Back to Journals » Cancer Management and Research » Volume 13
Targeted Deep Sequencing Reveals Unrecognized KIT Mutation Coexistent with NF1 Deficiency in GISTs
Authors Wu J, Zhou H, Yi X, He Q, Lei T ![]() , Tan F, Liu H
, Tan F, Liu H ![]() , Li B
, Li B
Received 14 September 2020
Accepted for publication 10 December 2020
Published 12 January 2021 Volume 2021:13 Pages 297—306
DOI https://doi.org/10.2147/CMAR.S280174
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Eileen O'Reilly
Jinchun Wu,1 Haiyan Zhou,2 Xiaoping Yi,3 Qiongzhi He,4 Tianxiang Lei,5 Fengbo Tan,5 Heli Liu,5 Bin Li1
1Department of Oncology, Xiangya Hospital, Central South University, Changsha 410008, People’s Republic of China; 2Department of Pathology, Xiangya Hospital, Central South University, Changsha 410008, People’s Republic of China; 3Department of Radiology, Xiangya Hospital, Central South University, Changsha 410008, People’s Republic of China; 4Geneplus-Beijing Institute, Beijing, People’s Republic of China; 5Department of General Surgery, Xiangya Hospital, Central South University, Changsha 410008, People’s Republic of China
Correspondence: Heli Liu; Bin Li Email [email protected]; [email protected]
Purpose: NF1-deficient GISTs account for about 1% of gastrointestinal stromal tumors (GISTs) and are usually considered as a subtype of KIT/PDGFRA wild-type GISTs that have no detectable KIT and PDGFRA mutations. Some KIT/PDGFRA wild-type GISTs actually have cryptic KIT mutations (mKIT). So we investigate whether concurrent mKIT existed in NF1-associated GISTs.
Patients and Methods: Three independent cohorts were retrospectively analyzed. KIT/PDGFRA wild-type GISTs in Xiangya Hospital between May 2017 and Oct 2019 were investigated by next-generation sequencing (NGS) approach targeted 1021 cancer-related genes regions. GISTs cases in Gene+ dataset from May 2017 to May 2020 were collected from the platform of this company. The genotypes of GISTs in MSKCC cohort were downloaded from cBioPortal.
Results: A total of 290 cases including 23 KIT/PDGFRA wild-type GISTs in Xiangya Hospital, 136 GISTs in Gene+ database, and 131 GISTs in MSKCC were enrolled. Twenty-six cases have NF1 mutations (mNF1), and 48% (12/26) of NF1-mutated GISTs have concurrent mKIT. Compared with MSKCC (2/10, 20%), a higher ratio of mKIT in NF1-associated GISTs was detected in Xiangya Hospital (3/5, 60%) and Gene+ (7/11, 64%) (p< 0.05). No mutation hotspot existed in mNF1. Most of mKIT centered within exon 11 (7/12, 58%) and others including exon 17 (3/12, 25%), exon 9(1/12, 8%), exon 13 (1/12, 8%) and exon 21 (1/12, 8%). No differences in age, gender, and location were detected between NF1-related GISTs with mKIT and those without mKIT. Three GIST cases of type I neurofibromatosis, skin neurofibromas and micro-GISTs (≤ 1 cm) were devoid of mKIT, but all the mini-GISTs (1∼ 2 cm) and clinic GIST lesions (> 2 cm) in two cases harbored mKIT.
Conclusion: mKIT was not unusual in NF1-associated GISTs, especially in Chinese populations. The gain-of-function mKIT possibly facilitated the progression of NF1-deficient lesions to clinic GISTs, however, the underlying mechanism warrants further studies.
Keywords: gastrointestinal stromal tumor, NF1, KIT, deep sequencing
Introduction
Gastrointestinal stromal tumors (GISTs) are mesenchymal tumors usually located in the gastrointestinal tract. Most GISTs are sporadic and have oncogenic KIT or PDGFRA mutations. About 10–15% GISTs lack KIT/PDGFRA mutation and are classified as KIT/PDGFRA wild-type GISTs.1 GISTs are a heterogeneous group that has various entities including SDH-defective, BRAF-mutated, NF1-deficient GISTs and other GISTs with rare gene abnormalities.1
Neurofibromin (NF1) gene encodes a protein named neurofibromin. It negatively regulates Ras family GTPases to maintain the inactive form (RAS-GDP) of RAS oncoprotein. NF1 gene mutation causes defective neurofibromin protein that leads to loss of its inhibitory role on the activation of RAS oncoprotein.2 Individuals with NF1 deficiency carry a 34-fold risk than the average population for developing GIST, and GISTs occur in about 5–25% of cases diagnosed as type I neurofibromatosis.3,4
With the advent of the technology of new generation sequencing (NGS), occult mNF1 was uncovered in about 60% quadruple-negative GIST, which suggested unrecognized NF1 deletion in clinical GISTs cases.5 Recently, NF1 somatic mutation was identified in a GIST patient without germline NF1 mutation, and somatic NF1 mutation in duodenal-jejunal flexure GIST frequently coexists with PDGFRA or mKIT.6–8 About 22% (5/23) of cryptic mKIT exists in KIT/PDGFRA/SDH/RAS-P wild-type GISTs, mostly within exon 11 and exon 9 of KIT, akin to the genotype of KIT in sporadic GISTs.1,9 Therefore, in this study, we retested our 23 archived Formalin-Fixed and Paraffin-Embedded (FFPE) samples of KIT/PDGFRA wild-type GISTs (negative of exons 9,11,13,14 and17 in KIT and exons 12, 14 and 18 in PDGFRA detected by routine Sanger sequencing) between May 2017 and Oct 2019 using targeted deep sequencing of 1021 cancer-related genes region. Meanwhile, we retrospectively analyze the profiles for the molecular status of genes aberration of GISTs by targeted deep sequencing with 73 genes panel or 1021 genes panel in Gene+ database and that of GISTs in MSKCC database using hybridization-capture-based sequencing with 410 genes that are druggable by approved therapies or are targets of experimental therapies. And then we make comparisons of the differences of gene landscapes between neurofibroma lesions in the skin and multiple lesions including micro- or mini-GISTs and GISTs more than 2 cm in intestines in three GIST cases with type I neurofibromatosis, aiming to investigate if mKIT existed in NF1-associated GISTs and possibly the pathogenesis of NF1-deficient GISTs.
