")
Back to Journals » International Journal of Nanomedicine » Volume 12
Targeted delivery of thymosin beta 4 to the injured myocardium using CREKA-conjugated nanoparticles
Authors Huang Z , Song Y, Pang Z , Zhang B, Yang H, Shi H, Chen J , Gong H, Qian J, Ge J
Received 8 January 2017
Accepted for publication 2 March 2017
Published 12 April 2017 Volume 2017:12 Pages 3023—3036
DOI https://doi.org/10.2147/IJN.S131949
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Linlin Sun
Zheyong Huang,1,* Yanan Song,1,* Zhiqing Pang,2 Bo Zhang,3 Hongbo Yang,1 Hongtao Shi,1 Jing Chen,1 Hui Gong,1,4 Juying Qian,1 Junbo Ge1,4
1Shanghai Institute of Cardiovascular Diseases, Zhongshan Hospital, Fudan University, 2School of Pharmacy, Fudan University, Key Laboratory of Smart Drug Delivery, Ministry of Education, Shanghai, 3Institute of Hematology, Union Hospital, Tongji Medical College, Huazhong University of Science & Technology, Wuhan, Hubei, 4Institute of Biomedical Science, Fudan University, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Purpose: Thymosin beta 4 (Tβ4) has multiple beneficial facets for myocardial injury, but its efficiency is limited by the low local concentration within the infarct. Here, we established a Tβ4 delivery system for cardiac repair based on the interaction between the abundant fibrin in the infarct zone and the fibrin-targeting moiety clot-binding peptide cysteine–arginine–glutamic acid–lysine–alanine (CREKA).
Methods and results: CREKA and Tβ4 were conjugated to nanoparticles (CNP–Tβ4). In vitro binding test revealed that CNP–Tβ4 had a significant binding ability to the surface of fibrin clots when compared to the control clots (NP–Tβ4). Based on the validation of fibrin expression in the early stage of ischemia injury, CNP–Tβ4 was intravenously administered to mice with acute myocardial ischemia–reperfusion injury. CNP–Tβ4 revealed a stronger fibrin-targeting ability than the NP–Tβ4 group and accumulated mainly in the infarcted area and colocalized with fibrin. Subsequently, treatment with CNP–Tβ4 resulted in a better therapeutic effect.
Conclusion: CRKEA modification favored Tβ4 accumulation and retention in the infarcted region, leading to augmented functional benefits. Fibrin-targeting delivery system represents a generalizable platform technology for regenerative medicine.
Keywords: thymosin beta 4, fibrin, CREKA, targeting delivery, ischemia reperfusion, cardiovascular
Introduction
Cardiovascular diseases, including myocardial infarction (MI) and heart failure, are the leading causes of death in the world. Acute occlusion of cardiac vessels leads to loss of dependent myocardium. Because the heart is incapable of sufficient muscle regeneration,1 necrotic myocardium is replaced by noncontractile, poorly conducting, fibrotic scar tissue, leading to pathological remodeling of the heart.2 Therefore, exogenously administered cells, growth factors or small molecule compounds have been recently used for stimulating the intrinsic myocardial repair process.
Thymosin beta 4 (Tβ4), a 43-amino-acid-sequestering peptide expressed in the embryonic heart, plays a critical role in epicardial development and coronary artery formation.3,4 Recent studies revealed that Tβ4 increased cardiac function by promoting the survival of cardiomyocytes,5–7 activating resident epicardial progenitor cells,4,8–12 and modulating inflammatory responses.13 Being a peptide, Tβ4 has additional advantages over conventional growth factors because of its small size, high activity, and minimized immunogenicity,14 and Tβ4 has been regarded as an attractive therapeutic agent for MI clinically (NCT00743769).
However, biomolecules that are administered through injections in soluble forms are susceptible to degradation,15 and systemic administration of Tβ4 may not maintain Tβ4 at a stable, effective blood concentration, especially as its half-life is very short.16,17 Previous studies have shown that multiple intraperitoneal (IP) injections or direct intracardiac (IC) injection of Tβ4 were required to be effective.5,8,9,18 However, these approaches aroused the concerns such as increased dosages of Tβ4, uneven distribution in the infarcted area, and invasive procedure. Although available data point to Tβ4 being a safe and well-tolerated molecule,19 Tβ4 was upregulated in certain types of malignancy.20,21 Therefore, there is an incentive to fabricate a targeting delivery system such that a high local concentration of Tβ4 can be built up within the infarct and side effects to other organs would be reduced.22,23
There is abundant extracellular matrix (ECM) in various tissue microenvironments. After injury, a dramatic change in the composition of the ECM occurs, providing mechanical support to the injured tissue and directly modulating the inflammatory and reparative responses.24–26 Recently, an ECM-targeting drug delivery strategy has been used to treat glioma27 and enhance tissue healing.28 However, to the best of our knowledge, little has been done to understand the feasibility of ECM as a therapeutic target for heart repair. After MI, increased vascular permeability results in extensive extravasation of plasma proteins that form a fibrin-based provisional matrix. Fibrin is the most important temporary ECM in the wounded area and plays an important role in blood clotting, fibrinolysis, cellular and matrix interactions, inflammation, wound healing, and angiogenesis.29–32 Intense fibrin expression was noted in the infarcted mouse myocardium after 3 h of reperfusion, clearly demarcating the area of myocardial injury, and after 72 h of reperfusion, fibrin was incorporated into a well-organized fibrin network in the infarct granulation tissue.26 The high abundance might accordingly favor excellent targeting efficiency when fibrin functions as the therapeutic target.33
Clot-binding peptide cysteine–arginine–glutamic acid–lysine–alanine (CREKA) is a tumor-homing peptide obtained by in vivo phage display. Both our and other studies had recently found that CREKA could specifically bind to fibrin in microthrombus and stroma of tumors.34–37 It occurred to us that CREKA peptide could mediate nanoparticle (NP) drug delivery system to fibrin in infarcted myocardium, which may increase the retention of Tβ4, improve its efficacy, and reduce its dosing systematically.
To justify this hypothesis, we developed a fibrin-targeting drug delivery system with NPs as the carrier, CREKA as the targeting moiety, and Tβ4 as the drug for myocardial injury. The targeting ability of CREKA–NP–Tβ4 (CNP–Tβ4) to fibrin was investigated in vitro and in vivo, and the therapeutic effect of CNP–Tβ4 was evaluated in the mouse model of myocardial ischemia–reperfusion (IR) injury compared with free Tβ4 and unmodified NP–Tβ4.
Materials and methods
Materials
CREKA was synthesized by the Chinese Peptide Company (Hangzhou, China). Tβ4 was obtained from Prospec (East Brunswick, NJ, USA). MPEG (molecular weight [MW] 3,000 Da) was ordered from NOF (Tokyo, Japan), and MAL–PEG (MW 3,400 Da) was custom synthesized by Nektar (Huntsville, AL, USA). R-COOH–PEG (MW 3,400 Da) was obtained from Laysan Bio (Arab, AL, USA). D,L-Lactide (purity: 99.5%) was purchased from Purac (Holland, the Netherlands). MPEG–PLA (MW 33,000 Da), MAL–PEG–PLA (MW 35,000 Da), and R-COOH–PEG–PLA (MW 33,400 Da) block copolymers were synthesized by ring-opening polymerization of lactide using MPEG, MAL–PEG, and COOH–PEG, respectively, as the initiator as described elsewhere.35 Sodium chlorate was ordered from Shanghai Chemical Reagent Company (Shanghai, People’s Republic of China). EDC hydrochloride (HCl) and NHS were purchased from Sigma–Aldrich (Washington DC, WA, USA). DiD was supplied by Sigma-Aldrich. DiR, a near-infrared dye, was obtained from Invitrogen (Carlsbad, CA, USA). Accustain Trichrome Stains (Masson) kit was obtained from Sigma-Aldrich. TRIzol reagent was obtained from Invitrogen. Fibrinogen/fibrin rabbit polyclonal primary antibody, Wt-1 rabbit polyclonal primary antibody, Ki67 rabbit polyclonal primary antibody, CD31 rabbit polyclonal primary antibody, SMαA rabbit polyclonal primary antibody, and Cardiac Troponin T mouse monoclonal primary antibody were obtained from Abcam (Cambridge, MA, USA). ApopTag® Fluorescein In Situ Apoptosis Detection Kit was ordered from Millipore (Billerica, MA, USA). Cy3-conjugated donkey anti-rabbit immunoglobulin G (IgG) secondary antibody and fluorescein isothiocyanate-conjugated donkey anti-mouse IgG secondary antibody were purchased from Jackson Immuno Research (West Grove, PA, USA). Human Thymosin β4 ELISA Kit was ordered from Cusabio Biotech (Barksdale, DE, USA). Male BALB/c mice (25±2 g) were purchased from the Shanghai SLAC Laboratory Animal Ltd. and housed under standard conditions with free access to food and water. Animal experiments were approved by the Animal Care and Use Committee of Zhongshan Hospital, Shanghai, People’s Republic of China and were in compliance with the Guide for the Care and Use of Laboratory Animals published by the National Research Council (US) Institute for Laboratory Animal Research.
