Back to Journals » International Journal of Nephrology and Renovascular Disease » Volume 11
Takayasu’s disease effects on the kidneys: current perspectives
Authors Li Cavoli G, Mulè G, Vallone MG ![]() , Caputo F
, Caputo F
Received 5 January 2018
Accepted for publication 4 May 2018
Published 15 August 2018 Volume 2018:11 Pages 225—233
DOI https://doi.org/10.2147/IJNRD.S146355
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Pravin Singhal
Gioacchino Li Cavoli,1 Giuseppe Mulè,2 Mario Giuseppe Vallone,3 Flavia Caputo1
1Nephrology, Dialysis and Renal Transplant Department, Civico and Di Cristina Hospital, Palermo, Italy; 2Internal Medicine, Cardiovascular and Renal Diseases Department, University of Palermo, Palermo, Italy; 3Radiology Department, Civico and Di Cristina Hospital, Palermo, Italy
Abstract: Takayasu arteritis (TA) is a chronic vasculitis disease of unknown etiology. Clinically significant renal disease is relatively common, and renovascular hypertension is the major renal problem. The assessment of TA activity is usually challenging because vascular inflammation may progress to fixed vascular injury without findings of active disease. Until now, the best therapeutic options have not been identified. This review highlights the current perspectives of renal involvement in TA.
Keywords: arteritis, kidney involvement, large vessel vasculitis
Introduction
Takayasu arteritis (TA) is an idiopathic inflammatory vasculitis, predominantly involving aorta and its major division branches, with unknown etiology and higher prevalence in women.1 The inflammatory changes produce vessel wall injury leading to necrosis, thrombosis, aneurysm formation, scarring, and obliteration of vessel lumen.2 Clinical significant renal disease is relatively common. Renovascular hypertension is the major renal problem caused by TA. The assessment of TA activity is usually challenging because vascular inflammation may progress to fixed vascular injury without overt findings of active disease. A current issue in the management of TA is the lack of outcome measures in clinical trials. Until now, the best therapeutic options have not been identified. This review highlights the current perspectives of renal involvement in TA.
Epidemiology
TA is an uncommon disease;3 its incidence and prevalence are probably underestimated. First reports were of Japanese and other Asian populations. The “pulseless disease,” the most known description of TA, was named after the ophthalmologist Mikito Takayasu who in 1908 described retinal vessel changes in a young Japanese woman with decreased pulses in branches of aortic arch.4 In the past century, TA was reported as a serious disease affecting women mostly in second–third decade of life, originating from Asia, but in recent years, it has been reported to afflict individuals of various ethnicities with worldwide distribution and improved prognosis over the previous decades. The most recent studies confirm the predominance of female patients;3 in several reports, the age at onset (or at diagnosis) is not all time <30 years but also in older ages of life (Table 1). In 2012, among 106 TA patients, Ohigashi et al reported 14 subjects (13 women and 1 man) with age at onset >40 years and no differences in clinical characteristics.5 TA incidence had been estimated to be 1–2 per million in Japan. The annual incidence of TA in the UK was reported to be 0.8 per million population and the prevalence 4.7 per million.6 In the period of 1997–2011 in southern Sweden, the annual incidence rate was reported to be 0.7 per million population.7 Birlik et al reported between the years 2006 and 2010 in Turkey a mean annual incidence of 1.11 per million.8 According to the Japanese TA registry, in 2011, the prevalence in Japan was 40 per million;9 in Europe, the TA prevalence had been reported to be from 4.7 to 33 per million and in the USA 0.9 per million.10 In 2014, a review in Arab populations of seven countries reported demographic findings comparable with those in other parts of the world.11–14
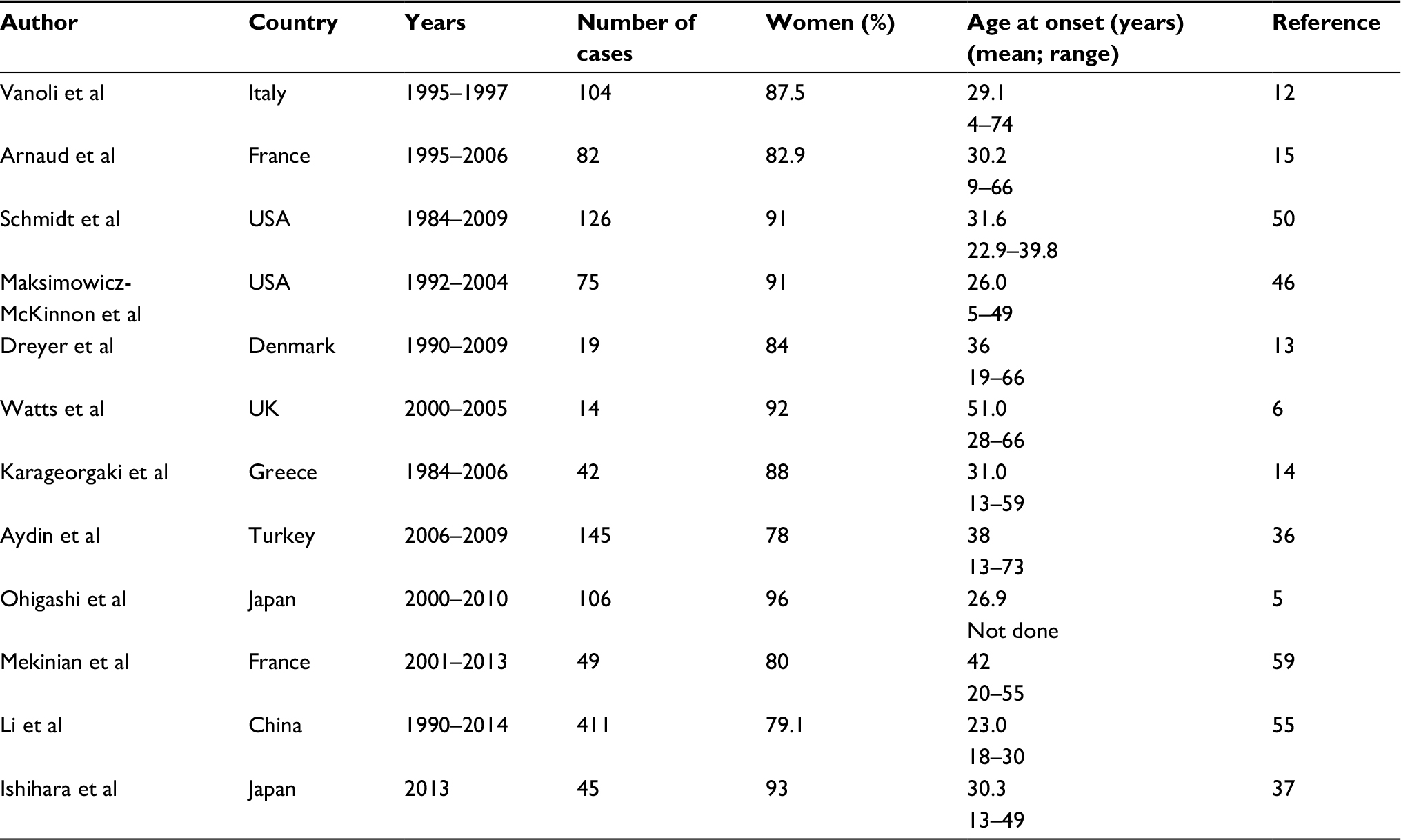 | Table 1 Recent studies on Takayasu’s arteritis |
Pathophysiology
Genetic studies