Patients and Methods
Patients and Samples
The flowchart below illustrates the study design and patient screening process of this study (Figure 1). From May 2017 to Oct 2019, a total of 576 GISTs cases in our center (Xiangya Hospital) were screened. One hundred and eighty-five patients with no available genetic data of routine Sanger sequencing and 11 patients lost to follow-up were all excluded. 380 GISTs were included and 23 KIT/PDGFRA wild-type GISTs were identified and enrolled in this study. All clinical and pathological information was retrospectively collected, and samples were tested using next-generation sequencing (NGS) approach targeted 1021 cancer-related genes regions. The mutational data of 136 GISTs and 131 GISTs in the Gene+ dataset and MSKCC of the American population were accordingly extracted from the data platform of China National Genebank (www.cngb.org) or downloaded from cBioPortal (http://cbioportal.org/msk-impact). The clinical information including gender, age, location and survival data were also collected. Overall survival (OS) was determined from the date of diagnosis to the date of death or loss to follow-up. Time to progression (TTP) was defined from the date of surgery or stable disease to the date of relapse or disease progression. All cases diagnosed with GISTs in Xiangya Hospital were confirmed by an experienced pathologist who is a member of our multiple disciplinary tumor boards for GISTs. The study was approved by the Institutional Review Board (IRB) of Xiangya Hospital of Central South University, and the written informed consent of patients from Xiangya Hospital was waived. The patients included in the Gene+ dataset have been fully informed and signed the informed consent to perform sequencing and mutational analysis of their genetic data while their privacies are protected, according to the protocol approved by the Institutional Review Board of Geneplus-Beijing. The signed informed consent was not required for the patients of MSKCC dataset which belong to the cBioPortal database since it was an open-access database.
 |
Figure 1 Case screening flow chart. Notes: A total of 576 GISTs cases in our center (Xiangya Hospital) from May 2017 to Oct 2019 were screened. One hundred and eighty-five patients with no available genetic data of routine Sanger sequencing and 11 patients lost to follow-up were all excluded. Twenty-nine patients with poor quality FFPE were also excluded. Finally, 23 cases of 52 wild-types KIT/PDGFRA GISTs were included (negative for exons 9,11,13,14, and 17 in KIT and exon 12, 14, and 18 in PDGFRA using Sanger sequencing). A total of 136 GISTs cases between May 2017 to May 2020 in the Gene+ database and the 131 cases of GISTs from the MSKCC database were also included in our study. |
Targeted Deep Sequencing and Data Collection
The FFPE tissues of 23 KIT/PDGFRA wild-type GISTs were analyzed using targeted deep sequencing of 1021 cancer-related genes region. Corresponding peripheral blood samples of these patients were collected as germline controls. The Next-Generation Sequencing was executed in accordance with the manufacturer’s instructions (Illumina, San Diego, CA, USA). The somatic mutations of KIT/PDGFRA wild-type GISTs were investigated for the genomics landscape description with the ComplexHeatmap R package. The molecular status of gene aberration of 136 GISTs patients in the Gene+ database was detected by targeted deep sequencing with 73 genes panel or 1021 genes panel. The 131 cases of GISTs in the MSKCC database were detected for gene abnormalities using hybridization-capture-based sequencing with 410 genes that are druggable by approved therapies or are targets of experimental therapies.
Statistical Analysis
Numerical variables were summarized as the mean (standard deviation) and median (interquartile range). Categorical variables were reported as counts (percentage). The comparison between the categorical variables was analyzed by a chi-square test. The comparison between continuous variables was analyzed by a Mann–Whitney test. Survival analysis was assessed by the Kaplan-Meier method, and the differences were analyzed by the long-rank test. All tests of hypotheses were two-tailed and conducted at a significance level of 0.05.
Results
The Mutation Type of NF1 in GISTs
Totally 290 GISTs patients including 23 KIT/PDGFRA wild-type cases in Xiangya Hospital, 136 cases in Gene+ database and 131 cases in MSKCC database were enrolled in this study. The incidence of mNF1 in KIT/PDGFRA wild-type GISTs of Xiangya Hospital was 23% (5/23). The prevalence of mNF1 in GISTs was about 8% both in the Gene+ database (11/136) and in the MSKCC database (10/131). In the NF1-associated GISTs cases, 46% (12/26) had mKIT. No difference in age, gender, and location was detected between the NF1-correlated GISTs with or without mKIT (Table 1). The mutation status of NF1 is displayed in Figure 2. The most frequent mutant area was exon 28 (5/26, 19.2%), followed by exon 3 (4/26, 15.4%) and exon 9 (3/26, 11.5%). No differences existed in the type of NF1 mutation between GISTs with KIT mutation and without KIT mutation (Table 2). Detailed descriptions of coexisting NF1 and KIT gene mutations in each of the 12 GISTs cases are shown in Table 3.