Preparation of CREKA–NP–Tβ4 (CNP–Tβ4)
Unmodified poly(ethylene glycol)–poly(lactic acid) (PEG–PLA) NPs were prepared using an emulsion/solvent evaporation technique, as previously reported.38 In brief, methoxy poly(ethylene glycol) (MPEG)–PLA (20 mg), maleimide (MAL)–PEG–PLA (2 mg), and Carboxyl (COOH)–PEG–PLA (16 mg) were dissolved in 1 mL of dichloromethane and then were added into 5 mL of 0.6% sodium cholate aqueous solution. The mixture was intensively emulsified by sonication (200 W, 5 s) 15 times in ice water using a probe sonicator (Scientz Biotechnology Co., Ltd., Ningbo, People’s Republic of China). After evaporating dichloromethane with a ZX-98 rotary evaporator (Shanghai Institute of Organic Chemistry, Shanghai, People’s Republic of China) at 37°C, the obtained NPs were resuspended in 0.5 mL of deionized water.
Tβ4 was conjugated to the surface of NPs using an 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide (EDC)/N-hydroxy-succinimide (NHS) technique.39 In brief, NPs were suspended in deionized water and incubated with excess EDC (200 mM) and NHS (100 mM) at room temperature for 30 min. The resulting N-hydroxysuccinimide-activated NPs were then centrifuged at 14,500 rpm for 45 min to remove the residual EDC and NHS. The activated NPs were allowed to react with 2 mg of Tβ4 under magnetic stirring for 4 h. Thereafter, the covalently linked NP–Tβ4 was concentrated by centrifugation at 14,500 rpm for 45 min to remove free Tβ4. CREKA–NP–Tβ4 was developed via MAL–thiol coupling reaction.40 NP–Tβ4 was suspended in phosphate-buffered saline (PBS; 0.01 M, pH =7.4) and reacted with 0.035 mg CREKA under magnetic stirring for 4 h at room temperature. Then, CNP–Tβ4 was concentrated by centrifugation at 14,500 rpm for 45 min to remove free CREKA. Fluorescence tracker 1,1-dioctadecyl-3,3,3,3-tetramethylindodicarbocyanine (DiD) or 1,10-dioctadecyl-3,3,30,30-tetramethylindo-tricarbocyanine iodide (DiR)-loaded NPs, including conventional NPs, NP–Tβ4, and CNP–Tβ4, were developed as blank conventional NPs, NP–Tβ4, and CNP–Tβ4 except that 30 μg of DiD or 200 μg of DiR was dissolved in 1 mL of dichloromethane in advance.
Characterization of CNP–Tβ4
Particle size and zeta potential of NPs were determined by dynamic light scattering using a zeta plus analyzer (Zetasizer Nano ZS; Malvern Instruments, Malvern, UK). The morphology of NPs was observed using a transmission electron microscope (TEM; H-600; Hitachi, Tokyo, Japan) after negative staining with 2% sodium phosphotungstate solution.
Peptide conjugation efficiency and peptide density on the NP surface
The concentration of Tβ4 in the supernatant was determined using a high-performance liquid chromatography (HPLC) system (Agilent 1200 series; Agilent, Santa Clara, CA, USA) with an analytical column (150×4.6 mm; pore size 5 μm; ZORBAX 300SB-C18; Agilent). The mobile phase contained a mixture of solvent A (0.1% trifluoroacetic acid in water) and solvent B (80% acetonitrile solution containing 0.09% trifluoroacetic acid; solvent A was increased from 25% to 37%, and solvent B was decreased from 75% to 25% in 12 min) at a flow rate of 1.0 mL/min. The sample injection volume was 20 μL, and the detector wavelength was 220 nm. The concentration of CREKA in the supernatant was analyzed using the same procedure except that the mobile phase was A:B = (92:8, v/v).
The Tβ4 and CREKA conjugation efficiency was calculated as follows: Conjugation efficiency = (total amount − the amount in supernatant)/total amount ×100%. The density on the NP surface was calculated as n = ([NA × Peptideconjugated/M])/([m/ρ] ÷ [4/3]π[D/2]3), where NA is the Avogadro’s constant, M the molar mass of peptide, m the weight of NPs, D the mean diameter of NPs, and ρ the NPs estimated to be 1.1 g/cm3.38
In vitro clot binding
Fibrin clots were formed on microscope slides. Each slide received 20 mL of fresh-frozen plasma (FFP), 2 mL of 0.4 M CaCl2, and 2 mL of thrombin (0.1 U/mL PBS). Slides were incubated at 37°C for 60 min, at which time the respective agent was added. The NP preparations (PBS, NP–Tβ4, and CNP–Tβ4) were then added to each clot and then incubated at 37°C for an additional 15 min. The clots were then washed thrice with PBS and imaged using a fluorescence microscope (Leica Microsystems, Wetzlar, Germany).
Pharmacokinetics of CNP–Tβ4 in mouse
For the pharmacokinetics study, adult male BALB/c mice weighing 25±2 g were intravenously injected with 0.1 mL of CNP–Tβ4 solution (containing 150 μg of Tβ4) and their blood was collected at preset time points via cheek pouch puncture. The concentration of Tβ4 in blood was measured by an enzyme-linked immunosorbent assay (ELISA) method and expressed as percentage of injected dose (% ID)/mL. The pharmacokinetics parameters including area under the concentration time curve (AUC0–t), mean residence time (MRT0–t), the maximum concentration (Cmax), and the half-life (t1/2) were calculated by the DAS 3.0 software.
Mouse model of myocardial IR
The animal experiments were approved by the Animal Care and Use Committee of Zhongshan Hospital and were in compliance with the “Guide for the Care and Use of Laboratory Animals” published by the National Academy Press (NIH Publication No 85-23, revised 1996). Adult male BALB/c mice (25±2 g) were purchased from the Shanghai SLAC Laboratory Animal Ltd. (Shanghai, China) and maintained under standard housing conditions.
We subjected wild-type BALB/c mice to transient ligation of left anterior descending (LAD) coronary artery for 90 min followed by reperfusion.41 Successful acute IR injury was confirmed not only by visual inspection of left ventricle (LV) color alteration but also by ST-segment-characterized electrocardiogram.
Detection of fibrin expression in the mouse model of IR
After different times of reperfusion (0 h, 3 h, 6 h, 12 h, 24 h, 2 days, 3 days, 4 days, 5 days, 6 days, 7 days, 10 days, 14 days), animals were euthanized by a lethal dose of thiopentone sodium followed by heart perfusion with saline and 4% paraformaldehyde. Then, hearts were embedded in optimal cutting temperature compound and fresh frozen. Sham-operated mice were killed and used as the control group. Each heart was cut for a transverse section (5-mm thick). Fibrin was detected by immunofluorescence using polyclonal rabbit anti-fibrinogen/fibrin primary antibody following the instruction of the manufacturer. Expression of fibrin was quantitatively assessed and expressed as a percentage of fibrin area of the LV area.
Ex vivo optical imaging of mouse model of IR
IR mice were randomly divided into three groups and injected with DiR-labeled NP–Tβ4, CNP–Tβ4 (the dose of DiR was 0.5 mg/kg), or PBS via the tail vein after reperfusion, respectively. The animals were perfused with 4% paraformaldehyde at various time points (12 h, 24 h, 3 days, and 7 days). The heart and other major organs, including lung, spleen, liver, kidney, and brain, were harvested, and ex vivo imaging of organs was captured using the in vivo IVIS Spectrum imaging system (PerkinElmer Inc., Waltham, MA, USA).
Histological analysis for frozen slices of hearts
After ex vivo optical imaging, hearts were embedded in optimal cutting temperature compound and then fresh frozen. Serial 5-μm thick CryoStar sections were obtained for laser scanning confocal microscopy imaging (LSM710; Carl Zeiss Meditec AG, Jena, Germany). Fibrin was detected and located by immunofluorescence using polyclonal rabbit anti-fibrinogen/fibrin primary antibody following the instruction of the manufacturer. Laser scanning confocal microscopy was performed on fresh-frozen heart sections to detect NPs and fibrin.