The TA etiology has not yet been clarified; it encompasses the correlation between environmental factors, especially infectious agents, and the genetic background in a susceptible individual.15 The progress in genetic studies has been hampered by the rarity of the disease. Some genetic studies have highlighted the interest on the human leukocyte antigen (HLA) gene and on tumor necrosis factor-alpha (TNF)-α gene. The genetic predisposition to many autoimmune diseases can be affected by HLA gene polymorphisms, particularly HLA-B alleles, possibly affecting susceptibility to TA.16,17 A recent meta-analysis confirmed that HLA-B*52 allele may contribute to susceptibility to TA in different ethnicities (pooled OR =3.91, 95% CI =3.22–4.74).18 Previous smaller genetic studies in Japanese population also found an association between TA and HLA-B*67.19 TNF-α is a potential proinflammatory cytokine with important inflammatory and immune activities, including those observed in TA.20 Inflammatory cells infiltrating arterial tissue in TA produce TNF. In addition, the therapy with TNF inhibitors are highly effective in patients with TA refractory to other therapies.21 The TNF gene is located on chromosome 6, within the class III region of the HLA. The G-to-A substitution in the promoter at position −308 in the TNF gene has been investigated in several studies.22 The above-described meta-analysis demonstrated a significant association of TA with TNF-α-308 A/G polymorphism for the A allele versus G allele and AA + AG versus GG;18 two genome-wide association studies in TA patients revealed the correlation between a single-nucleotide polymorphism and interleukin (IL)-12 B and documented a new one with FCGR2A/3A.23 This latter association was more recently replicated in Chinese population.24 The FCGR2A and FCGR3A genes are members of the Ig gene superfamily that encodes immunoglobulin Fc receptors. Variations of this gene family affect receptor functions such as phagocytosis and clearing of immune complexes.
Immunological studies
It is critical for the immune system to recognize foreign antigens by receptors on the surface of cells of macrophages, neutrophils, and eosinophils. Some studies have reported the association between the FCGR gene polymorphisms and several human autoimmune diseases, including giant cell arteritis (GCA).25 Therefore, GCA might share common genetic susceptibility loci with TA, implying that there might exist similar etiology mechanism for the two kinds of large vessel arteritis. Interestingly, according to the Chapel Hill Consensus Conference, GCA and Takayasu’s arteritis may represent different appearances of the same disease as both occur mostly in women and have similar histopathological findings.26 It is generally believed that the TA pathogenesis is implicated with the cell-mediated immune response. Immunohistochemical analyses of infiltrates in the aortic tissue in TA acute phases show a heterogeneous population of inflammatory cells27–29 with an uptake of the 18F-fludeoxyglucose when positron emission tomography (PET) is performed in these patients. Th1 cells induce macrophages activation and giant cell formation and contribute to the inflammatory lesions by activating neutrophils; γδ T cells and natural killer cells interact via their natural killer group 2, member D receptor with major histocompatibility complex class I-related chain A (MICA) antigens on vascular smooth muscle cells, resulting in cytotoxicity via their production of perforin. Further analysis in lesional tissue shows a limited use of T-cell receptor αβ genes. This may suggest that a specific, but not identified, antigen may be implicated in TA disease, and the initial inflammatory process may involve the vasa vasorum and the media–adventitia junction. Dendritic cells are believed to make a link between innate and adaptive immunity. These cells are colocalized with T cells in the adventitia and around the vasa vasorum of involved arteries from TA patients. The inflammatory process leads to the damage of the vessel wall with laminar necrosis and elastic fiber fragmentation (ie, elasticophagia), resulting in the replacement of arterial structures by fibrosis and neovascularization from the intima to the adventitia.30 More recently, it has been shown that TA patients present predominantly Th1 and Th17 responses upon the stimulation of peripheral blood mononuclear cells.31 The formation of a granulomatous inflammation in the arterial wall (adventitia and media) is the first phase of the vascular lesion. The typical feature of TA is the severe inflammation of the vasa vasorum. In contrast to atherosclerosis, the heat-shock protein 65, which responds to γδ T lymphocytes, is strongly induced in the media and vasa vasorum of TA patients, suggesting the participation of γδ T lymphocytes in the TA pathogenesis. Sometimes, the inflammation can extend to all layers of the aortic wall. In the chronic stage, strong calcification of the intima is another feature of TA. However, some authors have described the production of autoantibodies in TA (eg, antiendothelial cell antibodies and antiferritin antibodies), and TA patients with active disease present an increased number of newly formed antibody-secreting plasmablasts in the peripheral blood, indicating a possible role of humoral immunity in TA pathogenesis.
