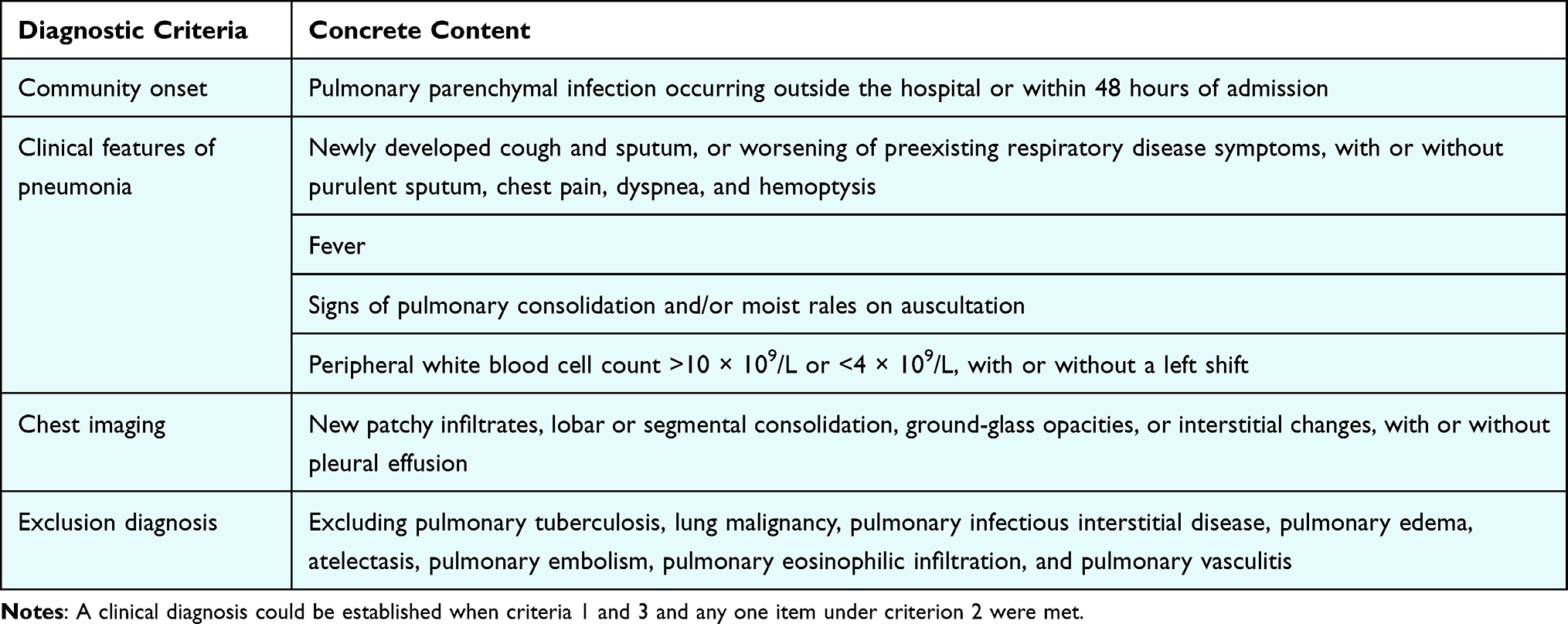 |
Table 1 CAP Diagnostic Criteria |
 |
Table 2 SCAP Diagnostic Criteria |
Data Collection
Clinical variables included baseline characteristics (name, group classification, sex, age) and laboratory indices such as CRP, IL-6, PCT, and white blood cell count. BALF was collected to assess the structure and diversity of the lower respiratory tract microbiome.
tNGS Library Preparation and Sequencing
BALF samples were collected by experienced bronchoscopists via the instillation of 20–50 mL of sterile saline. To mitigate oropharyngeal microbial interference, the initial 10 mL of aspirated fluid was strictly discarded. For laboratory processing, total DNA/RNA was extracted from a 300 μL BALF aliquot using the Pathogen Targeted Sequencing Kit (KingMed Diagnostics, Guangzhou, China), which incorporates a robust bead-beating step for effective mechanical lysis of thick-walled pathogens.
Target enrichment was achieved through a two-step multiplex PCR stratagem, employing optimized primers covering highly conserved regions of 225 common clinical pathogens. Sequencing was executed on the Illumina NextSeq 550 platform, ensuring a high-resolution output with a targeted read depth of at least 0.5 million reads per sample.
To ensure the integrity of microbial data, stringent quality control and decontamination protocols were implemented. Environmental and reagent-derived contaminants were monitored by processing negative controls (sterile water) in parallel with each clinical batch from the extraction phase through to sequencing. Computational decontamination was subsequently performed by filtering out taxa identified in negative controls unless their abundance exceeded a 5-fold threshold relative to the control. Additionally, common laboratory contaminants were removed based on an integrated internal database. Bioinformatics identification was conducted via the Pathogen-Identify pipeline (KingMed Diagnostics), with a pathogen being considered positive only if it yielded a minimum of 3 unique reads, ensuring a balance between sensitivity and clinical specificity.The bioinformatics analysis specifically targeted and quantified 127 clinically relevant pathogenic microorganisms included in the tNGS panel to compare microbial profiles between the two groups.
Statistical Analysis
Descriptive statistics were used to summarize clinical characteristics and diversity indices, with data presented as mean ± standard deviation (SD) or median (interquartile range, IQR) as appropriate. Data normality was assessed using the Shapiro–Wilk test to determine the suitability of parametric versus non-parametric methods.