Cardiac functional assessments
IR mice were randomly divided into four groups and injected with PBS, Tβ4 (150 μg Tβ4), NP–Tβ4 (containing 150 μg Tβ4), or CNP–Tβ4 (containing 150 μg Tβ4) via the tail vein injection right after reperfusion, respectively. After 4 weeks, the anesthetized mice were investigated for cardiac function by random-blind ultrasonography by multiple measurements of cardiac contraction. Cardiac functional parameters were performed via echocardiography as described previously.5
Histochemical and immunohistochemical assessments
At 7, 14, and 28 days after systemic injection of PBS, Tβ4, NP–Tβ4, or CNP–Tβ4, hearts were harvested and cut into 5-μm cryosections or paraffin-embedded sections. Fibrous tissue was identified by staining sections with an Accustain Trichrome Stains (Masson) kit. The TUNEL assay was performed using ApopTag® Fluorescein In Situ Apoptosis Detection Kit. The reactivation of Wilms tumor 1 (Wt-1)-positive epicardium-derived cells (EPDCs) was evaluated by staining sections with rabbit polyclonal anti-Wt-1 protein primary antibody, and cardiac cells were identified by staining sections with mouse monoclonal anti-Cardiac Troponin T primary antibodies against cardiac troponin T. Cell proliferation was evaluated by staining sections with rabbit polyclonal anti-Ki67 primary antibody. Vasculogenesis was evaluated by staining sections with rabbit polyclonal anti-cluster of differentiation 31 (CD31) primary antibodies and anti-smooth-muscle action (SMαA) antibodies.
Western blot analysis
One week after systemic injection of PBS, Tβ4, NP–Tβ4, or CNP–Tβ4, mice were sedated. Hearts were perfused with saline to remove blood, and the infarcted core tissue was removed. The remaining intact area of the hearts was separated, placed in 1 mL of TRIzol reagent, and frozen in liquid nitrogen. The protein fraction was isolated from the interphase of the TRIzol purification as recommended by the manufacturer. In all, 20 μg of the total protein was loaded and analyzed by Western blot analysis with protein-specific primary antibodies (Wt-1, CD31, and SMαA).
Statistical analysis
Animal numbers used in each treatment group are shown in Table 1. Results are presented as mean ± standard deviation (sd) unless specified otherwise. Comparisons between any two groups were performed using two-tailed unpaired Student’s t-test. Comparisons between more than two groups were performed using one-way analysis of variance (ANOVA) followed by post-hoc Bonferroni test. Differences were considered statistically significant when P<0.05.
 | Table 1 Study protocol and rat numbers used in each experiment |
Results
Characterization of NPs
As shown in Figure 1, particle sizes for NPs, NP–Tβ4, and CNP–Tβ4 were 105.0±1.6 nm, 117.6±1.0 nm, and 121.4±2.6 nm, respectively. Zeta potentials for NPs, NP–Tβ4, and CNP–Tβ4 were −10.0±0.86 mV, −8.38±0.61 mV, and −8.58±0.39 mV, respectively. CNP–Tβ4 showed a regular size of ~100 nm, which was observed by a TEM (Figure 1B). Under our experimental conditions (molar ratio of COOH–PEG–PLA to Tβ4 =1:1, reaction time =4 h), the Tβ4 conjugation efficiency was ~86.8%±4.5% and Tβ4 density on the NP surface was ~3,731.8±134.9 molecules per NP. The covalent attachment of CREKA to NPs was realized by means of the sulfhydryl groups of thiolated CREKA and the MAL groups on the surface of NPs. Under our experimental conditions (molar ratio of MAL–PEG–PLA to sulfhydryl of CREKA =1:1 and incubation time for conjugation reaction =4 h), the CREKA conjugation efficiency was ~98.5%±2.0% and the CREKA density on the surface of NPs was ~588.9±10.1 molecules per NP.
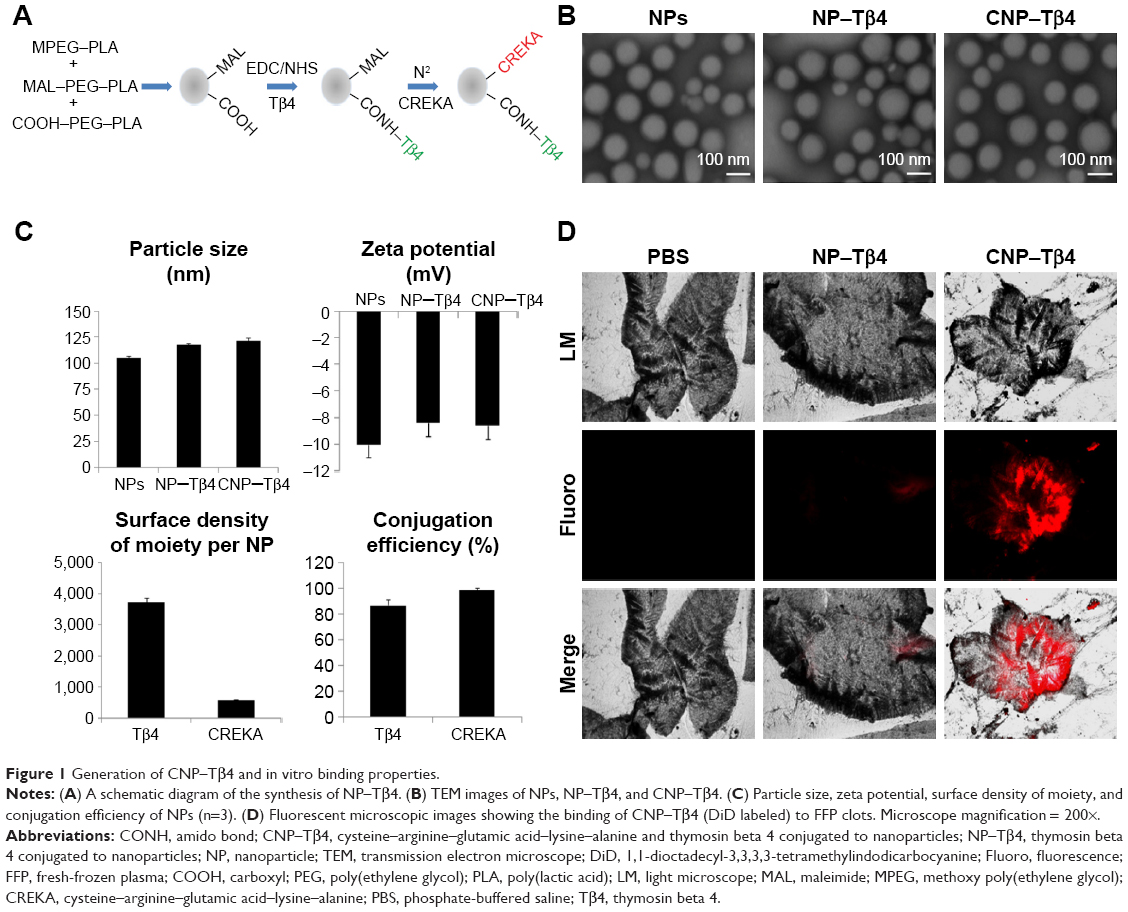 | Figure 1 Generation of CNP–Tβ4 and in vitro binding properties. |
In vitro clot binding of CNP–Tβ4
The targeting ability of the synthesized agents in vitro was tested with FFP clots. For imaging of fluorescence microscope, fibrin clots were formed on microscope slides. Fibrin clots, which were incubated with CNP–Tβ4, revealed fluorescence on the surface of clots, whereas there was no significant binding to the control clots (NP–Tβ4; Figure 1D).
Pharmacokinetics of CNP–Tβ4 in mice
The pharmacokinetics of CNP–Tβ4 was investigated by an intravenous injection of CNP–Tβ4 in healthy BALB/c mice. As shown in Figure 2, both CNP–Tβ4 and NP–Tβ4 displayed a significant decrease in plasma Tβ4 clearance compared with Tβ4. There were no significant differences in AUC0–t, MRT0–t, and t1/2 between NP–Tβ4 and CNP–Tβ4, indicating that CREKA conjugation on NPs did not significantly change the pharmacokinetics of NP–Tβ4.