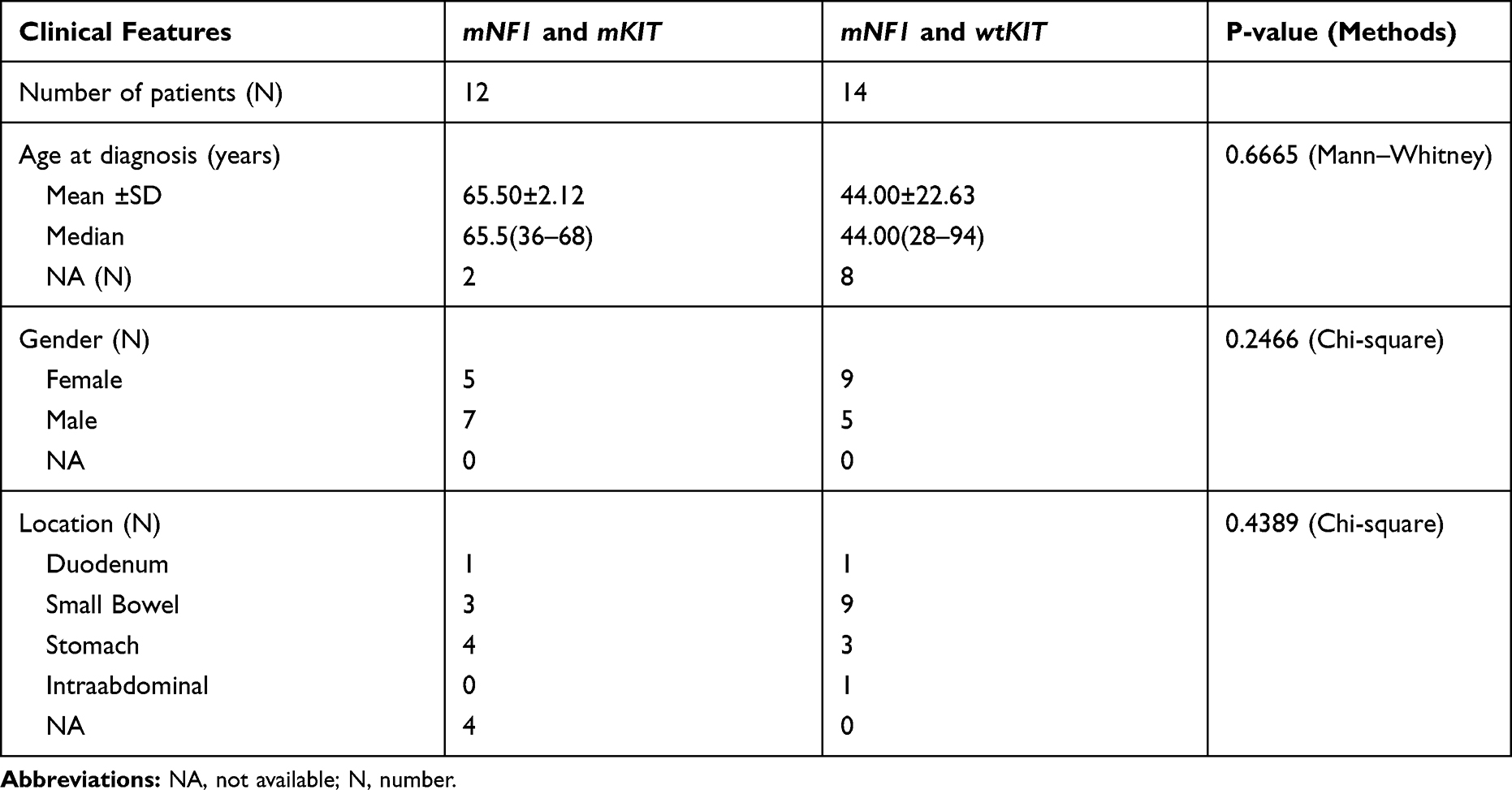 |
Table 1 Clinical Characteristics of GIST with NF1 Mutation |
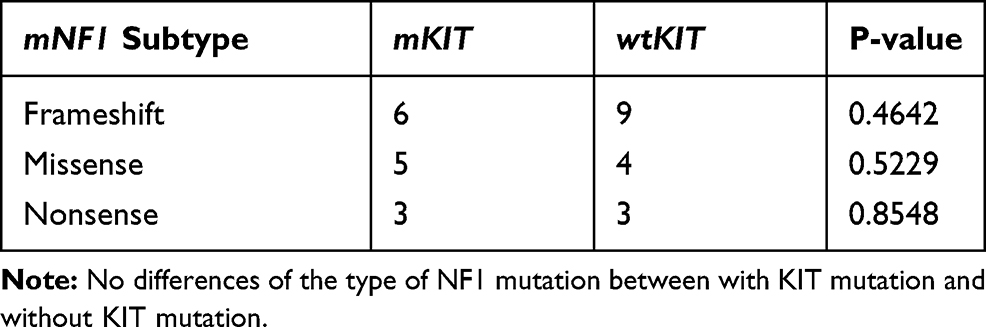 |
Table 2 The Mutation Types and Their Differences Between mNF1 with mKIT and mNF1 with wtKIT |
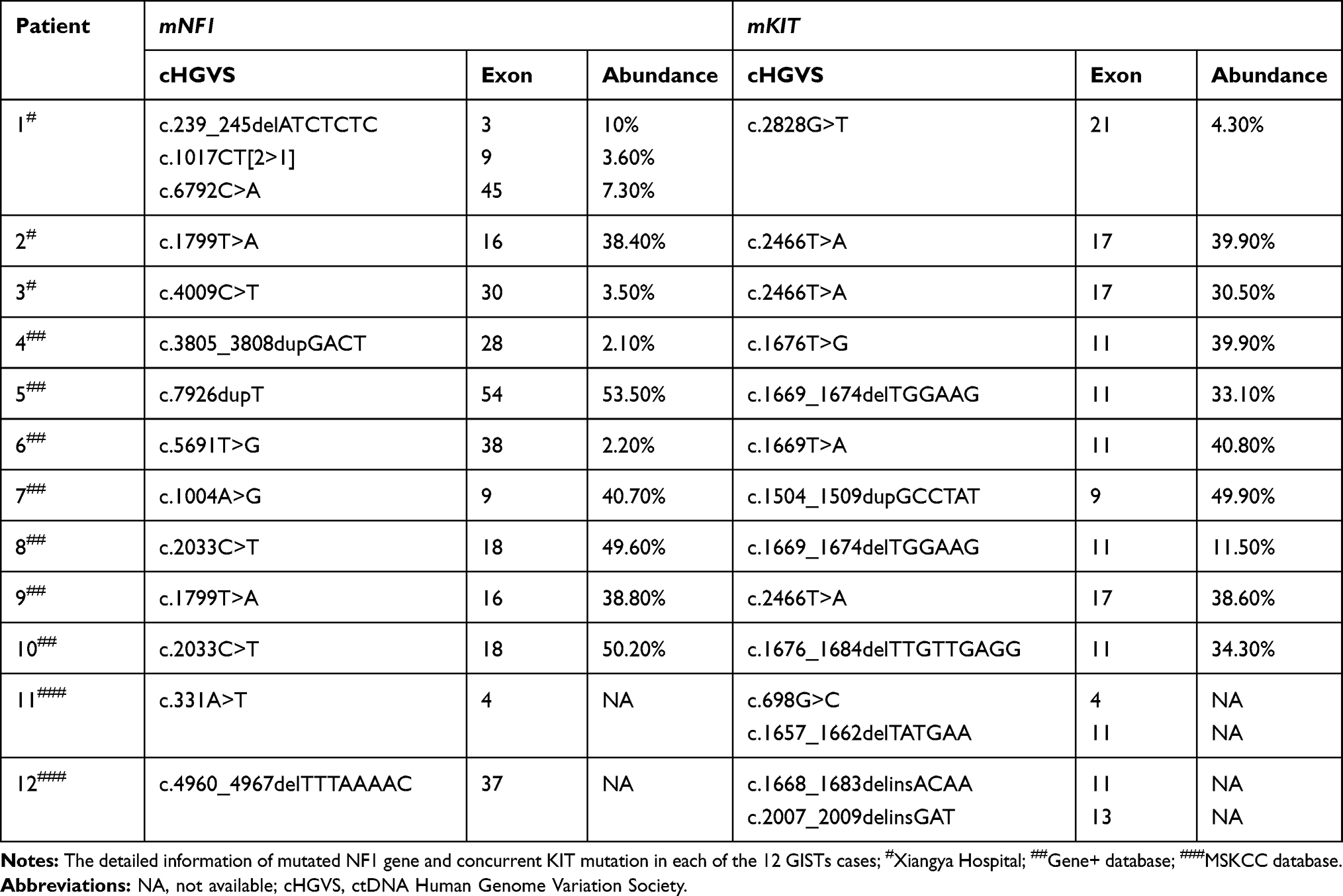 |
Table 3 Concurrent Mutation of NF1 and KIT in GISTs Patients |
 |
Figure 2 The mutation loci and subtypes within the NF1 gene and their corresponding pathogenicity in GISTs. Notes: #Xiangya Hospital; ##Gene+ database; ###MSKCC database; ±, Neutral; –, Deleterious; Red symbols of #, ## and ###represent n=2. |
Concurrent Gene Abnormalities in NF1-Deficiency GISTs
Growing evidence showed that extra molecule events are required for GISTs which developed from NF1-deficient lesion or neurofibroma,10 so we analyze the genomic landscapes of 26 GIST patients with mNF1. The most common abnormal genes concurrent with mNF1 were KIT oncogene with a frequency of 46% and TP53 gene with a frequency of 15%, followed by the mutations of other genes, including NOTCH2, RB1, and TSC2 et al, as shown in Supplementary 1.
KIT Mutation in NF1-Associated GISTs
Compared with the proportion of mKIT in the American population in the MSKCC database (20%, 2/10), a higher ratio of mKIT in NF1-associated GISTs was detected in the Chinese cohort including that of our center (60%, 3/5) and in the Gene+ database (64%, 7/11)(p<0.05) (Table 4). mKIT focus in the juxtamembrane domain (JM) or kinase domain showed hotspot mutation of KIT to be within exon 11 (7/12, 58%), similar to the gene subtype of sporadic KIT-mutated GISTs. Other mutation locations of KIT were exon 17 (3/12, 25%), exon 9(1/12, 8%), exon 13 (1/12, 8%), exon 21 (1/12, 8%) and exon 4(1/12, 8%) respectively.