To evaluate differences in clinical features and inflammatory markers between groups, between-group comparisons employed either the Student’s t-test (for normally distributed data) or the Mann–Whitney U-test (for non-normally distributed data). These tests were chosen to identify significant deviations in host inflammatory responses associated with CAP and SCAP. To assess microbial community patterns, principal coordinates analysis (PCoA) based on the Bray–Curtis distance matrix was performed, and differences in community structure (beta diversity) were tested using PERMANOVA to determine if infection severity significantly alters the overall lung microbiota. Linear discriminant analysis effect size (LEfSe) was further applied to identify specific microbial taxa as potential biomarkers for disease severity. Prior to LEfSe analysis, taxa were filtered to include only those with a prevalence of >10% across all samples and a relative abundance of >0.01% to minimize the influence of rare or transient species. The threshold for the LDA score was set at >2.0. To investigate the interaction between the host and the microbiome, correlations between diversity indices and clinical parameters were evaluated using Pearson or Spearman correlation analyses, selected based on the data distribution and the nature of the association (linear or monotonic).
In this study, mixed infection was defined as the simultaneous detection of two or more distinct pathogen species in a single BALF sample. To ensure diagnostic rigor, the positive threshold for each pathogen was strictly set at a minimum of 3 unique reads. To differentiate etiologic pathogens from colonizers and commensals, we integrated molecular, ecological, and clinical data: (1) Commensals were identified as low-abundance taxa within high-diversity samples that matched known oropharyngeal flora; (2) Potential colonizers were identified when specific taxa were detected without corresponding host inflammatory response or radiological evidence. (3) Etiologic pathogens were prioritized when high read counts aligned with elevated inflammatory markers and clinical symptoms. Furthermore, all identified mixed infections and suspected pathogens were independently reviewed by two clinical physicians to confirm their clinical relevance and exclude potential contaminants.
Alpha Diversity Analysis
Alpha diversity indices were calculated using the vegan package in R. Specifically, Shannon and Simpson indices were computed with the diversity function to reflect community species diversity and, for Shannon, evenness. Chao1 richness was obtained as the second element of the estimateR output (S. chao1), which estimates potential species richness while accounting for rare taxa; in this study, the mean ± SD was 3.807 ± 2.168. Observed operational taxonomic units (Observed OTUs) were calculated as the count of taxa with nonzero abundance per sample using rowSums (x > 0), reflecting observed richness, and showed results consistent with the Chao1 index. Together, these indices provide complementary perspectives on the respiratory microbiome’s diversity and richness in pneumonia patients, supporting subsequent between-group comparisons and correlation analyses.
Statistical Software, Significance, and Power
All analyses were performed in R version 4.5.0. A two-sided P<0.05 was considered statistically significant. Sample size was determined based on an expected effect size of 0.5, power of 0.8, and α=0.05, yielding a minimum of 64 participants per group. The study enrolled 97 cases in the severe group and 224 in the non-severe group, meeting the required statistical power. For multiple correlation tests, the Benjamini–Hochberg procedure was used to control the false discovery rate (FDR), with adjusted significance set at q<0. 05.
Effect Size Interpretation
Effect sizes were interpreted using conventional thresholds: for Cohen’s d, small (0.2), medium (0.5), and large (0.8); for the correlation coefficient r, small (0.1), medium (0.3), and large (0.5). In accordance with these criteria, results in this study yielding values near the lower thresholds were described as modest or small-to-moderate effects to avoid overestimation of clinical significance.