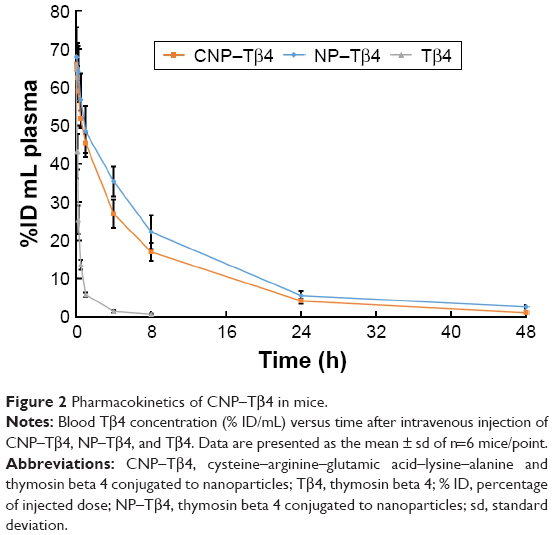 | Figure 2 Pharmacokinetics of CNP–Tβ4 in mice. |
Validation of fibrin expression in the mouse model of IR
An abundant existence of fibrin in the infarcted area after reperfusion was demonstrated by immunofluorescent staining and distributed in extracellular spaces as a kind of ECM (Figure 3). Quantitative analysis demonstrated that the fibrin content in the infarcted area peaked during the early phase of healing (3–48 h reperfusion) but markedly decreased after 7 days of reperfusion (n=5 at each time point). The time frame of fibrin expression was nearly coincident with the effective acting time of Tβ4,25 indicating that fibrin might serve as the target of CREKA-functioned NPs.
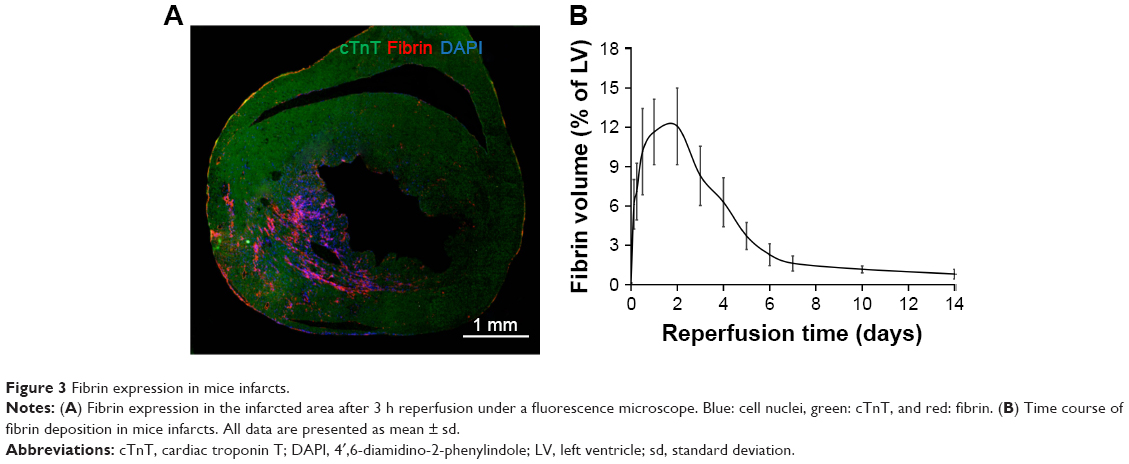 | Figure 3 Fibrin expression in mice infarcts. |
In vivo distribution of CNP–Tβ4 in the mouse model of IR
The in vivo distribution and accumulation of DiR-labeled CNP–Tβ4 were determined in the IR model of mice with the ex vivo imaging of heart. As shown in Figure 4A, the signal in the heart of CNP–Tβ4-treated mice increased slowly and reached the peak value at 24 h. Compared with the NP–Tβ4 group, the fluorescence of the CNP–Tβ4 group was significantly higher at any time point post administration, which ranged from 12 h to 7 days (P<0.05, n=5), suggesting that the modification of NPs with CREKA could improve the accumulation of NPs in the infarcted area. As to other major organs, CNP–Tβ4 distribution was almost the same as NP–Tβ4, mainly distributed in the mononuclear phagocyte system (MPS)-related organs, including liver and spleen (Figure 4B).
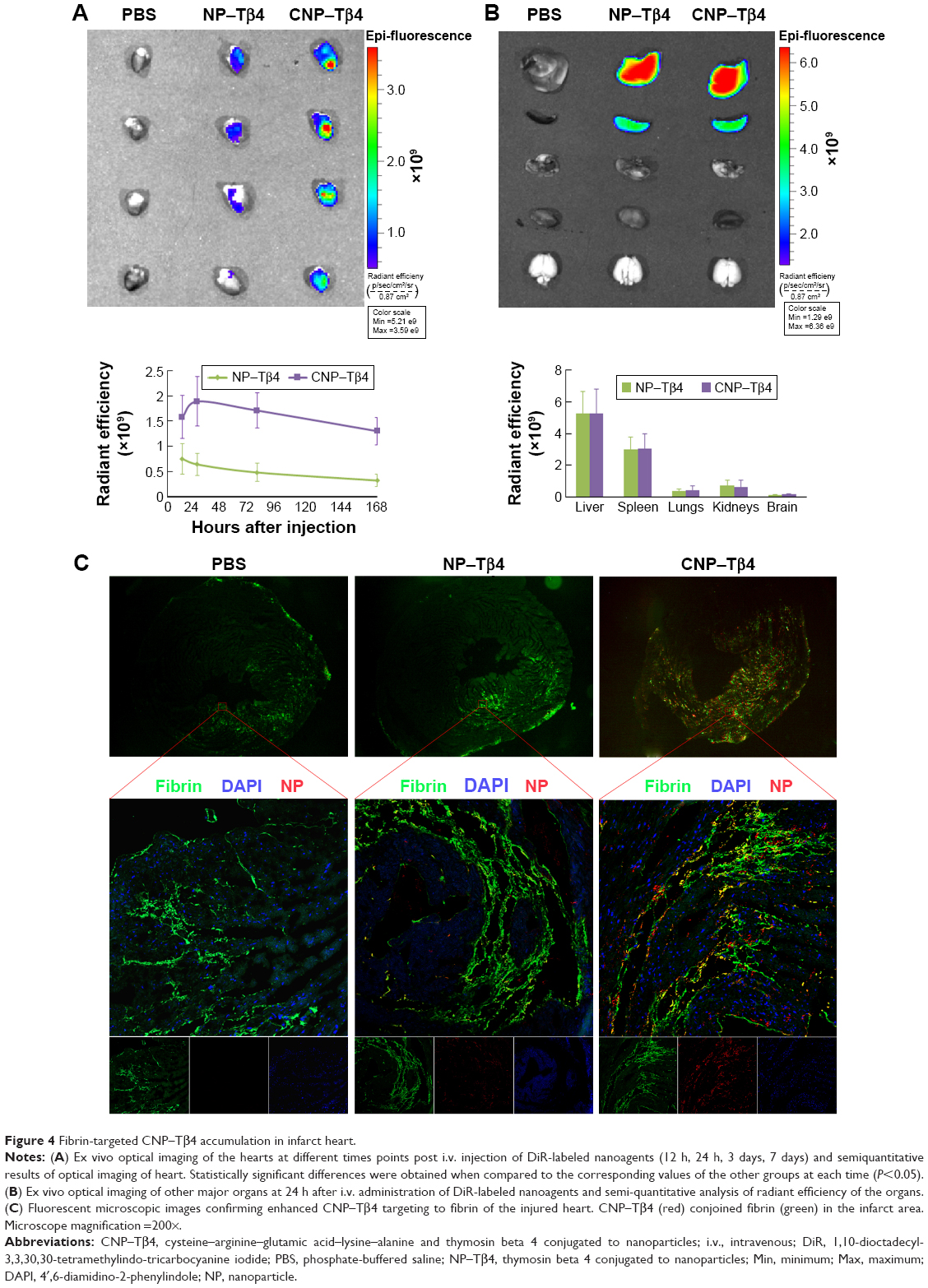 | Figure 4 Fibrin-targeted CNP–Tβ4 accumulation in infarct heart. |
Co-localization of NPs in the injured area of heart
As fibrin was abundantly distributed in the infarcted area of heart, especially in the early stage of IR, it was an optimal therapeutic target for its accessibility and abundance. As shown in Figure 4C, the CNP–Tβ4 group revealed stronger signal than the NP–Tβ4 group and accumulated mainly in the infarcted area and colocalized with fibrin. In the NP–Tβ4 group, the signals of NPs were also found in the infarcted area because of enhanced vascular permeability and retention (EPR) effect. These results indicated that CREKA conjugation to the NPs could significantly facilitate the accumulation of NPs to the targeted site and a better therapeutic effect could be expected.