Pathogenetic mechanisms
The pathogenetic mechanisms putatively responsible for the development of TA have been summarized by Arnaud et al17 in the following sequence of events (Figure 1): 1) the process begins with the expression of the 65-kDa heat-shock protein and the induction of the MICA on vascular cells in the aortic tissue, triggered by an unknown stimulus; 2) acute vascular inflammation after γδ T cells and natural killer cells has recognized MICA on vascular smooth muscle cells and has released perforin, chemokines, and proinflammatory cytokines; and 3) finally, T cells recognize some antigens presented by a shared epitope, associated with specific major histocompatibility complex alleles on the dendritic cells, activated through their Toll-like receptors. Th1 lymphocytes drive the formation of giant cells through the production of interferon-γ and activate macrophages with release of vascular endothelial growth factor and platelet-derived growth factor.
 | Figure 1 Putative pathogenetic mechanisms of Takayasu’s arteritis. Reprinted from Autoimmun Rev, (11), Arnaud L, Haroche J, Mathian A, Gorochov G, Amoura Z; T Pathogenesis of Takayasu’s arteritis: a 2011 update, Copyright (©2011), with permission from Elsevier.17 Abbreviations: AECA, anti-endothelial cell antibodies; FAS-L, FAS ligand; γδ, gamma-delta cell; HLA, human leukocyte antigen; HSP65, heat-shock protein 65; ICAM-1, intercellular adhesion molecule 1; IFN, interferon; IL, interleukin; MICA, major histocompatibility complex class I-related chain A; NKG2D, natural killer group 2, member D receptor; PDGF, platelet-derived growth factor; TGF, transforming growth factor; TLR, Toll-like receptors; VEGF, vascular endothelial growth factor. |
Classification criteria
According the 2012 International Chapel Hill Consensus Conference,26 both GCA and TA are chronic granulomatous large vessel vasculitis; primary difference is the age of patients (<40 years in TA patients and >50 years in GCA patients). The age at onset has been used to distinguish between these two diseases, but to date, no established biological markers specific for the diagnosis of GCA and TA have been reported. There are differences in the epidemiology of large vessel vasculitis. In Japan, TA is prevalent and GCA is rare, whereas in Europe and the USA, GCA is more prevalent than TA. Based on the overlapping in the arterial histopathology and imaging findings between TA, GCA, and IgG4-related diseases involving the aorta, some authors consider these conditions a spectrum of the same disease.28–30 First diagnostic criteria for TA were developed by Ishikawa in 1988.31 Later, two sets of classification criteria, the 1990 American College of Rheumatology Classification Criteria for TA (Table 2) and the 2006 classification criteria for childhood TA proposed by the European League Against Rheumatism (Table 3), have provided clinical and radiological features for diagnosis.32 The usefulness of these criteria is limited in clinical practice, particularly in the middle-aged population,33 and according to some authors, there is a need for new diagnostic criteria.34
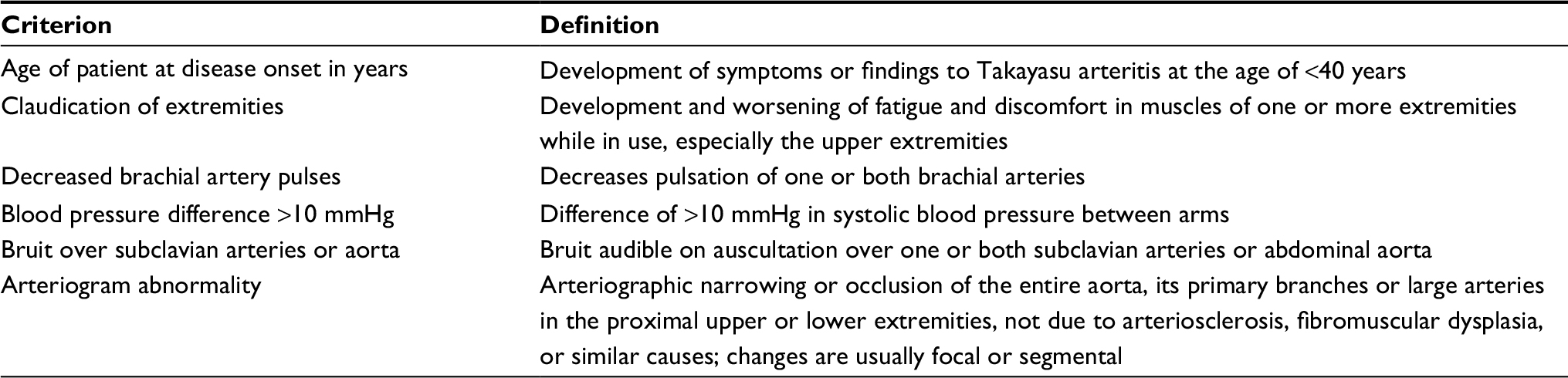 | Table 2 Criteria for the classification of Takayasu’s arteritis, the American College of Rheumatology 1990 Notes: Takayasu’s arteritis is identified if at least three of the six criteria are present. The presence of three or more criteria yields a sensitivity of 90.5% and a specificity of 97.8%. Data from Arend et al.32 |
 | Table 3 Classification criteria for Takayasu arteritis (EULAR 2006)33 Note: Data from Ozen et al.33 Abbreviations: CRP, C-reactive protein; ESR, erythrocyte sedimentation rate; EULAR, European League Against Rheumatism 2006. |
Disease activity
Diagnostic issues