Results
Clinical Characteristics of CAP Vs SCAP
Among 321 patients, 224 had CAP and 97 had SCAP. Patients with SCAP were older than those with CAP (mean 69.39 vs 58.59 years; P<0.001) and had a higher proportion of males (P=0.030). SCAP was associated with higher rates of hypertension (P=0.003) and coronary artery disease (P<0.001). The prevalence of diabetes was numerically higher in SCAP but did not reach statistical significance (P=0.067). There was no significant difference in the prevalence of cerebral infarction between groups (P=0.104) (See Table 3).
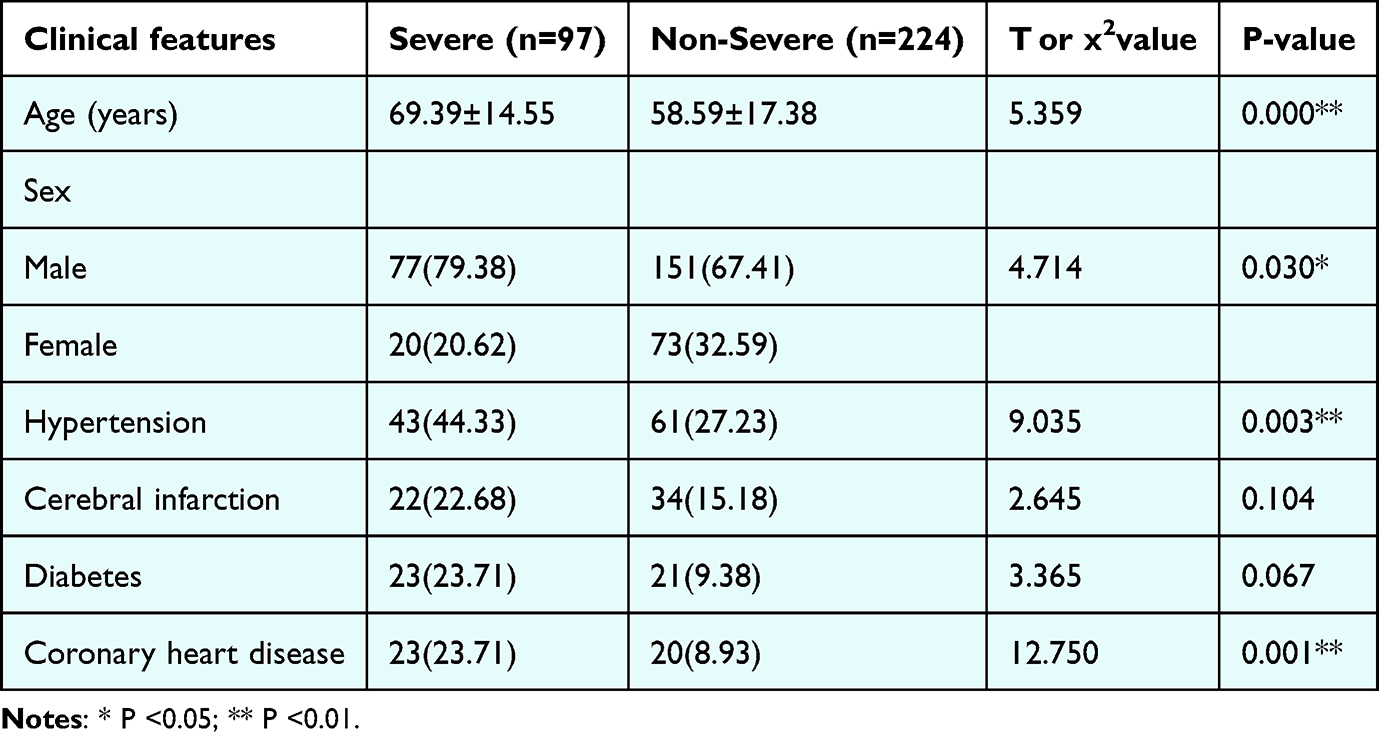 |
Table 3 Comparison of General Clinical Data Between the Two Groups [n (%)] |
Pathogen Identification and Distribution
A total of 98 pathogens were detected in the BALF samples using tNGS (excluding microorganisms with a detection rate of 0). Overall, bacteria were the most prevalent, accounting for 466 cases, followed by viruses (n=439), fungi (n=253), and other pathogens (n=40, mainly including Mycoplasma pneumoniae (29 cases), Tropheryma whipplei (7 cases), Chlamydia psittaci (2 cases), Ureaplasma urealyticum (1 case), and Chlamydia pneumoniae (1 case)). The complete list of identified pathogens and their quantitative distribution across groups are summarized in Table 2. The results indicate that in BALF samples, bacteria and viruses are the main detected pathogens, accounting for most of the total detected cases, reflecting the prevalence of bacteria and viruses in respiratory tract infections (See Table 4).
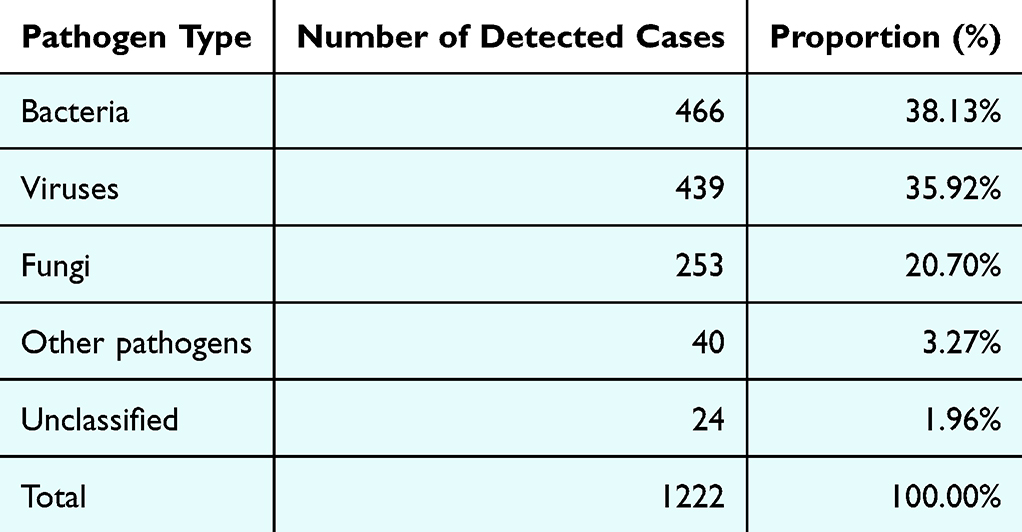 |
Table 4 Detection Distribution of Common Respiratory Pathogens |
Comparison of Inflammatory Biomarkers Between CAP and SCAP
Patients with SCAP had significantly higher levels of CRP, IL-6, PCT, ESR, white blood cell count, neutrophil percentage, and lymphocyte percentage than those with non-severe CAP (all P < 0.01) (See Table 5).
 |
Table 5 Comparison of Inflammatory and Immune Parameters Between Severe and Non-Severe Patients |
Microbial Community Composition
PCoA based on Bray–Curtis distances revealed a clear trend toward separation between the severe and non-severe groups. The first principal coordinate (PC1) explained 23.4% of the total variance and the second (PC2) explained 15.8%, for a cumulative 39.2%. Samples from the severe group were mainly distributed on the positive side of PC1, whereas non-severe samples were more dispersed but largely clustered on the negative side of PC1. PERMANOVA confirmed a statistically significant between-group difference in community structure (F = 4.67, R2 = 0.014, P = 0.001), indicating that disease severity explains only a small fraction of the total microbial variance, while other unmeasured factors likely contribute to the majority of the community shifts (See Figure 1).
 |
Figure 1 Principal coordinates analysis (PCoA) based on Bray-Curtis distances ofrespiratory microbiota from severe (n = 97) and non-severe (n = 224) patients. PCI and PC2 explain 23.4% and 15.8% of the variance (cumulative 39.2%). Severe samples cluster on the positive side of PC1, while non-severe samples are mostly on the negative side. PERMANOVA: F = 4.67, R2 = 0.014. P = 0.001. Blue dots represent CAP samples and Orange dots represent SCAP samples. Ellipses indicate 95% confidence intervals. |
Differential Microbiota Analysis
LEfSe analysis identified 47 taxa with significant differences between severe and non-severe groups (Linear discriminant analysis, LDA score > 2.0). Ten genera showed higher abundance in the severe group, while nine genera were higher in the non-severe group. Severe-group enriched pathogens included Pseudomonas aeruginosa (LDA = 4.2), Acinetobacter baumannii (LDA = 3.8), Klebsiella pneumoniae (LDA = 3.6), and Staphylococcus aureus (LDA = 3.2). Non-severe–group enriched taxa included Streptococcus pneumoniae (LDA = 3.4), Haemophilus influenzae (LDA = 2.9), and Moraxella catarrhalis (LDA = 2.6) (See Figure 2).