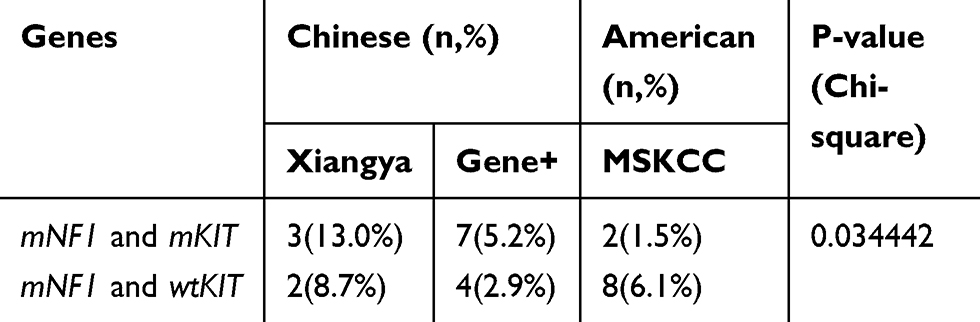 |
Table 4 NF1 Concurrent with KIT Mutation |
The mutation types of KIT contained in-frame deletions (4/7, 57%), deletion and insertion (2/7, 29%) and missense mutation (1/7, 14%) in exon 11, N822K in exon 17 (3/3, 100%), A502_Y503dup in exon 9 (1/1, 100%) and I669_T670delinsMI in exon 13 (1/1, 100%). In the MSKCC dataset, both two cases of NF1-deficiency GISTs harbored double mutations, in which one of the shared mutations was still within the exon 11 of KIT (Figure 3).
 |
Figure 3 KIT mutations loci and subtype. Abbreviations: TM, transmembrane domain; JM, juxtamembrane domain; N, number. Notes: The distribution of various mutation subtypes identified by NGS within the exons of KIT gene; #Xiangya Hospital; ##Gene+ database; ###MSKCC database; ±, Neutral; –, Deleterious; Red symbols of ## and ###represent n=2. |
KIT Mutation in Various Lesions in Type I Neurofibromatosis
With the purpose of exploring the role of KIT in the pathogenesis of NF1-deficient GISTs, we collected various samples range from skin neurofibromas, micro-GIST lesions (≤1 cm), mini-GIST lesions between 1 cm and 2 cm, and GIST lesions (>2 cm) in three GISTs patients with type I neurofibroma. All these 3 cases had multiple café-au-lait spots and superficial neurofibromas which were considered as the diagnosis of type I Neurofibromatosis. Among the three cases, mKIT existed in 2 cases with GISTs that were more than 2 cm but not in the skin neurofibromas subtype and micro-GISTs (Figure 4). Evidently, as shown in Figure 4, the number of gene mutations gradually increased with the clinical progression of neurofibromas to overt GISTS in all three cases.
 |
Figure 4 The genomic landscapes of skin neurofibroma and GISTs in 3 cases of type I neurofibromatosis. Notes: (A–C) represent individual cases 1–3, respectively. In panel A (case 1) and panel B (case 2), compared with the lesions of skin neurofibromas and micro-GISTs (≤1 cm) that merely harbor mNF1, GISTs lesions (>2 cm) have NF1 aberration with a concurrent mKIT. In all three cases, the number of gene mutations is gradually increasing with the neurofibromas lesions to micro-GISTs or/and mini-GISTs to clinic GISTs. |
Survival of the Patients with mNF1 and Those without mNF1
At the time of drafting this paper, only one patient with wild-type NF1 genotype had a recurrence, while all other patients recruited at our center still have stable disease. In the MSKCC dataset, the overall survival (OS) of the patients with wild-type NF1 genotype showed shorter than that of the patients with NF1 mutation, but the difference in OS between these two groups had no statistical significance. (Supplementary 2).
Discussion
In this study, we investigated the frequency of NF1 mutation in GISTs. The incidence of mNF1 in KIT/PDGFRA wild-type GISTs of Xiangya Hospital was 23%. The prevalence of mNF1 in GISTs in both Gene+ database and MSKCC database were 8%, indicating no racial correlation. Among NF1-associated GISTs, 46% of cases had concurrent KIT oncogene mutation. In our center and Gene+ database, both of which had a Chinese population, mKIT had a higher frequency of 60% (3/5) and 64% (7/11) in NF1-deficiency GISTs than that of the MSCKCC database with the American population (20%, 2/10) (p<0.05). No hotspot mutations were detected in NF1, and there was no difference of mutation subtype in the NF1 gene between those with or without mKIT. The mutation of KIT in NF1-associated GISTs centered on the JM or TKI domain, similar to that in the sporadic mKIT GISTs.1
In the three GIST patients with type I neurofibromatosis, NGS was performed in a various range of neurofibromas lesions in the skin, micro-GISTs, or mini-GISTs and clinic GISTs more than 2 cm in the intestines. mKIT was not detected in neurofibromas of the skin and micro-GISTs, but it was identified in mini-GISTs and clinic GISTs bigger than 2 cm in two cases.