Clinical features, inflammatory markers, and imaging characteristics are usually used to analyze the disease activity of large vessel vasculitis (LVV), but the evaluation of activity in TA is a difficult issue because there are no validated clinical and instrumental criteria to quantify the disease activity. TA assessment is based on the onset of new physical and laboratory findings. Conversely, the development of new vascular lesions is observed even in asymptomatic TA patients. A lack of effective criteria for TA activity has limited the clinical research and clinical management in TA. The National Institutes of Health (NIH) criteria for active disease, which define the clinical status on the basis of four findings, features of vascular ischemia or inflammation, elevated erythrocyte sedimentation rate (ESR) or C-reactive protein (CRP) level, systemic features, and angiographic changes, have been used for evaluating TA activity.35 Later, some authors highlighted that NIH criteria could not accurately reflect the progression of vascular inflammation because in half of these patients the clinical findings do not correlate with acute-phase reactants (APRs), vessel biopsy is rarely available, and radiology studies are harder to use in the follow-up, suggesting that these criteria do not identify smoldering vessel inflammation and not all TA patients with systemic inflammatory flares develop anatomical progression of the vascular involvement.
Recent studies
In 2005, the Disease Extent Index-Takayasu (DEI.Tak) was developed for the follow-up of TA, assessing only laboratory parameters (systemic, cutaneous, mucous membranes, chest, abdomen, kidney, central nervous, genitourinary and cardiovascular systems, eyes, ear–nose–throat, ESR, and CRP) and clinical findings without the requirement for imaging techniques.36 Following studies have validated the effectiveness of DEI.Tak for evaluating the TA activity with the development of the Indian Takayasu Clinical Activity Score (ITAS2010), a disease activity index that incorporates symptoms and clinical signs with APRs.37,38 ITAS2010 includes only six systems in a simplified adoption of the Birmingham Vasculitis Activity Score. In 2009, the Outcome Measures in Rheumatology Vasculitis Working Group initiated the LVV task force for developing a core set of outcome measures for clinical trials in LVV.39,40 Serum pentraxin-3 (PTX3), a long pentraxin produced by immune and vascular cells, and matrix metalloproteinase-9, produced in response to inflammatory cytokines, have been proposed as sensitive biomarkers for assessing TA activity, even in the absence of an increase in CRP. PTX3 was the most reliable single marker,41,42 but these data require confirmatory studies. Recent studies indicate that IL-6 is the best biomarker associated with TA activity.43
TA and renal arteries
Visceral arterial involvement is reported in 11%–68% of the TA patients, usually in the form of steno-occlusive lesion. The renal arteries are frequently involved, usually with renovascular hypertension. The incidence is from 24% to 68%. The renal artery involvement is often bilateral and frequently ostial and proximal, usually with coexistent stenosis of the perirenal aorta; conversely, renovascular dilatative and aneurysmal lesions are rare. Recently, magnetic resonance imaging and computed tomography (CT), noninvasive techniques, have been used for the detection of active disease and to demonstrate mural involvement. These mural changes correlate with TA activity and with clinical remission. The complexity of pathologic changes in the aortic and renal wall makes surgical revascularization technically difficult. Endovascular reconstruction is rarely performed; the aneurysms often involve the perirenal aortic wall and the visceral arteries; therefore, the endovascular reconstruction is impossible. There are few reports about the use of drug-eluting stent in TA renal artery stenosis. Agarwal et al reported a chronic total occlusion of renal artery in a 17-year-old girl with TA, treated with a drug-eluting stent. The authors reported a full renal reperfusion, improvement in arterial pressure, and patency of the stent over 9 months after the procedure.44 Hecht et al reported about a 12-year-old girl with TA, treated with a balloon angioplasty of the both renal arteries with a paclitaxel-eluting balloon, achieving the elimination of the renal artery stenosis.45 The drug-eluting technology is a good option but needs more studies over the time.