 |
Figure 2 Microbiota Differential Analysis (Bar Chart of LEfSe LDA Score Distribution). LDA scores (log10-transfbrmed) derived from LEfSe show the taxa significantly enriched in the CAP group (blue bars) and the SCAP group (red bars). Threshold: LDA >2.0. |
Comparison of BALF Microbiota Diversity
A systematic descriptive analysis characterized the distribution of alpha-diversity indices and the differences between groups (See Figure 3). The severe group showed significantly higher Shannon diversity than the non-severe group, with a median of 0.648 and an interquartile range of 0.258–1.006 compared with a median of 0.376 and an interquartile range of 0.065–0. 755 (P < 0.001). Simpson diversity was also higher in the severe group, at 0.384 with an interquartile range of 0.109–0.553, compared with 0.182 with an interquartile range of 0.023–0.453 in the non-severe group (P < 0.001). In contrast, Simpson dominance was significantly lower in the severe group, at 0.616 with an interquartile range of 0.447–0.891, compared with 0.818 with an interquartile range of 0.547–0.977 (P < 0.001), indicating reduced dominance by a few taxa and a more even community structure. Richness was likewise higher in the severe group, as shown by the Chao1 index (4 with an interquartile range of 3–6 vs 3 with an interquartile range of 2–5, P < 0.001) and by the observed operational taxonomic units (4 with an interquartile range of 3–6 vs 3 with an interquartile range of 2–5, P < 0.001). According to Cohen’s criteria, the effect sizes of 0.191–0.252 represent small-to-moderate effects. While these differences are statistically significant, they indicate a discernible but modest shift in microbial structure associated with disease severity, rather than a strong divergent pattern (See Table 6).
 |
Table 6 Comparison of Microbiota Diversity in Bronchoalveolar Lavage Fluid |
 |
Figure 3 Comparison of respiratory microbiota a-diversity and composition between CAP patients and SCAP patients. (a–e) Box plots showing the respiratory microbiota diversity indices: Shannon index (a), Simpson index (b), Chaol index (c), Observed OTUs index (d), and Simpson dominance index (e) in the CAP group and SCAP group. Higher values indicate higher divers. |
Correlations Between Microbiota Diversity and Inflammatory Markers
Correlation analyses revealed a consistent but modest pattern of associations between microbiota diversity indices and seven key clinical inflammatory indices (CRP, IL-6, PCT, ESR, WBC, neutrophil proportion, lymphocyte proportion), with 18 significant associations identified (P < 0.05). The Chao1 index and observed OTUs exhibited significant associations: positive correlations with CRP (r = 0.141, P = 0.012), IL-6 (r = 0.176, P = 0.002), WBC (r = 0.155, P = 0.005), and a mild-to-moderate positive correlation with neutrophil proportion (r = 0.273, P < 0.001), alongside a negative correlation with lymphocyte proportion (r = −0.232, P <0. 001). The Shannon index was primarily associated with immune cell proportions, correlating positively with neutrophil proportion (r =0. 200, P < 0.001) and negatively with lymphocyte proportion (r = −0.139, P = 0.012). The Simpson index correlated positively with IL-6 (r = 0.118, P = 0.035) and neutrophil proportion (r = 0.179, P = 0.001). Collectively, these patterns indicate that greater microbiota diversity is significantly, though weakly, associated with heightened inflammatory activity and immune dysregulation. In contrast, Simpson dominance showed the opposite pattern—negative correlations with IL-6 (r = −0.163, P = 0.003) and neutrophil proportion (r = −0.206, P < 0.001), and a positive correlation with lymphocyte proportion (r = 0.153, P = 0.006)—further supporting a dysregulated community structure in severe cases. (See Table 7 and Figure 4).