CNP–Tβ4 administration improves cardiac functional recovery after IR
Because of the effects of Tβ4 to enhance early myocyte survival and improve cardiac function, we tested whether CNP–Tβ4 might aid in cardiac repair in vivo after myocardial damage. We created IR injury in mice by transient coronary artery ligation and recanalized artery after 90 min to perfuse NPs to the infarcted area. After recanalization, we treated mice with PBS, Tβ4 (150 μg), NP–Tβ4 (containing 150 μg Tβ4), or CNP–Tβ4 (containing 150 μg Tβ4) via the tail vein injection. After 4 weeks, the mice were investigated for cardiac function by random-blind ultrasonography and trichrome staining.
As shown in Figure 5A–D, there was no improvement in cardiac fractional shortening or ejection fraction of the Tβ4 group, which treated mice with 150 μg Tβ4 via the tail vein injection. However, there was a significant improvement in the CNP–Tβ4 group, which suggested a better therapeutic effect with accumulation of CNP–Tβ4 in the targeted site. In addition, the NP–Tβ4 group had improved cardiac function because of the EPR effect, but did not reach a significant difference. Active targeting with CREKA facilitated the benefits. Moreover, the end-diastolic diameter (EDDs) and end-systolic diameter (ESDs) were significantly higher in the control group, indicating that treatment with CNP–Tβ4 resulted in decreased cardiac dilation after IR injury, consistent with improved function.
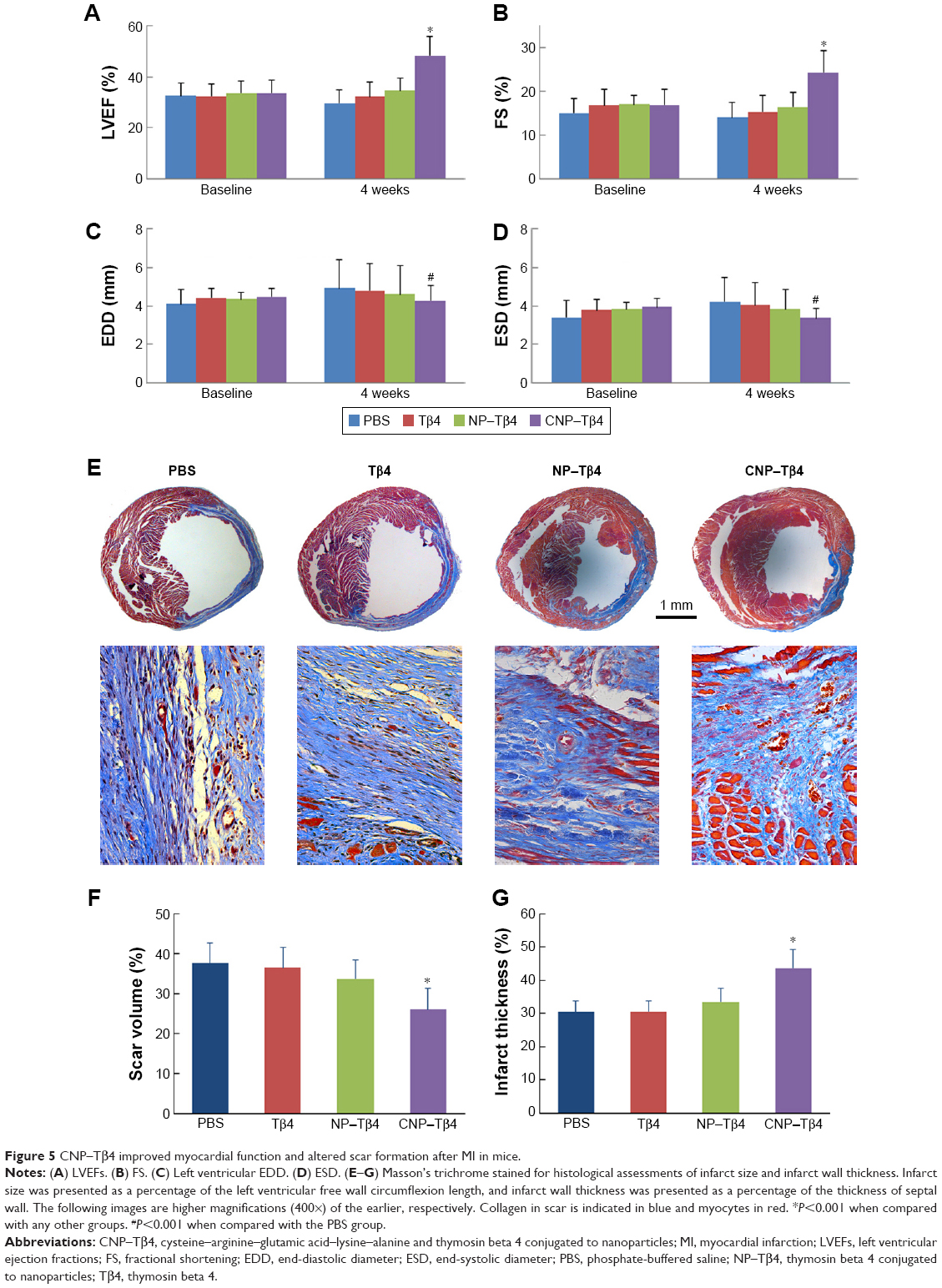 | Figure 5 CNP–Tβ4 improved myocardial function and altered scar formation after MI in mice. |
Trichrome stain revealed structural improvements at week 4 after injury (Figure 5E–G). The size of scar was significantly reduced by 30.9% in mice treated with CNP–Tβ4. The infarcted region of the wall was significantly thicker in the CNP–Tβ4 group than in any other group (Figure 5G).
CNP–Tβ4 administration promotes cardiac survival after IR
The numbers of TUNEL-positive cardiomyocytes in the peri-infarct region were remarkable in the control group and the Tβ4-treated group after 7 days of MI. NP–Tβ4 treatment markedly reduced TUNEL-positive cells (P<0.05 compared with the control group), and CNP–Tβ4 treatment enhanced the protective effect (P<0.001 compared with each other group; Figure 6A and B). With CNP–Tβ4 treatment, the infarcted heart revealed an increase in cell proliferation in the peri-infarct region, which was determined by the presence of Ki67-positive cells after 7 days of post-IR as compared to other groups (Figure 6C and D).
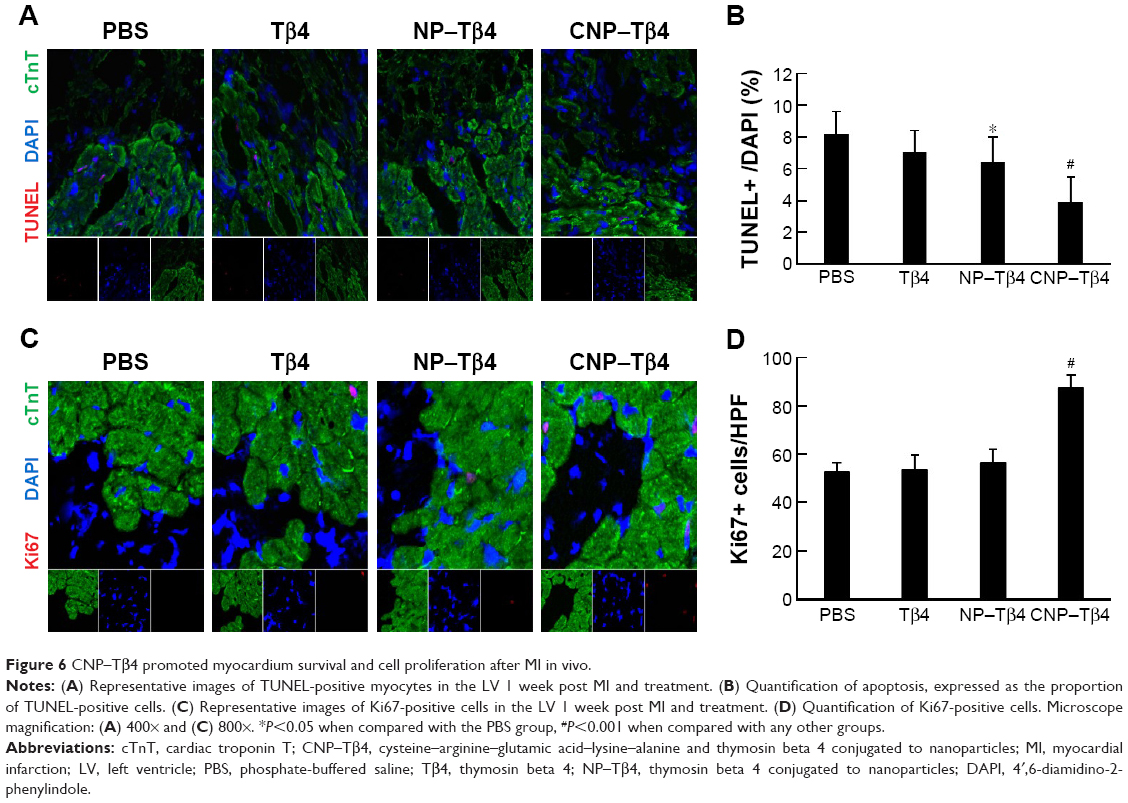 | Figure 6 CNP–Tβ4 promoted myocardium survival and cell proliferation after MI in vivo. |
CNP–Tβ4 reactivates the quiescent adult epicardium
Results of previous studies showed that Tβ4 might activate the adult epicardium and initiate the expression of embryonic development genes in adult mice. Wt-1 is a biomarker for the development of embryonic heart and is not normally expressed in an adult heart. We exploited the ability of CNP–Tβ4 to reactivate the adult epicardium in order to restore Wt-1 expression. Figure 7A and B shows that CNP–Tβ4 raises the expression of Wt-1 and significantly increases the number of Wt-1 positive cells after 7 days. Western blot analysis showed that Wt-1 expression was 1.49-fold higher than that in the control group (Figure 7C and D).