Takayasu’s arteritis and kidney involvement
Kidney vascular involvement in TA is frequent and worsens the progression of the disease. Glomerular or tubular–interstitial damage in TA is a rare, chronic, slowly progressive complication, and acute renal dysfunction is even rarer. The glomerular disease is exceptional. Only few clinical reports are currently available about the glomerular changes associated with TA. Mesangial proliferation is the predominant histological picture.46,47 In 2007, de Pablo et al retrospectively analyzed the renal biopsies in 25 autopsy cases of TA. They detected 10 cases with diffuse mesangial proliferative glomerulonephritis (GN), four cases with other forms of GN (focal segmental glomerulosclerosis, global and focal glomerulosclerosis, segmental necrotizing GN, and amyloidosis), and 11 cases with nonspecific changes.48 In 2014, Koda et al described a case of membranous nephropathy in a 54-year-old man with nephrotic syndrome and a not-previously-diagnosed TA.49 TA commonly involves the renal arteries. Stenosis and occlusion are the most reported abnormalities in renal arteries in TA; renovascular hypertension and ischemic nephropathy are the most common clinical manifestations. In a retrospectively study of a US cohort of 126 TA subjects in the period of 1984–2009, renal artery changes were detected in 24 of 41 patients (58%) with new-onset hypertension.50 In Asiatic countries, especially in China, Korea, and India, TA is the most frequent cause of pediatric renal artery stenosis.51 The prevalence of renal artery involvement among TA subjects varies from 11.5% to 62%; it is higher in Asian populations than in populations of North America, Northern Europe, and Africa.52
Surgical interventions in Takayasu’s arteritis
Over the last decades, significant improvements have been made in the surgical interventions in TA; however, after endovascular procedures and stent insertion, restenosis rates are higher. The most common TA changes treated are arterial stenoses/occlusions, aortic regurgitation, and aneurysms. The principal indications are uncontrolled renovascular hypertension, aortic coarctation, and ischemic heart or symptomatic cerebrovascular diseases.53 During the active phase of the disease, both endovascular interventions and surgical procedures should be avoided because the biological inflammation at the time of intervention increases the complication rate. Patients with clinically inactive disease and treated with effective periprocedural immunosuppressant therapy have significantly improved outcomes. There are no guidelines about the choice between open surgery and endovascular approaches, and the efficacy of surgical management has not yet been established because the course and prognosis vary widely according to the affected artery.54 Among 411 Chinese TA patients in the period of 1990–2014, the bypass surgery was the most frequent surgical intervention for TA-related lesions (54 of 411, 13.1%), followed by percutaneous transluminal angioplasty (PTA; 25 of 411, 6.1%) for renal artery stenosis or occlusion, 22 (5.4%) PTA for stenosis or occlusion in other arteries, 35 (8.5%) stent implantation for stenosis, and 11 (2.7%) other operations for TA-related lesions.55 Disease progression and need for subsequent reintervention in subjects surgically treated for TA are frequent occurrences in those who require revascularization for end-organ ischemia.56
Medical therapy
Conventional immunosuppressant therapy
The difficulty of early disease recognition and the lack of standard and reliable parameters reflecting disease activity make TA a difficult disease to deal with. Although the life expectancy of TA patients was estimated to be low, the introduction of glucocorticosteroids (GCs) and immunosuppressive therapy has strongly improved the prognosis. There is evidence that long-term corticosteroid therapy contributes to the improvement of the angiographic findings. However, up to now, no randomized controlled trials have evaluated medical therapy in TA, and the reported results of different treatments differ widely. GCs represent the first-line therapy of TA, but ~50% of all TA subjects have chronic active disease and the GC therapy alone does not provide sustained remissions. These patients relapse upon GC dose tapering or GC therapy discontinuation. For TA patients resistant to GCs, immunosuppressive drugs (cyclophosphamide, methotrexate [MTX], azathioprine, and mycophenolate mofetil) are usually added to GCs to induce disease remission, to prevent the development of new angiographic arterial changes, and to spare GCs. The rates of remission induction in TA patients for MTX, azathioprine, and mycophenolate mofetil in the period of 1994–2007 were reported to be 81.0%, 76.7%, and 90.0%, respectively, but there is no randomized study comparing the efficacy of different immunosuppressive drugs in the treatment of TA.59 However, relapses remain frequent despite the use of adjunctive immunosuppressive therapies, and the treatment of refractory TA remains an unresolved clinical issue. In the period of 2000–2007, six Turkish TA patients with an age range of 12–17 years were treated with cyclophosphamide induction and corticosteroids followed by MTX. The authors reported that this treatment was an effective and safe treatment for childhood TA.57 In the period of 2003–2007, 10 Japanese patients with TA resistant to GCs (five after at least one immunosuppressive drug and five as first immunosuppressive therapy) were treated with mycophenolate mofetil for an average of 23.3 months; in nine patients, the clinical activity disappeared with significant tapering of the prednisone dose.58