 |
Table 7 Correlation Between Respiratory Microbiota Diversity and Immune Factors |
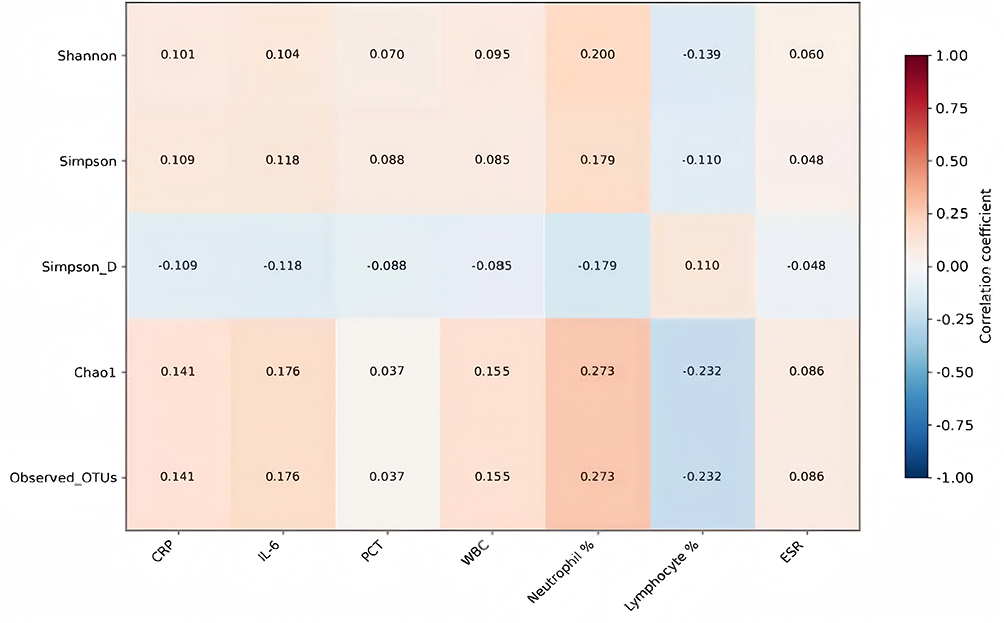 |
Figure 4 Heatmap of Correlations between Diversity Indices and Clinical. Indicator sit comprehensively shows the correlation strength between diversity indices and clinical indicators. Among them, Chaol and Observed OTUs exhibit a stronger red (positive correlation) pattern with inflammatory indicators, and a blue (negative correlation) pattern with lymphocyte proportion. The most significant correlations are concentrated in the neutrophil proportion column, where all diversity indices show positive correlations. |
Overall Distribution of Pathogens
We performed a comprehensive statistical analysis of pathogen detection across all patients (See Figure 5), presents the distribution of positive cases for the top 10 predominant pathogens showing significant differences between the two patient groups. Among frequently detected pathogens, “Candida albicans” exhibited notably higher positive cases in the non-severe group (approximately 80 cases) compared to the severe group (approximately 45 cases). In contrast, the positive cases of pathogens such as Herpes simplex virus type 1 (HSV-1) and “Klebsiella pneumoniae” were relatively more frequent in the severe group. These findings indicate distinct pathogen detection profiles between severe and non-severe patients, reflecting characteristic differences in their microbial spectra.
 |
Figure 5 Pathogenic spectrum in the two patient groups. Note: The inclusion of “Macrolide antibiotics” in the figure refers to antibiotic resistance profiles rather than a specific pathogen and has been clarified in the figure legend. |
Furthermore, we analyzed the incidence of co-infections in both groups (See Figure 6a). Within the severe group, 34.0% of patients (approximately 33 cases) were found to harbor more than six pathogens, whereas 13.8% of patients (approximately 30 cases) in the non-severe group had more than six pathogens detected. Among those with five pathogens, 10.3% (about 10 cases) were in the severe group versus 11.6% (26 cases) in the non-severe group. For four pathogens, 20.6% (20 cases) were severe, compared to 15.6% (34 cases) non-severe. Patients with three pathogens comprised 17.5% (17 cases) of the severe group and 21% (46 cases) of the non-severe group. Those with two detected pathogens accounted for 9.3% (9 cases) of the severe group and 24.6% (54 cases) of the non-severe group, and with only one pathogen detected, 8.2% (8 cases) belonged to the severe group versus 13.4% (29 cases) non-severe.
 |
Figure 6 (a) Distribution of observed species (OTUs) in the two patient groups, (b) Correlation analysis between co-infections and hospital stay duration in severe patients, (c) Correlation analysis between co-infections and hospital stay duration in non-severe patients. Note: The horizontal axis represents the number of pathogens detected; the vertical axis represents the duration of hospital stay. |
Notably, the length of hospital stay increased with the number of pathogens detected in patients with SCAP. A strong positive correlation was observed between the number of detected pathogens and hospital stay duration (r = 0.731, P < 0.05), as shown in (See Figure 6b and c).
Discussion
Compared with the extensively studied gut microbiome, research on the lung microbiome remains in its early stages. The lung was long considered “sterile”, but with the advent of next-generation sequencing (NGS), accumulating evidence demonstrates that the lung harbors rich microbial communities that play functional roles in the progression of pulmonary diseases.13,14 The respiratory micro-ecosystem has unique features: its biomass is far smaller than that of the gut, yet its diversity is higher, it is more sensitive to environmental fluctuations, and it is strongly influenced by oral and upper airway microbiota. Accordingly, developing methodological frameworks tailored to the lung microbiome is essential to advance the field.15,16 Under physiological conditions, respiratory homeostasis is maintained through inter-microbial competitive inhibition, modulation by microbial metabolites, and coordination with host immunity.17 In SCAP, however, this ecological equilibrium is disrupted and becomes a core driver of disease progression. On this basis, we analyzed the BALF microbiota and clinical characteristics of SCAP patients.
We observed significantly increased alpha diversity in SCAP, with Shannon, Simpson (diversity), Chao1, and observed OTUs all higher than in the non-severe group, alongside a marked decrease in Simpson dominance. These patterns indicate that severe cases harbor more complex communities lacking a clear dominant taxon. PCoA and LEfSe further revealed systematic differences in beta diversity and community composition between groups. The severe group was enriched for Candida albicans, Epstein–Barr virus (EBV), herpes simplex virus type 1 (HSV-1), Klebsiella pneumoniae, and Acinetobacter baumannii, whereas the non-severe group was characterized by Candida albicans, EBV, Fusobacterium nucleatum, “micro-single-celled bacterium”, and a pharyngitis-associated Streptococcus group. These findings underscore the profound ecological shifts accompanying SCAP. While conventional culture-based tests remain the traditional benchmark for pathogen identification, their diagnostic sensitivity in our cohort was likely hindered by prior broad-spectrum antibiotic exposure and the inherently slow growth of certain fastidious microbes. Our study leveraged tNGS to overcome these limitations, capturing a more comprehensive microbial landscape that includes non-culturable organisms and those masked by antibiotic pressure. Although the lack of paired, systematic culture data for the entire cohort represents a limitation, the clinical adjudication process by independent physicians ensured that the identified microbial signatures remained clinically grounded. This approach underscores the role of tNGS as a powerful complementary tool, providing broader ecological insights and etiologic clarity beyond the reach of conventional diagnostics. As sequencing transitions from research to clinical practice, NGS has become increasingly utilized. In diagnostic microbiology laboratories, common applications include mNGS and tNGS. In BALF from patients with lung infections, tNGS has shown detection rates comparable to mNGS (82.17% vs. 86.51%), with consistent performance across bacteria, fungi, and viruses.18 With growing demand for more cost-effective and widely accessible testing, tNGS is gaining importance. By enriching target microbial genes using specific probes or primers prior to sequencing, tNGS offers strong specificity, high sensitivity, and excellent efficiency, effectively overcoming limitations of conventional methods and enabling precise identification of low-abundance pathogens. Notably, tNGS maintains high sensitivity (70.8%–95.0%) even in low-pathogen-load samples such as respiratory specimens.19 In addition, tNGS performs particularly well in detecting drug-resistant Mycobacterium tuberculosis, identifying resistance genes and informing clinical decision-making—an advantage that mNGS struggles to match.20 Collectively, these findings highlight the significance and practical value of tNGS in the diagnosis of respiratory infectious diseases.