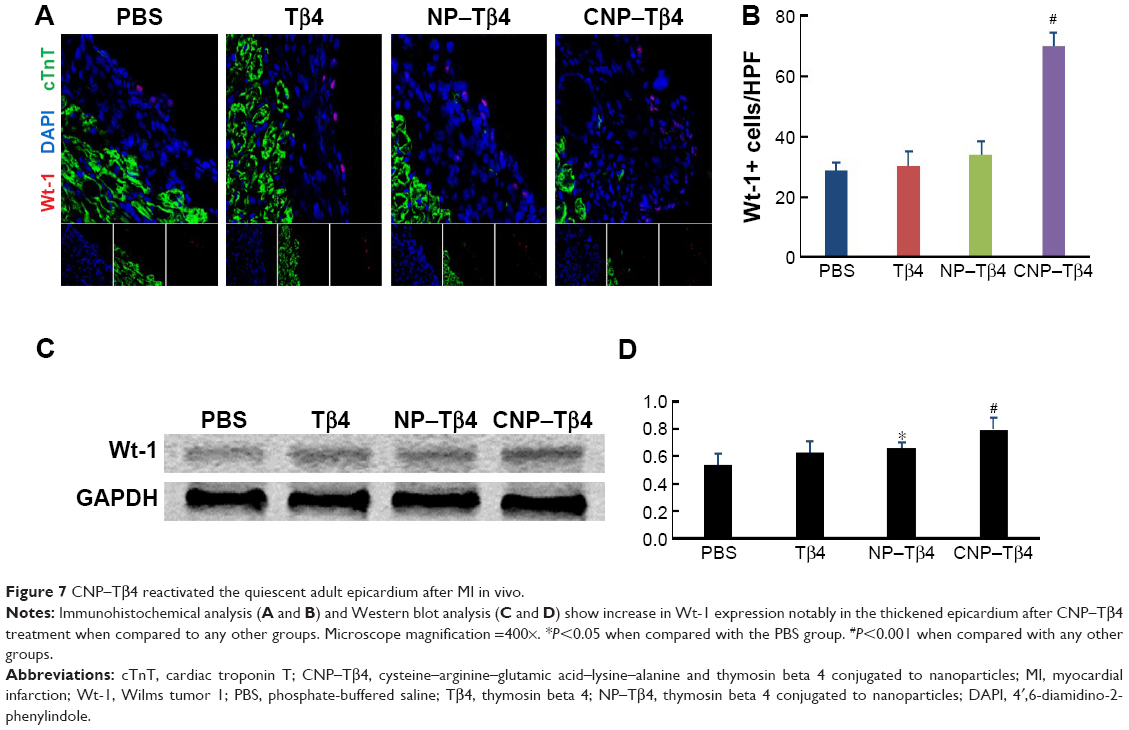 | Figure 7 CNP–Tβ4 reactivated the quiescent adult epicardium after MI in vivo. |
CNP–Tβ4 enhances the neovascularization after IR
We next investigated whether CNP–Tβ4 could promote neovascularization after MI by measuring arteriole density and capillary density in tissue sections and staining for expression of the smooth muscle marker smooth muscle α-actin (SMαA) and the endothelial-cell marker CD31 after 2 weeks post infarction. As shown in Figure 8, arteriole density (ie, the number of SMαA+ vessels) was similar in the control group, Tβ4 group, and NP–Tβ4 group and significantly increased in the CNP–Tβ4 group (P<0.001 compared to each other group). CD31+ vessel density was significantly higher in sections from the hearts of CNP–Tβ4-treated mice than in sections from the hearts of the control group (P<0.001), but that in sections from the hearts of the Tβ4 group and NP–Tβ4 group was similar to that in sections from the hearts of the control group. The results were corroborated by significantly increased CD31 and SMαA protein expression (1.52-fold and 1.44-fold, respectively, compared to the control group, after 7 days post infarction).
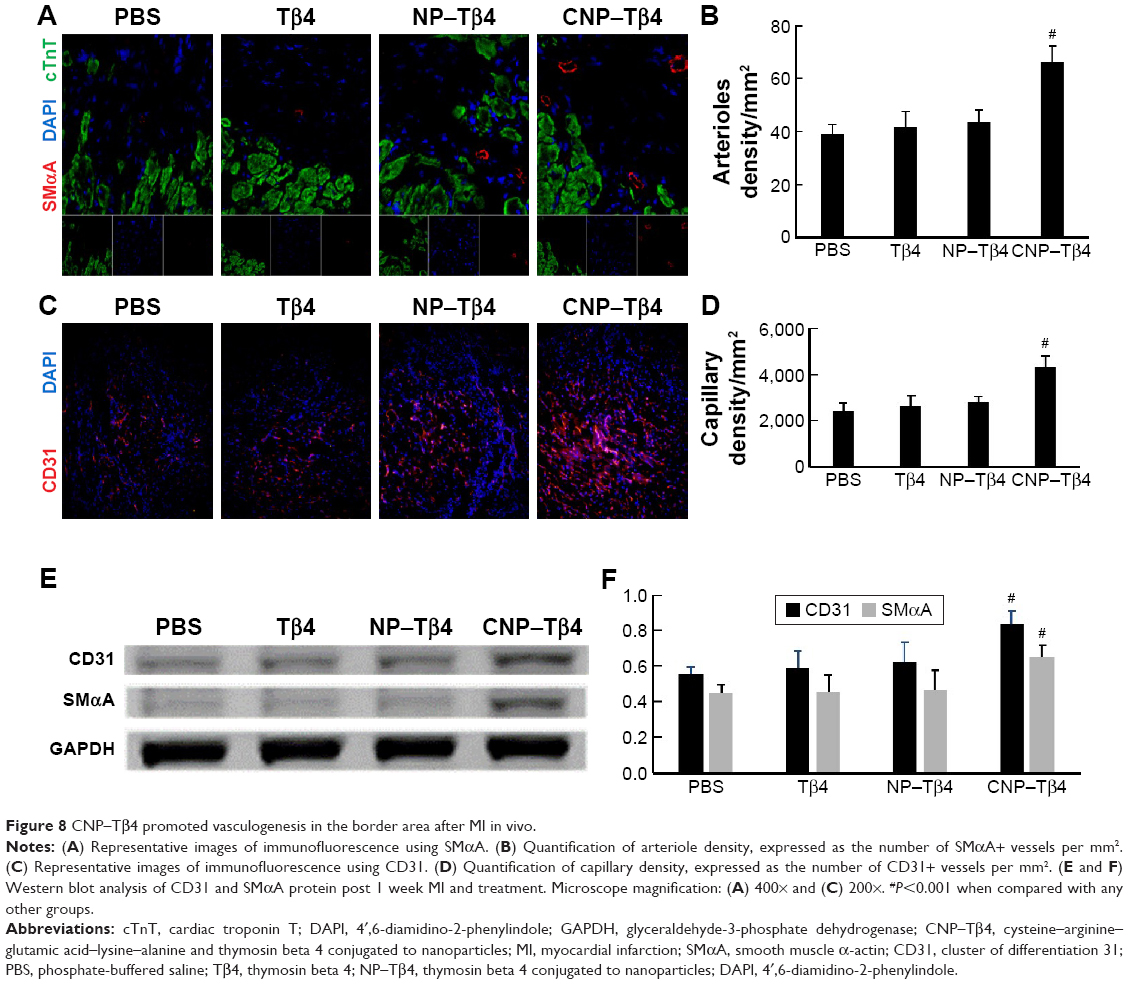 | Figure 8 CNP–Tβ4 promoted vasculogenesis in the border area after MI in vivo. |
Discussion
In this study, we developed a fibrin-targeting drug delivery system CNP–Tβ4 for myocardial injury treatment, which used NPs as the drug carrier and CREKA as the targeting moiety. The fibrin-targeting efficacy of CNP–Tβ4 was validated both in vitro and in vivo. CNP–Tβ4 treatment was associated with significant structural and functional improvements at week 4 after cardiac injury in mice, whereas NP–Tβ4 alone produced only marginal functional benefit, emphasizing the utility of active fibrin targeting.