New therapeutic options
TNF-α antagonists and leflunomide are new options in subjects resistant to conventional immunosuppressive treatments. In 2013, nine Russian women with TA refractory to standard immunosuppressant drugs were treated with TNF inhibitors. The treatment (eight patients with infliximab and one with adalimumab) resulted in complete remission in five patients and incomplete remission in three patients with tapering of prednisone to ≤10 mg daily in all patients. The treatment was safe and well-tolerated (except for allergic reactions after infusions of infliximab in one patient). Four patients developed relapse of vasculitis when the dosing interval of infliximab was increased to 6–8 weeks.21 A nationwide French study retrospectively analyzed the long-term outcome (median follow-up of 24 months, range 2–95 months) of biologically targeted treatments in the period of 2001–2013; 49 TA subjects resistant to conventional therapy were treated with TNF-α antagonists (infliximab, etanercept, and adalimumab; 80% of treatments) and tocilizumab (TCZ; 20%). According to the authors, TNF-α antagonists and TCZ led to remission with 93% improvement (ie, complete and partial responders) at 12 months, and 75% of patients being complete responders, with a relatively good safety profile. TNF-α antagonists and TCZ seemed to have equivalent efficacy and tolerance.59 In 2016, among 12 Brazilian TA patients, treated with leflunomide, five had a sustained remission and continued leflunomide therapy, while seven had to change to another therapy due to failure to prevent relapses in six patients and toxicity in one patient. The mean follow-up time was 43.0±7.6 months.60 According to the authors, leflunomide induces remission in active TA similarly to TNF antagonists but seems to be inferior to biological agents in preventing disease relapses in TA. TCZ and rituximab have been shown to lead to improved disease activity in small numbers of TA, including refractory to anti-TNF treatment. In 2012, four Italian patients with active LVV (two with GCA and two with TA), treated with TCZ, showed a satisfactory clinical and laboratory response, with significant improvements in PET/CT findings, without serious adverse events.61 In 2017, a single-center prospective study analyzed the efficacy and safety of TCZ in 16 Chinese TA patients (15 patients with previous conventional immune suppressants); three patients withdrew TCZ after one infusion due to unbearable neck pain; the other 13 patients were treated for a median of 13 (7–20) months with reduction of ESR, CRP, and mural thickness of common carotid artery and subclavian artery.62 The favorable outcome of B-cell depletion therapy through rituximab treatment in refractory TA was reported by Hoyer in 2012 (three subjects) and Caltran et al in 2014 (two subjects).63 The authors suggested that B cells and plasmablasts/plasma cells may represent novel targets for effective therapies for TA. In 2010, de Souza et al retrospectively analyzed 30 Brazilian TA patients treated with antiplatelet therapy (median aspirin daily 200 mg in 29 patients and ticlopidine in one patient). The authors reported that antiplatelet drugs had a protective effect against ischemic events without bleeding complications. There was no significant difference concerning the use of anticoagulant drugs, prednisone, immunosuppressive treatments, or anti-TNF-α agents between subjects with or without ischemic events.64
Prognosis
There are few reports about the TA long-term prognosis. In the past century, relevant morbidity and mortality were seen worldwide in young patients.48,49 Up to date, the disease usually has a long-term indolent course, and acute events (ischemic heart, symptomatic cerebrovascular diseases, or sudden visual loss) are uncommon and variably observed in different populations. In a Japanese population, a decrease in the mortality of TA patients has been reported, in the period of 2000–2010. In France in a population of different ethnicity, the survival rates were reported: 100% and 95.0%, respectively, at 5 years and 10 years in whites, 100% at both 5 years and 10 years in blacks, and only 67.4% at both 5 years and 10 years in North African patients. The authors suggested major prognostic differences according to ethnicity.
Conclusion
Although there is often a delay in the diagnosis of TA, due to different presentation and nonspecific symptoms, the development of noninvasive diagnostic imaging tools, earlier diagnosis, and more effective medical treatments have improved the long-term prognosis of TA in the last decades. However, the TA activity assessment is often difficult since there is no effective outcome measure reflecting significant ongoing arterial wall inflammation. To date, there is a growing need for TA biomarkers.