This study delineated differences between CAP and SCAP in pivotal clinical markers. Patients in the SCAP group were older, with a higher proportion of elderly individuals, underscoring age as a key risk factor for severe disease. In line with prior reports, SCAP patients exhibited significantly higher CRP and PCT levels than CAP patients, indicating a more intense inflammatory response.21 We also observed higher levels in SCAP of PCT, ESR, white blood cell count, neutrophil proportion, and lymphocyte proportion. Numerous studies have shown that perturbations of the respiratory microbiota can dysregulate host immune control—via growth factors, cytokines, and other mediators that promote the release of IL-6, IL-8, and related inflammatory factors—thereby exacerbating SCAP progression; thus, the onset and evolution of severe pneumonia may be linked to shifts in the lung microbiome.22 Nevertheless, given the small effect sizes observed, these microbial indices should be interpreted as complementary biological markers rather than primary tools for differential diagnosis. Building on this rationale, we assessed correlations between microbial diversity and immune markers. Chao1 and observed OTUs displayed the strongest association patterns, while the Shannon index was chiefly related to immune cell proportions. The Simpson index was positively correlated with IL-6 and neutrophil proportion, whereas Simpson dominance showed an opposite pattern relative to the other indices. Together, these relationships suggest a subtle but significant association, between microbiome diversity and disease severity. While these findings reflect the underlying ecological dysbiosis in severe pneumonia, the small effect sizes indicate that microbiome diversity should be viewed as a complementary indicator of host-microbe interplay rather than a robust standalone biomarker for clinical severity.
Despite meaningful progress, respiratory microbiome research still faces multiple hurdles. Sampling is a critical bottleneck: compared with the skin and gut, the respiratory tract has much lower biomass, making specimen collection and detection inherently challenging.23 Moreover, because the upper airway is directly exposed to the external environment and is the first site of contact for inhaled contaminants and pathogens, upper-airway signals can confound analyses and influence prognostic interpretation.
A further limitation lies in the scope of tNGS. By design, tNGS interrogates only predefined pathogens; its breadth is constrained by the number of primers that can be included (typically up to 20,000), and practical considerations such as pathogen specificity and primer melting temperatures reduce the usable set even further. A notable discrepancy exists between our results and a concurrent study utilizing mNGS, which reported no significant difference in alpha diversity between CAP/SCAP patients and controls.24 This divergence likely stems from the fundamental methodological differences: while mNGS provides an unbiased snapshot of the entire microbial environment, it often suffers from a lower signal-to-noise ratio in clinical samples. In contrast, the multiplex PCR amplification used in tNGS selectively enriches predefined pathogens, effectively “unmasking” low-abundance co-infecting species that remain below the detection limit of mNGS. This enrichment effect potentially inflates diversity metrics, suggesting that the observed increase may partly stem from methodological discrepancies rather than reflecting a purely biological shift. Furthermore, it raises the question of whether this increased alpha diversity in SCAP indicates a state of dysbiosis. In SCAP, our findings suggest that dysbiosis manifests as a “pathogenic enrichment”—a consequence of acute multi-pathogen invasion and host immune compromise rather than a primary cause of the disease. Therefore, the observed dysbiosis appears to be a reactive state following the infection. Consequently, tNGS remains less suitable for unknown pathogens compared to mNGS. Moreover, different combinations of co-infections identified by tNGS may uniquely impact clinical severity, which requires further investigation using standardized definitions to clarify their synergistic roles in SCAP pathogenesis. Consequently, tNGS cannot detect unknown pathogens and is less suited to emerging agents or complex environmental mixtures—capabilities that mNGS is better positioned to provide.25 Moreover, different combinations of co-infections (bacterial-viral vs. bacterial-bacterial) identified by tNGS may uniquely impact clinical severity, which requires further investigation using standardized definitions to clarify their synergistic roles in SCAP pathogenesis. In addition, tNGS performance and generalizability depend on clear specifications for sample type, target panel, positivity thresholds, and supporting databases; without standardized criteria, widespread implementation is difficult.26
For these reasons, tNGS is not recommended as a stand-alone test but should be combined with conventional diagnostic methods. Such an integrated approach leverages the broad applicability of traditional assays together with the high specificity and sensitivity of tNGS, providing clinicians with more complete evidence to make accurate diagnoses and to institute timely, appropriate therapy—thereby improving patient outcomes. In sum, this study delineates distinctive features of the respiratory microbiota in SCAP, showing that increased diversity is significantly, though modestly, associated with heightened inflammatory responses and immune imbalance.