The first prerequisite for biologically targeting therapy is to find a good target element, which should exclusively distribute in the injured region within therapeutic time window. The fibrin-based provisional matrix is the “first order” and most important provisional matrix after MI.25,33 In agreement with previous studies,26 our results revealed that fibrin was expressed in the acute phase and lasted during the proliferative phase, which lasted from 48 h to 5 days25 after MI in rodents. Abundant fibrin was visualized in the infarcted area after 3 h of reperfusion, peaked during 24–48 h, and markedly decreased after 7 days of reperfusion. Owing to the multiple benefits of Tβ4, including early injury responses (prosurvival and anti-inflammatory activities) and long-term regenerative functions (reactivation of the quiescent adult epicardium and neovascularization) for IR, the time frame of fibrin expression was nearly coincident with the effective acting time of Tβ4. Additionally, fibrin was specifically and predominantly expressed in the infarcted area. The spatiotemporal characteristics of fibrin expression after injury, including specific appearance in the infarcted area spatially and in the early repair phase temporarily, render fibrin a perfect target for Tβ4 delivery. Recently, type I collagen had been used as the target to retain vascular endothelial growth factor (VEGF) in the border zone.42 As a target, fibrin is theoretically superior to collagen, which exists in normal myocardium, but decreases in the infarct zone after MI.43
CREKA has been identified as a tumor-homing peptide that recognizes clotting products in the vessels of a variety of tumor types by in vivo phage library screening. It was shown to specifically and quantitatively bind to fibrin in the blood vessels and stroma of tumor.34 In recent years, this peptide has been used in the targeting detection of fibrin in both atherosclerosis35 and microthrombus.36 Meanwhile, the surface density of ligands on NPs is an important factor that determines multivalent effects of ligands-modified NPs, which can dramatically enhance targeting specificity and efficiency of drug delivery and has been successfully verified in tumor imaging and therapy.44,45 In this study, >580 CREKA molecules were distributed over the surface of each NP with a size of 100 nm. The high density of CREKA molecules on NPs will provide a sufficient multimetric association between NPs and fibrin in the infarcted area, which would greatly increase the local concentration of CNP–Tβ4 at the targeted site and hence improve the potential benefit. Additionally, PEG-modified NPs could increase the drug delivery capacity and prolong drug plasma half-time,46 increasing the opportunity to achieve the targeted site.
Delivery of CNP–Tβ4 clearly alleviated cardiac dysfunction and reduced scar size, which might be closely related to its effect in the acute and healing phase. In the present study, CNP–Tβ4 was delivered into the infarcted zone immediately after IR injury and retained an adequate concentration over time. In the acute phase, CNP–Tβ4 reduced myocardial apoptosis and improved cardiomyocyte viability after ischemia. During the healing phase, CNP–Tβ4 reactivated quiescent adult epicardium and enhanced blood vessel growth, which may subsequently attenuate infarct extension as a result of secondary ischemic myocyte loss. During the post-healing period, the angiogenesis triggered by CNP–Tβ4 may be sustained, which would result in improvement of cardiac function. Meanwhile, Tβ4 and NP–Tβ4 were less efficient in preserving cardiac function than CNP–Tβ4, because of poor homing to the target organ and the “washout” effect,47 whereas CNP–Tβ4 was retained in the infarcted area. The high binding ability of CNP–Tβ4 to the infarcted area increased the retention of Tβ4 and thus improved its therapeutic efficiency.
Tβ4 had multiple beneficial effects of myocardial repair; however, the conventional methods of administration, including multiple IP injections or direct intramyocardial injection, were inconvenient and invasive. This has become a limitation for its clinical transformation. Peripheral systemic intravenous administration is the most convenient route of drug delivery, offering the advantages of a nonsurgical and non-interventional method that can be performed repeatedly. However, it suffers from paltry homing to the target organ, because Tβ4 within the infarct mainly originated from the EPR effect after ischemic injury, and the low local concentration might be further decreased by the “washout” effect. The fibrin-homing features of CNP–Tβ4 may revive the intravenous or intracoronary injection as a feasible route for Tβ4 therapy in myocardial injury. Another advantage of CNP–Tβ4 targeting system is the decrease in the overall dose of Tβ4. IP administration of Tβ4 resulted in the use of 600 μg Tβ4 in total,5,9 as compared with 150 μg used in our study. It will be essential to lower the underlying possibility of inducing cancerous growth when realizing its full potential benefits.
Available data showed that Tβ4 was a safe and well-tolerated molecule, and the relation of malignancy to Tβ4 was not verified.19 Similarly, malignant tumor was not observed in the myocardium in our study. However, the CNP–Tβ4 targeting delivery system notably elevated the concentration of Tβ4 in local infarct, and Tβ4 was upregulated in certain types of malignancy.20,21 Therefore, the potential risk of carcinogenicity in Tβ4-targeting delivery system needs further study.
The fibrin-targeting NP platform described here has a potentially wide application value in regenerative medicine, far more than Tβ4 therapy in myocardial repair. Provided therapeutic agents (eg, exogenously administered cells, growth factors, peptides, or small molecule compounds) could be connected with CREKA effectively and efficiently, the fibrin-targeting system could be capable of localizing systemically delivered cells/drugs to the injured heart, enhancing their efficacy and reducing their dosing.33,48 In addition, fibrin is the common provisionally pathological matrix after tissue injury; it is logical to assume that the strategy could also be applicable in any other organ injury or lesions within a defined time window.
Conclusion
The present study established a fibrin-targeting drug delivery system CNP–Tβ4 for myocardial injury treatment in mice. CREKA modification favored Tβ4 accumulation and retention in the infarcted region, leading to augmented functional benefits. Fibrin-targeting delivery system represents a generalizable platform technology for regenerative medicine.
Acknowledgments
The authors gratefully acknowledge financial support from the National Natural Science Foundation of China (grants 81600199, 81570223, 81370003, 81500201, and 81521001) and the Science and Technology Commission of Shanghai Municipality (grant 13JC1401703). The authors thank Xiangdong Yang for his contribution to the interpretation of data.
Disclosure
The authors report no conflicts of interest in this work.