Disclosure
The authors report no conflicts of interest in this work.
References
Numano F. Differences in clinical presentation and outcome in different countries for Takayasu’s arteritis. Curr Opin Rheumatol. 1997;9:12–15. | ||
Hotchi M. Pathological studies on Takayasu arteritis. Heart Vessels Suppl. 1992;7:11–17. | ||
Alibaz-Öner F, Aydın SZ, Direskeneli H. Recent advances in Takayasu’s arteritis. Eur J Rheumatol. 2015;2:24–30. | ||
Terao C. History of Takayasu arteritis and Dr. Mikito Takayasu. Int J Rheum Dis. 2014;17:931–935. | ||
Ohigashi H, Haraguchi G, Konishi M, et al. Improved prognosis of Takayasu arteritis over the past decade – comprehensive analysis of 106 patients. Circ J. 2012;76:1004–1011. | ||
Watts R, Al-Taiar A, Mooney J, Scott D, Macgregor A. The epidemiology of Takayasu arteritis in the UK. Rheumatology (Oxford). 2009;48:1008–1011. | ||
Mohammad AJ, Mandl T. Takayasu arteritis in southern Sweden. J Rheumatol. 2015;42:853–858. | ||
Birlik M, Kücükyavas Y, Aksu K, et al. Epidemiology of Takayasu’s arteritis in Turkey. Clin Exp Rheumatol. 2016;34:S33–S39. | ||
Terao C, Yoshifuji H, Mimori T. Recent advances in Takayasu arteritis. Int J Rheum Dis. 2014;17:238–247. | ||
Onen F, Akkoc N. Epidemiology of Takayasu arteritis. Presse Med. 2017;46:e197–e203. | ||
Mustafa KN. Takayasu’s arteritis in Arabs. Clin Rheumatol. 2014;33:1777–1783. | ||
Vanoli M, Daina E, Salvarani C, et al. Takayasu’s arteritis: a study of 104 Italian patients. Arthritis Rheum. 2005;53:100–107. | ||
Dreyer L, Faurschou M, Baslund B. A population-based study of Takayasu’s arteritis in eastern Denmark. Clin Exp Rheumatol. 2011;29:S40–S42. | ||
Karageorgaki ZT, Bertsias GK, Mavragani CP, et al. Takayasu arteritis: epidemiological, clinical, and immunogenetic features in Greece. Clin Exp Rheumatol. 2009;27:S33–S39. | ||
Arnaud L, Haroche J, Limal N, et al. Takayasu arteritis in France: a single-center retrospective study of 82 cases comparing white, North African, and black patients. Medicine (Baltimore). 2010;89:1–17. | ||
Gutierrez-Arcelus M, Rich SS, Raychaudhuri S. Autoimmune diseases – connecting risk alleles with molecular traits of the immune system. Nat Rev Genet. 2016;17:160–174. | ||
Arnaud L, Haroche J, Mathian A, Gorochov G, Amoura Z. Pathogenesis of Takayasu’s arteritis: a 2011 update. Autoimmun Rev. 2011;11:61–67. | ||
Chen S, Luan H, Li L, et al. Relationship of HLA-B*51 and HLA-B*52 alleles and TNF-α-308A/G polymorphism with susceptibility to Takayasu arteritis: a meta-analysis. Clin Rheumatol. 2017;36:173–181. | ||
Terao C, Yoshifuji H, Ohmura K, et al. Association of Takayasu arteritis with HLA-B*67:01 and two amino acids in HLA-B protein. Rheumatology (Oxford). 2013;52:1769–1774. | ||
Seko Y. Takayasu arteritis: insights into immunopathology. Jpn Heart J. 2000;41:15–26. | ||
Novikov PI, Smitienko IO, Moiseev SV. Tumor necrosis factor alpha inhibitors in patients with Takayasu’s arteritis refractory to standard immunosuppressive treatment: cases series and review of the literature. Clin Rheumatol. 2013;32:1827–1832. | ||
D’Alfonso S, Richiardi PM. A polymorphic variation in a putative regulation box of the TNFA promoter region. Immunogenetics. 1994;39:150–154. | ||
Saruhan-Direskeneli G, Bicakcigil M, Yilmaz V, et al. Interleukin (IL)-12, IL-2 and IL-6 gene polymorphisms in Takayasu’s arteritis from Turkey. Hum Immunol. 2006;67:735–740. | ||
Qin F, Wang H, Song L, et al. Single nucleotide polymorphism rs10919543 in FCGR2A/FCGR3A region confers susceptibility to Takayasu arteritis in Chinese population. Chin Med J (Engl). 2016;129:854–859. | ||
Morgan AW, Robinson JI, Barrett JH, et al. Association of FCGR2A and FCGR2A-FCGR3A haplotypes with susceptibility to giant cell arteritis. Arthritis Res Ther. 2006;8:R109. | ||
Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013;65:1–11. | ||
Inder SJ, Bobryshev YV, Cherian SM, et al. Immunophenotypic analysis of the aortic wall in Takayasu’s arteritis: involvement of lymphocytes, dendritic cells and granulocytes in immuno-inflammatory reactions. Cardiovasc Surg. 2000;8:141–148. | ||
Maksimowicz-McKinnon K, Clark TM, Hoffman GS. Takayasu arteritis and giant cell arteritis: a spectrum within the same disease? Medicine (Baltimore). 2009;88:221–226. | ||
Kim YJ, Park YS, Koo BS, et al. Immunoglobulin G4-related disease with lymphoplasmacytic aortitis mimicking Takayasu arteritis. J Clin Rheumatol. 2011;17:451–452. | ||
Koster MJ, Warrington KJ. Classification of large vessel vasculitis: Can we separate giant cell arteritis from Takayasu arteritis? Presse Med. 2017;46:e205–e213. | ||
Ishikawa K. Diagnostic approach and proposed criteria for the clinical diagnosis of Takayasu’s arteriopathy. J Am Coll Cardiol. 1988;12:964–972. | ||
Arend WP, Michel BA, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of Takayasu arteritis. Arthritis Rheum. 1990;33:1129–1134. | ||
Ozen S, Ruperto N, Dillon MJ, et al. EULAR/PReS endorsed consensus criteria for the classification of childhood vasculitides. Ann Rheum Dis. 2006;65:936–941. | ||