GISTs are mesenchymal tumors mostly located in the gastrointestinal tract. The majority of GISTs have KIT or PDGFRA mutations, and the rest about 10–15% harbor wild-type KIT/PDGFRA, hence considered as a heterogeneous entity mainly including SDH-defective and RAS-pathway abnormalities of K/N/H‑RAS, BRAF, and NF1 mutation. NF1 is a tumor suppressor gene that negatively modulates RAS signaling that accounts for less than 1% of total KIT/PDGFRA wild-type GISTs.1
Recently, using massively parallel sequencing, Gasparotto et al reported that about 60% (13/22) of quadruple-negative tumors harbored biallelic inactivation of NF1.5 In concordance with this, our center also disclosed about 22% of GIST patients have mNF1 in KIT/PDGFRA wild-type GISTs.
mKIT was first identified in GIST patients in 1998, leading to the understanding of the underlying mechanism of KIT-driven GISTs and exploration of abnormal genes that are concurrent with mKIT.17 The first report about the molecular profile of NF1-associated GISTs was provided by Kinoshita and colleagues. There was no mKIT in their case series of 29 GISTs lesions in seven NF-1 patients; neither was there any NF1 aberration in sporadic mKIT GISTs.18,19
The mutual exclusion of the mutations of KIT and NF1 in GISTs was reported in accumulative clinic studies.19 Indeed, haplodeficiency of neurofibromin was enough to promote the growth of a specific subtype of ICC expressing both KIT and S-100 protein in the small intestine, inferring that GIST progression in patients with type I Neurofibromatosis do not need gain-of-function mutation of KIT.14
However, recent studies via targeted deep sequencing uncovered about 20% (5/26) have occult KIT abnormalities in a series of KIT/PDGFRA wild-type GISTs cases originally classified by Sanger sequencing, and 4 out of 5 cases were in the exon 11 of KIT.9 Similarly, Gao and colleagues reported that 19 out of 146 KIT/PDGFRA wild-type GISTs had the mutation of KIT according to NGS results,20 which was also mostly within the exon 11 of KIT (14/19,74%). These results indicated that the incidence of mKIT was underreported in the KIT/PDGFRA wild-type GISTs patients in the clinic when using conventional Sanger sequencing.
In nine cases of type I neurofibromatosis, Takazawa et al reported about 8% of mKIT in 36 GIST lesions and all GISTs were of low-risk grade.10,14 Yantiss et al reported that three patients with NF1 and multiple small intestinal stromal tumors were investigated and three tumors in different locations from one patient had identical point mutations in KIT exon 11, which has been previously reported in several studies of germline KIT mutation in JM or kinase domain developing GISTs.16 One NF1 case with all three GIST lesions had an identical mutation in KIT exon 11 with in-frame deletions.13 Mussi and colleagues detected 2 cases of 28 NF1-deficiency GISTs who had mKIT with one of the cases of deletion in exon 11 and another one case of duplication in exon 9 in primary lesion. Intriguingly, one case in this report had exon 17 mutation of KIT in the metastatic lesion but not in the primary tumor.11
In our study, we found that 46% cases of NF1-deficient GISTs harbored concurrent mKIT in total. Two of the three patients in our center had the frequency of more than 30% of Exon 17 (p.N822K) as identified in NGS, which were negative for KIT mutation in the conventional Sanger method (detection of exons 9,11,13,14 and 17 in KIT and exon 12, 14 and 18 in PDGFRA). The possible explanation was tumor heterogeneity, as reported in the cases presented by Namgung and Bajor.12,25 Similar to our center, all the GISTs patients in the Gene+ database were of the Chinese population and had a higher frequency of 63% (10/16) of KIT mutation in NF1-deficiency GISTs than that of the American cohort in the MSCKCC database (20%, 2/10) (p<0.05). The exact reason for this discrepancy is not clear. We speculate that racial and ethnic differences among the cohorts may play a role.
Apart from one of mKIT was in the extracellular domain of exon 4 in KIT, the others were within the region of JM or kinase domain. In line with the previous studies,21 the mutation in exon 11 was the most common subtype of mKIT, followed by exon 17, exon 9, exon 13, and exon 21. The mutation types of KIT contained in-frame deletions, deletion and insertion, and missense mutation in exon 11, as well as point mutations in exon 17 and duplication in exon 9. In the MSKCC dataset, both two cases of NF1-deficiency GISTs harbored double mutations within KIT, one of the shared mutations was still within the exon 11 of KIT (Figure 3). These results uncovered that the mutation scenery of KIT in NF1-associated GISTs was similar to that of sporadic mKIT GISTs, hinting at the role of KIT in the development of NF1-aberrant GISTs, but it was still largely unknown.
KIT protein is a transmembrane type III receptor tyrosine kinase, consisting of an extracellular region and a cytoplasmic region which constitute a juxtamembrane domain and two kinase domains. Type I neurofibromatosis presents alterations in the NF1 gene located in the long arm of chromosome 17 (17q11.2) and contains 50 exons, which code for the neurofibromin protein. GISTs linked to NF-1 frequently prove loss of heterozygosity at 14q and 22q similar to sporadic GISTs mutated by KIT and PDGFRA.22 Further study showed that the KIT ligand signal can stimulate the proliferation of neurofibrosarcoma cells.23 Heterozygous inactivation of the NF1 gene enhances c-kit-induced Ras output by reducing neurofibromin levels and RAS-GAP activity in cell lineages, which interprets the coordination mechanism underlying the mKIT with deficient NF1 for GIST progression.13,24 Namgung presented two GIST lesions of one type I neurofibromatosis patient with E11 mutation in KIT in the extramural portion of the largest tumor, but wtKIT in the intramural portion of the largest tumor and the other tumor.12 Secondary point mutation of D820N in exon 17 was shown in the metastatic lesion of one NF1 patient, but the primary tumor was wild type.11 Both of them suggested that mKIT probably developed at a later step of NF1-deficient tumor genesis.25 This hypothesis was also observed in our study showing that mKIT was in the mini-GISTs more than 1 cm, but not in micro-GISTs and skin neurofibromas. Further studies were required to depict the role and the mechanism of mKIT in the pathogenesis of GISTs in some NF1 patients.16,26
The treatment with imatinib in the GISTs associated with NF1 inactivation is ineffective and not recommended.1 Three NF1 cases with mKIT showed a poor reaction to imatinib treatment.11 Several cases showed that NF1-associated GISTs have a good response to sunitinib or Regorafenib.27,28 So the optimal treatment in the subtype of concurrent mutations of NF1 and KIT remains to be elucidated. Besides, it is reported that the risk of recurrence and mortality is very similar between NF1 and non-NF1 patients after surgical resection of GISTs.29 In accordance with this, most cases included in our study were post-surgery, and an adjuvant TKI regimen was given to these GIST patients with medium- to high-risk grade. The TTP and OS of these subgroups between NF1 deficiency and NF1-intact GISTs were similar. However, this conclusion remains equivocal due to the short follow-up period and the limited sample size.