References
Porrello ER, Mahmoud AI, Simpson E, et al. Transient regenerative potential of the neonatal mouse heart. Science. 2011;331(6020):1078–1080. | ||
Reinecke H, Minami E, Zhu WZ, Laflamme MA. Cardiogenic differentiation and transdifferentiation of progenitor cells. Circ Res. 2008;103(10):1058–1071. | ||
Smart N, Risebro CA, Melville AA, et al. Thymosin beta-4 is essential for coronary vessel development and promotes neovascularization via adult epicardium. Ann N Y Acad Sci. 2007;1112:171–188. | ||
Smart N, Risebro CA, Melville AA, et al. Thymosin beta4 induces adult epicardial progenitor mobilization and neovascularization. Nature. 2007;445(7124):177–182. | ||
Bock-Marquette I, Saxena A, White MD, Dimaio JM, Srivastava D. Thymosin beta4 activates integrin-linked kinase and promotes cardiac cell migration, survival and cardiac repair. Nature. 2004;432(7016):466–472. | ||
Hinkel R, El-Aouni C, Olson T, et al. Thymosin beta4 is an essential paracrine factor of embryonic endothelial progenitor cell-mediated cardioprotection. Circulation. 2008;117(17):2232–2240. | ||
Srivastava D, Saxena A, Michael Dimaio J, Bock-Marquette I. Thymosin beta4 is cardioprotective after myocardial infarction. Ann N Y Acad Sci. 2007;1112:161–170. | ||
Smart N, Risebro CA, Clark JE, et al. Thymosin beta4 facilitates epicardial neovascularization of the injured adult heart. Ann N Y Acad Sci. 2010;1194:97–104. | ||
Smart N, Bollini S, Dube KN, et al. De novo cardiomyocytes from within the activated adult heart after injury. Nature. 2011;474(7353):640–644. | ||
Peng H, Xu J, Yang XP, et al. Thymosin-beta4 prevents cardiac rupture and improves cardiac function in mice with myocardial infarction. Am J Physiol Heart Circ Physiol. 2014;307(5):H741–H751. | ||
Smart N, Dube KN, Riley PR. Epicardial progenitor cells in cardiac regeneration and neovascularisation. Vascul Pharmacol. 2013;58(3):164–173. | ||
Riley PR, Smart N. Thymosin beta4 induces epicardium-derived neovascularization in the adult heart. Biochem Soc Trans. 2009;37(pt 6):1218–1220. | ||
Evans MA, Smart N, Dube KN, et al. Thymosin beta4-sulfoxide attenuates inflammatory cell infiltration and promotes cardiac wound healing. Nat Commun. 2013;4:2081. | ||
Huther A, Dietrich U. The emergence of peptides as therapeutic drugs for the inhibition of HIV-1. AIDS Rev. 2007;9(4):208–217. | ||
Lin CC, Metters AT. Hydrogels in controlled release formulations: network design and mathematical modeling. Adv Drug Deliv Rev. 2006;58(12–13):1379–1408. | ||
Ruff D, Crockford D, Girardi G, Zhang Y. A randomized, placebo-controlled, single and multiple dose study of intravenous thymosin beta4 in healthy volunteers. Ann N Y Acad Sci. 2010;1194:223–229. | ||
Mora CA, Baumann CA, Paino JE, Goldstein AL, Badamchian M. Biodistribution of synthetic thymosin beta 4 in the serum, urine, and major organs of mice. Int J Immunopharmacol. 1997;19(1):1–8. | ||
Chiu LL, Radisic M. Controlled release of thymosin beta4 using collagen-chitosan composite hydrogels promotes epicardial cell migration and angiogenesis. J Control Release. 2011;155(3):376–385. | ||
Crockford D, Turjman N, Allan C, Angel J. Thymosin beta4: structure, function, and biological properties supporting current and future clinical applications. Ann N Y Acad Sci. 2010;1194:179–189. | ||
Hong KO, Lee JI, Hong SP, et al. Thymosin beta4 induces proliferation, invasion, and epithelial-to-mesenchymal transition of oral squamous cell carcinoma. Amino Acids. 2016;48(1):117–127. | ||
Wirsching HG, Krishnan S, Florea AM, et al. Thymosin beta 4 gene silencing decreases stemness and invasiveness in glioblastoma. Brain. 2014;137(pt 2):433–448. | ||
Kamaly N, Yameen B, Wu J, et al. Degradable controlled-release polymers and polymeric nanoparticles: mechanisms of controlling drug release. Chem Rev. 2016;116(4):2602–2663. | ||
Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv Drug Deliv Rev. 2014;66:2–25. | ||
Mercer SE, Odelberg SJ, Simon HG. A dynamic spatiotemporal extracellular matrix facilitates epicardial-mediated vertebrate heart regeneration. Dev Biol. 2013;382(2):457–469. | ||
Dobaczewski M, Gonzalez-Quesada C, Frangogiannis NG. The extracellular matrix as a modulator of the inflammatory and reparative response following myocardial infarction. J Mol Cell Cardiol. 2010;48(3):504–511. | ||
Dobaczewski M, Bujak M, Zymek P, Ren G, Entman ML, Frangogiannis NG. Extracellular matrix remodeling in canine and mouse myocardial infarcts. Cell Tissue Res. 2006;324(3):475–488. | ||
Zhang B, Shen S, Liao Z, et al. Targeting fibronectins of glioma extracellular matrix by CLT1 peptide-conjugated nanoparticles. Biomaterials. 2014;35(13):4088–4098. | ||
Martino MM, Briquez PS, Guc E, et al. Growth factors engineered for super-affinity to the extracellular matrix enhance tissue healing. Science. 2014;343(6173):885–888. | ||
Joshi N, Kopec AK, Ray JL, et al. Fibrin deposition following bile duct injury limits fibrosis through an alphaMbeta2-dependent mechanism. Blood. 2016;127(22):2751–2762. | ||
Pankajakshan D, Krishnan LK. Design of fibrin matrix composition to enhance endothelial cell growth and extracellular matrix deposition for in vitro tissue engineering. Artif Organs. 2009;33(1):16–25. | ||
Petzelbauer P, Zacharowski PA, Miyazaki Y, et al. The fibrin-derived peptide Bbeta15-42 protects the myocardium against ischemia-reperfusion injury. Nat Med. 2005;11(3):298–304. | ||
Laurens N, Koolwijk P, De Maat MPM. Fibrin structure and wound healing. J Thromb Haemost. 2006;4(5):932–939. | ||
Huang Z, Song Y, Pang Z, et al. Fibrin-targeting delivery: a novel platform for cardiac regenerative medicine. J Cell Mol Med. 2016;20(12):2410–2413. | ||
Zhang B, Wang H, Shen S, et al. Fibrin-targeting peptide CREKA-conjugated multi-walled carbon nanotubes for self-amplified photothermal therapy of tumor. Biomaterials. 2016;79:46–55. | ||
Deshpande D, Kethireddy S, Gattacceca F, et al. Comparative pharmacokinetics and tissue distribution analysis of systemically administered 17-beta-estradiol and its metabolites in vivo delivered using a cationic nanoemulsion or a peptide-modified nanoemulsion system for targeting atherosclerosis. J Control Release. 2014;180:117–124. | ||
Song Y, Huang Z, Xu J, et al. Multimodal SPION-CREKA peptide based agents for molecular imaging of microthrombus in a rat myocardial ischemia-reperfusion model. Biomaterials. 2014;35(9):2961–2970. | ||
Chung EJ, Cheng Y, Morshed R, et al. Fibrin-binding, peptide amphiphile micelles for targeting glioblastoma. Biomaterials. 2014;35(4):1249–1256. | ||
Olivier JC, Huertas R, Lee HJ, Calon F, Pardridge WM. Synthesis of pegylated immunonanoparticles. Pharm Res. 2002;19(8):1137–1143. | ||
Guo J, Gao X, Su L, et al. Aptamer-functionalized PEG-PLGA nanoparticles for enhanced anti-glioma drug delivery. Biomaterials. 2011;32(31):8010–8020. | ||
Zhang B, Wang H, Liao Z, et al. EGFP-EGF1-conjugated nanoparticles for targeting both neovascular and glioma cells in therapy of brain glioma. Biomaterials. 2014;35(13):4133–4145. | ||
Korf-Klingebiel M, Reboll MR, Klede S, et al. Myeloid-derived growth factor (C19orf10) mediates cardiac repair following myocardial infarction. Nat Med. 2015;21(2):140–149. | ||
Zhang J, Ding L, Zhao Y, et al. Collagen-targeting vascular endothelial growth factor improves cardiac performance after myocardial infarction. Circulation. 2009;119(13):1776–1784. | ||
Cleutjens JP, Kandala JC, Guarda E, et al. Regulation of collagen degradation in the rat myocardium after infarction. J Mol Cell Cardiol. 1995;27(6):1281–1292. | ||
Li MH, Choi SK, Thomas TP, et al. Dendrimer-based multivalent methotrexates as dual acting nanoconjugates for cancer cell targeting. Eur J Med Chem. 2012;47(1):560–572. | ||
Hong S, Leroueil PR, Majoros IJ, Orr BG, Baker JR Jr, Banaszak Holl MM. The binding avidity of a nanoparticle-based multivalent targeted drug delivery platform. Chem Biol. 2007;14(1):107–115. | ||
Gref R, Minamitake Y, Peracchia MT, Trubetskoy V, Torchilin V, Langer R. Biodegradable long-circulating polymeric nanospheres. Science. 1994;263(5153):1600–1603. | ||
Grossman PM, Han Z, Palasis M, Barry JJ, Lederman RJ. Incomplete retention after direct myocardial injection. Catheter Cardiovasc Interv. 2002;55(3):392–397. | ||
Huang Z, Shen Y, Sun A, et al. Magnetic targeting enhances retrograde cell retention in a rat model of myocardial infarction. Stem Cell Res Ther. 2013;4(6):149. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.