Kong X, Ma L, Wu L, et al. Evaluation of clinical measurements and development of new diagnostic criteria for Takayasu arteritis in a Chinese population. Clin Exp Rheumatol. 2015;33:S-48–S-55. | ||
Kerr GS, Hallahan CW, Giordano J, et al. Takayasu arteritis. Ann Intern Med. 1994;120:919–929. | ||
Aydin SZ, Yilmaz N, Akar S, et al. Assessment of disease activity and progression in Takayasu’s arteritis with Disease Extent Index-Takayasu. Rheumatology (Oxford). 2010;49:1889–1893. | ||
Ishihara T, Haraguchi G, Kamiishi T, Tezuka D, Inagaki H, Isobe M. Sensitive assessment of activity of Takayasu’s arteritis by pentraxin 3, a new biomarker. J Am Coll Cardiol. 2011;57:1712–1713. | ||
Sivakumar MR, Misra R, Bacon PA. The Indian perspective of Takayasu arteritis and development of a disease extent index (DEI.Tak) to assess Takayasu arteritis. Rheumatology. 2005;44:iii6–iii7. | ||
Misra R, Danda D, Rajappa SM, et al. Development and initial validation of the Indian Takayasu Clinical Activity Score (ITAS2010). Rheumatology (Oxford). 2013;52:1795–1801. | ||
Aydin SZ, Merkel PA, Direskeneli H. Outcome measures for Takayasu’s arteritis. Curr Opin Rheumatol. 2015;27:32–37. | ||
Ishihara T, Haraguchi G, Tezuka D, Kamiishi T, Inagaki H, Isobe M. Diagnosis and assessment of Takayasu arteritis by multiple biomarkers. Circ J. 2013;77:477–483. | ||
Dagna L, Salvo F, Tiraboschi M, et al. Pentraxin-3 as a marker of disease activity in Takayasu arteritis. Ann Intern Med. 2011;155(7):425–433. | ||
Savioli B, Abdulahad WH, Brouwer E, Kallenberg CGM, de Souza AWS. Are cytokines and chemokines suitable biomarkers for Takayasu arteritis? Autoimmun Rev. 2017;16:1071–1078. | ||
Agarwal G, Vats HS, Raval AN, Yevzlin AS, Chan MR, Gimelli G. Chronic total occlusion and successful drug-eluting stent placement in Takayasu arteritis-induced renal artery stenosis. Clin Med Res. 2013;11:233–236. | ||
Hecht T, Esmaeili A, Behnke-Hall K. Balloon angioplasty of the bilateral renal arteries by Takayasu arteritis with a paclitaxel-eluting balloon. Cardiol Young. 2015;25:1431–1434. | ||
Maksimowicz-McKinnon K, Clark TM, Hoffman GS. Limitations of therapy and a guarded prognosis in an American cohort of Takayasu arteritis patients. Arthritis Rheum. 2007;56:1000–1009. | ||
Boubaker K, Kaaroud H, Goucha R, Kheder A. Atteinte rénale au cours de la maladie de Takayasu. [Renal injury in Takayasu’s arteritis]. Nephrol Ther. 2014;10:451–456. French. | ||
de Pablo P, García-Torres R, Uribe N, et al. Kidney involvement in Takayasu arteritis. Clin Exp Rheumatol. 2007;25:S10–S14. | ||
Koda R, Yoshino A, Imanishi Y, et al. A case of membranous glomerulonephropathy associated with Takayasu’s arteritis. Case Rep Nephrol Urol. 2014;4:60–69. | ||
Schmidt J, Kermani TA, Bacani AK, et al. Diagnostic features, treatment, and outcomes of Takayasu arteritis in a US cohort of 126 patients. Mayo Clin Proc. 2013;88:822–830. | ||
Zhu G, He F, Gu Y, et al. Angioplasty for pediatric renovascular hypertension: a 13-year experience. Diagn Interv Radiol. 2014;20:285–292. | ||
Chen Z, Li J, Yang Y, et al. The renal artery is involved in Chinese Takayasu’s arteritis patients. Kidney Int. 2018;93:245–251. | ||
Mason JC. Takayasu arteritis: surgical interventions. Curr Opin Rheumatol. 2015;27:45–52. | ||
Miyata T, Sato O, Koyama H, Shigematsu H, Tada Y. Long-term survival after surgical treatment of patients with Takayasu’s arteritis. Circulation. 2003;108:1474–1480. | ||
Li J, Sun F, Chen Z, et al. The clinical characteristics of Chinese Takayasu’s arteritis patients: a retrospective study of 411 patients over 24 years. Arthritis Res Ther. 2017;19:107. | ||
Ham SW, Kumar SR, Rowe VL, Weaver FA. Disease progression after initial surgical intervention for Takayasu arteritis. J Vasc Surg. 2011;54:1345–1351. | ||
Ozen S, Duzova A, Bakkaloglu A, et al. Takayasu arteritis in children: preliminary experience with cyclophosphamide induction and corticosteroids followed by methotrexate. J Pediatr. 2007;150:72–76. | ||
Shinjo SK, Pereira RM, Tizziani VA, Radu AS, Levy-Neto M. Mycophenolate mofetil reduces disease activity and steroid dosage in Takayasu arteritis. Clin Rheumatol. 2007;26:1871–1875. | ||
Mekinian A, Comarmond C, Resche-Rigon M, et al. Efficacy of biological-targeted treatments in Takayasu arteritis: multicenter, retrospective study of 49 patients. Circulation. 2015;132:1693–1700. | ||
de Souza AW, de Almeida Agustinelli R, de Cinque Almeida H, et al. Leflunomide in Takayasu arteritis – a long term observational study. Rev Bras Reumatol Engl Ed. 2016;56:371–375. | ||
Salvarani C, Magnani L, Catanoso M, et al. Tocilizumab: a novel therapy for patients with large-vessel vasculitis. Rheumatology (Oxford). 2012;51:151–156. | ||
Zhou J, Chen Z, Li J, et al. The efficacy of tocilizumab for the treatment of Chinese Takayasu’s arteritis. Clin Exp Rheumatol. 2017;35:171–175. | ||
Hoyer BF, Mumtaz IM, Loddenkemper K, et al. Takayasu arteritis is characterised by disturbances of B cell homeostasis and responds to B cell depletion therapy with rituximab. Ann Rheum Dis. 2012;71(1):75–79. | ||
de Souza AW, Machado NP, Pereira VM, et al. Antiplatelet therapy for the prevention of arterial ischemic events in Takayasu arteritis. Circ J. 2010;74:1236–1241. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.