This study has several limitations. Firstly, it was a retrospective study that is inherently limited by some confounding factors or bias, including clinical information. Secondly, NGS with different gene panels among these three cohorts from our center, Gene+, and MSKCC datasets affects the comparability of their differences. Thirdly, limited samples and a short follow-up time are inconclusive to show the effect of NF1 mutation on the survival of GIST cases.
Conclusion
Collectively, our study disclosed a high frequency of unrecognized mKIT in NF1-associated GISTs. Further underlying mechanisms of KIT oncogene cooperation with deficient NF1 in the pathogenesis of GISTs warrant further probing. NGS holds the potential for screening undiscovered mKIT from a subset of GISTs originally regarded as KIT/PDGFRA wild-type tumors using regular Sanger sequencing. Due to its comparatively higher sensitivity and tumor heterogeneity, it provides a better understanding of mKIT in NF1-associated GISTs as well as identifying potentially eligible patients for optimal therapy.
Ethics Statement
The retrospective study was approved by the Institutional Review Board (IRB) of Xiangya Hospital of Central South University, and informed consent of patients from Xiangya Hospital was waived (IRB No. 202,009,686). The signed informed consent was obtained from the patients of the Gene+ dataset, and not required for the patients of MSKCC dataset which belongs to the cBioPortal database since it was publicly available. The study was directed in accordance with the recommendations delineated in the World Medical Association Declaration of Helsinki. All personal data within this study were remained confidential and not for commercial use.
Acknowledgment
We want to give our thanks to Omar Abdihamid who check the words, grammar and English expression in the manuscript and attached legends.
Funding
This work was granted by the National Natural Science Foundation of China (81200366, 81572281, 81702278, 81974367) and Province Natural Science Foundation of Hunan (No. 14JJ6004) and the Key Subject Education Department of Hunan ([2012]594) and Scientific Research Project of Hunan Provincial Department of Education (No. 18K001).
Disclosure
Qiongzhi He is an employee of Geneplus-Beijing Institute. The authors declare that they have no other potential conflicts of interest for this work.
References
1. von Mehren M, Joensuu H. Gastrointestinal stromal tumors. J clin oncol. 2018;36(2):136–143. doi:10.1200/JCO.2017.74.9705
2. Philpott C, Tovell H, Frayling IM, Cooper DN, Upadhyaya M. The NF1 somatic mutational landscape in sporadic human cancers. Hum Genomics. 2017;11(1):13. doi:10.1186/s40246-017-0109-3
3. Giuly JA, Picand R, Giuly D, Monges B. Von Recklinghausen disease and gastrointestinal stromal tumors. Am J Surg. 2003;185(1):86–87. doi:10.1016/S0002-9610(02)01111-X
4. Uusitalo E, Rantanen M, Kallionpää RA, et al. Distinctive cancer associations in patients with neurofibromatosis type 1. J clin oncol. 2016;34(17):1978–1986. doi:10.1200/JCO.2015.65.3576
5. Gasparotto D, Rossi S, Polano M, et al. Quadruple-negative gist is a sentinel for unrecognized neurofibromatosis type 1 syndrome. Clin Cancer Res. 2017;23(1):273–282. doi:10.1158/1078-0432.CCR-16-0152
6. Belinsky MG, Rink L, Cai KQ, et al. Somatic loss of function mutations in neurofibromin 1 and MYC associated factor X genes identified by exome-wide sequencing in a wild-type GIST case. BMC Cancer. 2015;15:887. doi:10.1186/s12885-015-1872-y
7. Burgoyne AM, De Siena M, Alkhuziem M, et al. Duodenal-jejunal flexure gi stromal tumor frequently heralds somatic nf1 and notch pathway mutations. JCO Precision Oncol. 2017;2017:2017. doi:10.1200/PO.17.00014
8. Li K, Tjhoi W, Shou C, et al. Multiple gastrointestinal stromal tumors: analysis of clinicopathologic characteristics and prognosis of 20 patients. Cancer Manag Res. 2019;11:7031–7038. doi:10.2147/CMAR.S197560
9. Astolfi A, Indio V, Nannini M, et al. Targeted deep sequencing uncovers cryptic kit mutations in KIT/PDGFRA/SDH/RAS-P wild-type GIST. Front Oncol. 2020;10:504. doi:10.3389/fonc.2020.00504
10. Salvi PF, Lorenzon L, Caterino S, Antolino L, Antonelli MS, Balducci G. Gastrointestinal stromal tumors associated with neurofibromatosis 1: a single centre experience and systematic review of the literature including 252 cases. Int J Surg Oncol. 2013;2013:398570. doi:10.1155/2013/398570