Future Perspectives and Recommendations
From a future perspective, Our findings regarding the distinctive microbial signatures in SCAP provide a potential framework for precision antibiotic stewardship. By identifying specific pathogen clusters and the associated immune inflammatory profiles, clinicians may be able to transition from broad-spectrum empiric therapy to targeted anti-pathogen strategies more rapidly. Furthermore, we recommend that future work should focus on validating the potential utility of microbiome diversity as a complementary biomarker of disease severity through larger, multi-center longitudinal cohorts. Such efforts will be essential to clarify the synergistic roles of co-infections in SCAP pathogenesis and to minimize the emergence of multidrug-resistant organisms while optimizing treatment efficacy for severe patients.
Limitations
Our study has several limitations that warrant consideration. First, Cross-sectional design: Captures a single time point and cannot establish causality or depict the dynamic evolution of the microbiome–host axis across the disease course. Second, Culture-based constraints: Where culture-dependent methods were used, fastidious or special-growth–requirement organisms may have been missed, likely underestimating true community diversity. Third, whether the identified microbial differences reflect true etiologic shifts or altered colonization patterns remains a key question. While the pathogens enriched in the SCAP group were predominantly treated as clinically causative agents by attending physicians—leading to targeted therapeutic adjustments—the low R2 in our beta diversity analysis (0.014) underscores that the lung microbiome is a highly complex ecosystem. This indicates that disease severity is only one of many driving forces, and other unmeasured factors likely contribute to the majority of community shifts. Last, a direct comparison between tNGS and conventional culture-based methods was not performed in this study. While culture remains the traditional benchmark, its low sensitivity—particularly in patients with prior antibiotic exposure—often limits its utility in characterizing the full microbial landscape. Although we employed stringent bioinformatic filtering and clinical physician oversight to differentiate potential pathogens from colonizers, the lack of paired culture data means we could not formally calculate the diagnostic sensitivity and specificity of our tNGS pipeline. Future prospective trials incorporating head-to-head comparisons with culture and other molecular methods (mNGS) are necessary to fully establish the clinical “gold standard” utility of tNGS in SCAP management.Last, although statistically powered (severe n=97; non-severe n=224), the cohort remains modest for dissecting complex host-microbe interaction networks and for robust subgroup analyses. Residual confounding: Incomplete control of antibiotic exposure history, length of stay, and comorbidities—all of which can reshape airway communities—may bias associations. To address these challenges, future research should prioritize,prospective longitudinal cohorts with serial sampling to map temporal dynamics of the microbiome, inflammation, and immunity from presentation through recovery. Multi-omics integration—metagenomics/tNGS alongside metatranscriptomics, metabolomics, and host immune profiling—with absolute quantification to move from association to mechanism. Low-biomass best practices: protected sampling, upper-airway decontamination, matched negative controls and spike-ins, and bioinformatic decontamination to mitigate contamination. Standardization of pre-analytical, analytical, and interpretive pipelines, including specimen handling, target panels and positivity thresholds for tNGS, and harmonized clinical metadata (timing/dose of antibiotics, comorbidity indices). Analytic frameworks for causal inference to disentangle microbiome–immunity–severity relationships. Interventional studies that leverage sequencing readouts to guide therapy (antibiotic stewardship, targeted anti-pathogen strategies, and adjunctive immunomodulation), with outcomes to validate clinical utility. External validation across centers and regions to enhance generalizability. Taken together, larger, rigorously designed prospective studies that combine advanced sequencing with robust control of confounders are needed to validate these findings, clarify the causal links between microbiome diversity and disease severity, and provide a foundation for microbiome-informed clinical interventions in SCAP.
Conclusion
In conclusion, this study demonstrates that patients with SCAP exhibit a significantly altered respiratory microbial landscape characterized by increased alpha diversity and distinct pathogenic enrichment compared to non-severe cases. Our findings reveal that these microbial shifts are subtly but significantly associated with heightened host systemic inflammation,including elevated levels of CRP and IL-6,and immune dysregulation. While the relatively small effect sizes suggest that microbiome diversity should be utilized as a complementary biological indicator rather than a standalone diagnostic biomarker, the integration of tNGS with clinical adjudication provides enhanced etiologic clarity, especially in cases where conventional cultures are limited. These results underscore the importance of the lung pathobiome in disease progression and provide a preliminary framework for moving toward microbiome-informed precision antimicrobial stewardship in severe respiratory infections.
Ethics Statement
The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
The study was conducted in accordance with the Declaration of Helsinki (as revised in 2013). This study was reviewed and approved by the Ethics Committees of Yuncheng Central Hospital and Shanxi Provincial People’s Hospital (YXLL2024200 and No.682[2025]). All patients or their family members were fully informed of the study details and signed the informed consent forms. Written informed consent was obtained from all patients; however, due to the severity of the illness and impaired decisional capacity (e.g., respiratory failure or sedation), informed consent for critically ill patients was obtained from their next of kin or legally authorized representatives.
Acknowledgments
We are deeply grateful to all individuals who offered practical help and unwavering support to this project.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
No external funding was received for the analysis or drafting of this manuscript.
Disclosure
The authors report no conflicts of interest in this work.
This manuscript has been previously available as a preprint on Research Square: https://doi.org/10.21203/rs.3.rs-8342132/v1. The authors confirm that this work has not been formally published elsewhere.
References
1. Qu J, Zhang J, Chen Y, et al. Aetiology of severe community acquired pneumonia in adults identified by combined detection methods: a multi-centre prospective study in China. Emerging Microbes Infect. 2022;11(1):556–16. doi:10.1080/22221751.2022.2035194
2. Ferreira-Coimbra J, Sarda C, Rello J. Burden of community-acquired pneumonia and unmet clinical needs. Adv Ther. 2020;37(4):1302–1318. doi:10.1007/s12325-020-01248-7
3. Niederman MS, Torres A. Severe community-acquired pneumonia. Eur Respir Rev. 2022;31(166):220123. doi:10.1183/16000617.0123-2022
4. Gonçalves-Pereira J, Froes F, G PF, et al. Community-acquired pneumonia mortality trends according to age and gender: 2009 to 2019. BMC Pulm Med. 2025;25(1):391. doi:10.1186/s12890-025-03875-8
5. Fujikura Y, Somekawa K, Manabe T, et al. Aetiological agents of adult community-acquired pneumonia in Japan: systematic review and meta-analysis of published data. BMJ Open Respir Res. 2023;10(1):e001800. doi:10.1136/bmjresp-2023-001800
6. Serigstad S, Markussen DL, Ritz C, et al. The changing spectrum of microbial aetiology of respiratory tract infections in hospitalized patients before and during the COVID-19 pandemic. BMC Infect Dis. 2022;22(1):763. doi:10.1186/s12879-022-07732-5
7. Waldeck F, Lemmel S, Panning M, et al. Comparing viral, bacterial, and coinfections in community-acquired pneumonia, a retrospective cohort study. Inter J Infect Dis. 2025;154:107841. doi:10.1016/j.ijid.2025.107841
8. Satam H, Joshi K, Mangrolia U, et al. Next-generation sequencing technology: current trends and advancements. Biology. 2023;12(7):997. doi:10.3390/biology12070997
9. Luo W, Zhang S, Sun J, et al. Microbial and clinical disparities in pneumonia: insights from metagenomic next-generation sequencing in patients with community-acquired and severe pneumonia. Front Microbiol. 2025;16:1538109. doi:10.3389/fmicb.2025.1538109
10. Ma R, Liu Z, Zhang L, et al. Epidemiological characteristics of severe community-acquired pneumonia in children admitted to two tertiary hospitals in Shihezi, Xinjiang Region, China in 2023: a cross-sectional analysis. J Thoracic Dis. 2024;16(10):6969–6982. doi:10.21037/jtd-24-1417
11. Zhang HH, Ou-Yang X, Liu XP, et al. [Value of targeted next-generation sequencing in pathogen detection for neonates with respiratory distress syndrome: a prospective randomized controlled trial]. Zhongguo Dang Dai Er Ke Za Zhi. 2025;27(10):1191–1198. Chinese. doi:10.7499/j.issn.1008-8830.2502073
12. Dai X, Xu K, Tong Y, et al. Application of targeted next-generation sequencing in bronchoalveolar lavage fluid for the detection of pathogens in pulmonary infections. Infect Drug Resist. 2025;18:511–522. doi:10.2147/IDR.S499265
13. Carney SM, Clemente JC, Cox MJ, et al. Methods in lung microbiome research. Am J Respir Cell Mol Biol. 2020;62(3):283–299. doi:10.1165/rcmb.2019-0273TR
14. O’Dwyer DN, Ashley SL, Gurczynski SJ, et al. Lung microbiota contribute to pulmonary inflammation and disease progression in pulmonary fibrosis. Am J Respir Crit Care Med. 2019;199(9):1127–1138. doi:10.1164/rccm.201809-1650OC
15. Li R, Li J, Zhou X. Lung microbiome: new insights into the pathogenesis of respiratory diseases. Signal Transduct Target Ther. 2024;9(1):19. doi:10.1038/s41392-023-01722-y
16. Wu S, Wang S, Wu Z, et al. Comparative analysis of the clinical characteristic and lung microbiota in adult and elderly patients with pulmonary tuberculosis. Sci Rep. 2025;15(1):1–10. doi:10.1038/s41598-025-04970-w
17. Li X, Chen M, Chen T, et al. The intricate interplay among microbiota, mucosal immunity, and viral infection in the respiratory tract. J Transl Med. 2025;23(1):488. doi:10.1186/s12967-025-06433-2
18. Li S, Tong J, Liu Y, et al. Targeted next generation sequencing is comparable with metagenomic next generation sequencing in adults with pneumonia for pathogenic microorganism detection. J Infect. 2022;85(5):e127–e129. doi:10.1016/j.jinf.2022.08.022
19. Köndgen S, Oh DY, Thürmer A, et al. A robust, scalable, and cost-efficient approach to whole genome sequencing of RSV directly from clinical samples. J Clin Microbiol. 2024;62(3):e0111123. doi:10.1128/jcm.01111-23
20. Gu D, Liu J, Wang J, et al. Integrating DNA and RNA sequencing for enhanced pathogen detection in respiratory infections. J Transl Med. 2025;23(1):325. doi:10.1186/s12967-025-06342-4
21. Lu Y, Gai W, Li M, et al. Psittacosis pneumonia features, distinguishing characteristics, and outcomes: a retrospective study. Infect Drug Resist. 2024;17:5523–5533. doi:10.2147/IDR.S482471
22. Budden KF, Shukla SD, Rehman SF, et al. Functional effects of the microbiota in chronic respiratory disease. Lancet Respir Med. 2019;7(10):907–920. doi:10.1016/S2213-2600(18)30510-1
23. Bartosik T. The microbiome in diseases of the nose and paranasal sinuses. HNO. 2025;73(9):610–614. doi:10.1007/s00106-025-01661-w
24. Zhao Y, Zhang W, Zhang X. Application of metagenomic next-generation sequencing in the diagnosis of infectious diseases. Front Cell Infect Microbiol. 2024;14:1458316. doi:10.3389/fcimb.2024.1458316
25. Li C, Wu J, Feng Y. Prospective study on diagnostic efficacy of targeted NGS vs. traditional testing for respiratory infections in myelosuppressed hematology patients. Front Med. 2025;12:1488652. doi:10.3389/fmed.2025.1488652
26. Liang A, Wu X, Zhu Y, et al. Targeted next-generation sequencing (tNGS): an upcoming application for pathogen identification in clinical diagnosis. J Infect Public Health. 2025;18(10):102936. doi:10.1016/j.jiph.2025.102936
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.