11. Mussi C, Schildhaus HU, Gronchi A, Wardelmann E, Hohenberger P. Therapeutic consequences from molecular biology for gastrointestinal stromal tumor patients affected by neurofibromatosis type 1. Clin Cancer Res. 2008;14(14):4550–4555. doi:10.1158/1078-0432.CCR-08-0086
12. Namgung H. Gastrointestinal stromal tumor with KIT mutation in neurofibromatosis type 1. J Korean Surg Soc. 2011;81(4):276–280. doi:10.4174/jkss.2011.81.4.276
13. Cheng SP, Huang MJ, Yang TL, et al. Neurofibromatosis with gastrointestinal stromal tumors: insights into the association. Dig Dis Sci. 2004;49(7–8):1165–1169. doi:10.1023/B:DDAS.0000037806.14471.a2
14. Takazawa Y, Sakurai S, Sakuma Y, et al. Gastrointestinal stromal tumors of neurofibromatosis type I (von Recklinghausen’s disease). Am J Surg Pathol. 2005;29(6):755–763. doi:10.1097/01.pas.0000163359.32734.f9
15. Tokunaga M, Nanjo S, Yoshita H, et al. Multiple synchronous sporadic gastrointestinal stromal tumors in the stomach and jejunum. Internal Medicine. 2018;57(12):1719–1723. doi:10.2169/internalmedicine.0229-17
16. Yantiss RK, Rosenberg AE, Sarran L, Besmer P, Antonescu CR. Multiple gastrointestinal stromal tumors in type I neurofibromatosis: a pathologic and molecular study. Modern Pathol. 2005;18(4):475–484. doi:10.1038/modpathol.3800334
17. Mei L, Smith SC, Faber AC, et al. Gastrointestinal stromal tumors: the gist of precision medicine. Trends in Cancer. 2018;4(1):74–91. doi:10.1016/j.trecan.2017.11.006
18. Kinoshita K, Hirota S, Isozaki K, et al. Absence of c-kit gene mutations in gastrointestinal stromal tumours from neurofibromatosis type 1 patients. J Pathol. 2004;202(1):80–85. doi:10.1002/path.1487
19. Miettinen M, Fetsch JF, Sobin LH, Lasota J. Gastrointestinal stromal tumors in patients with neurofibromatosis 1: a clinicopathologic and molecular genetic study of 45 cases. Am J Surg Pathol. 2006;30(1):90–96. doi:10.1097/01.pas.0000176433.81079.bd
20. Gao J, Li J, Li Y, et al. Intratumoral KIT mutational heterogeneity and recurrent KIT/PDGFRA mutations in KIT/PDGFRA wild-type gastrointestinal stromal tumors. Oncotarget. 2016;7(21):30241–30249. doi:10.18632/oncotarget.7148
21. Szucs Z, Thway K, Fisher C, et al. Molecular subtypes of gastrointestinal stromal tumors and their prognostic and therapeutic implications. Future Oncol. 2017;13(1):93–107. doi:10.2217/fon-2016-0192
22. Miettinen M, Lasota J, Sobin LH. Gastrointestinal stromal tumors of the stomach in children and young adults: a clinicopathologic, immunohistochemical, and molecular genetic study of 44 cases with long-term follow-up and review of the literature. Am J Surg Pathol. 2005;29(10):1373–1381. doi:10.1097/01.pas.0000172190.79552.8b
23. Badache A, Muja N, De Vries GHD. Expression of Kit in neurofibromin-deficient human Schwann cells: role in Schwann cell hyperplasia associated with type 1 neurofibromatosis. Oncogene. 1998;17(6):795–800. doi:10.1038/sj.onc.1201978
24. Ryan JJ, Klein KA, Neuberger TJ, et al. Role for the stem cell factor/KIT complex in Schwann cell neoplasia and mast cell proliferation associated with neurofibromatosis. J Neurosci Res. 1994;37(3):415–432. doi:10.1002/jnr.490370314
25. Bajor J. Gastrointestinal stromal tumors in neurofibromatosis type 1. Orv Hetil. 2009;150(4):149–153. doi:10.1556/oh.2009.28478
26. Shinomura Y, Kinoshita K, Tsutsui S, Hirota S. Pathophysiology, diagnosis, and treatment of gastrointestinal stromal tumors. J Gastroenterol. 2005;40(8):775–780. doi:10.1007/s00535-005-1674-0
27. Kalender M, Sevinc A, Tutar E, Sirikci A, Camci C. Effect of sunitinib on metastatic gastrointestinal stromal tumor in patients with neurofibromatosis type 1: a case report. World j Gastroenterol. 2007;13(18):2629–2632. doi:10.3748/wjg.v13.i18.2629
28. Fujimi A, Nagamachi Y, Yamauchi N, et al. Gastrointestinal stromal tumor in a patient with neurofibromatosis type 1 that was successfully treated with regorafenib. Internal Medicine. 2019;58(13):1865–1870. doi:10.2169/internalmedicine.2321-18
29. Nishida T, Tsujimoto M, Takahashi T, Hirota S, Blay JY, Wataya-Kaneda M. Gastrointestinal stromal tumors in Japanese patients with neurofibromatosis type I. J Gastroenterol. 2016;51(6):571–578. doi:10.1007/s00535-015-1132-6
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.