Back to Journals » International Journal of Nanomedicine » Volume 15
Tailoring Nanoparticle-Biofilm Interactions to Increase the Efficacy of Antimicrobial Agents Against Staphylococcus aureus
Authors Fulaz S ![]() , Devlin H
, Devlin H ![]() , Vitale S
, Vitale S ![]() , Quinn L
, Quinn L ![]() , O'Gara JP
, O'Gara JP ![]() , Casey E
, Casey E ![]()
Received 31 March 2020
Accepted for publication 9 May 2020
Published 7 July 2020 Volume 2020:15 Pages 4779—4791
DOI https://doi.org/10.2147/IJN.S256227
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Stephanie Fulaz,1,* Henry Devlin,1,* Stefania Vitale,1 Laura Quinn,1 James P O’Gara,2 Eoin Casey1
1UCD School of Chemical and Bioprocess Engineering, University College Dublin, Dublin, Ireland; 2Department of Microbiology, School of Natural Sciences, National University of Ireland, Galway, Ireland
*These authors contributed equally to this work
Correspondence: Eoin Casey
UCD School of Chemical and Bioprocess Engineering, University College Dublin, Belfield, Dublin 4, Dublin, Ireland
Email [email protected]
Background: Considering the timeline required for the development of novel antimicrobial drugs, increased attention should be given to repurposing old drugs and improving antimicrobial efficacy, particularly for chronic infections associated with biofilms. Methicillin-susceptible Staphylococcus aureus (MSSA) and methicillin-resistant S. aureus (MRSA) are common causes of biofilm-associated infections but produce different biofilm matrices. MSSA biofilm cells are typically embedded in an extracellular polysaccharide matrix, whereas MRSA biofilms comprise predominantly of surface proteins and extracellular DNA (eDNA). Nanoparticles (NPs) have the potential to enhance the delivery of antimicrobial agents into biofilms. However, the mechanisms which influence the interactions between NPs and the biofilm matrix are not yet fully understood.
Methods: To investigate the influence of NPs surface chemistry on vancomycin (VAN) encapsulation and NP entrapment in MRSA and MSSA biofilms, mesoporous silica nanoparticles (MSNs) with different surface functionalization (bare-B, amine-D, carboxyl-C, aromatic-A) were synthesised using an adapted Stöber method. The antibacterial efficacy of VAN-loaded MSNs was assessed against MRSA and MSSA biofilms.
Results: The two negatively charged MSNs (MSN-B and MSN-C) showed a higher VAN loading in comparison to the positively charged MSNs (MSN-D and MSN-A). Cellular binding with MSN suspensions (0.25 mg mL− 1) correlated with the reduced viability of both MSSA and MRSA biofilm cells. This allowed the administration of low MSNs concentrations while maintaining a high local concentration of the antibiotic surrounding the bacterial cells.
Conclusion: Our data suggest that by tailoring the surface functionalization of MSNs, enhanced bacterial cell targeting can be achieved, leading to a novel treatment strategy for biofilm infections.
Keywords: Staphylococcus aureus, mesoporous silica nanoparticles, EPS matrix, antimicrobial, vancomycin, nanoparticle-biofilm interactions
Introduction
Antibiotic resistance coupled with biofilm formation is a serious health problem causing a huge economic burden to the healthcare system.1 Currently, drug-resistant bacteria cause 700,000 deaths per year and, according to the World Health Organization, by 2050, this number will reach 10 million deaths per year.2,3 Bacterial biofilms are the main cause of chronic and persistent infections and healthcare-associated infections,4,5 accounting for approximately 80% of all chronic and recurrent infections6 such as endocarditis, osteomyelitis and pneumonia.7 They pose a serious health threat owing mainly to the ability to withstand the host immune response, antibiotics and environmental stressors.8,9 The cells embedded in the biofilm are up to 1000 times less susceptible to antibiotics than their planktonic counterparts.10 This reduction in antibiotic susceptibility is associated with the altered physiology of biofilm cells, which are embedded in an extracellular matrix, consisting of extracellular polymeric substances (EPS) produced by the bacterial cells.11,12 The major components of the EPS are polysaccharides, proteins and eDNA. This highly hydrated matrix offers chemical and mechanical support to the bacterial community.11,12 The complex interlink of the EPS components leads to poor antibiotic penetration due to diffusion limitation.13 The EPS components and acidic microenvironments14 within the matrix can also interact with and inactivate drug molecules reducing the local concentration around the bacterial cells.15
Among all drug-resistant infections, methicillin-resistant Staphylococcus aureus (MRSA) is the most frequent, with a high incidence worldwide.16,18 In the USA alone, MRSA accounts for 11,285 deaths per year. The treatment of MRSA infections is mainly based on vancomycin and daptomycin, with emerging susceptibility issues to each.19 MRSA and Methicillin- sensitive S. aureus (MSSA) clinical isolates have been shown to use different mechanisms to form biofilms. It has previously been identified that the attainment or loss of methicillin resistance impacts the biofilm phenotype formed,20 with MSSA more likely to form biofilms consisting of icaADBC-encoded PIA (polysaccharide intercellular adhesion) or PNAG (Poly-N-acetylglucosamine) polysaccharide,21,22 whilst the ica operon appears to be redundant for MRSA biofilm formation, which is instead dependent on surface proteins (such as the fibronectin-binding proteins) and extracellular DNA (eDNA) released from cells that have undergone autolysis.22
Considering that the development of new drugs is a very long and costly process,23,24 the repurposing of existing drugs is gaining traction.25 On the other hand, strategies aimed to increase antimicrobial efficacy can help reduce the development of drug resistance, mainly by leading to a decrease in the drug concentration needed for the treatment and clearance of biofilm infections. Nanoparticles have been investigated as potential drug-carriers, as they have the ability to enhance the penetration of antimicrobial agents into the deeper regions of the biofilm.26 A promising approach is the use of nanoparticles to improve therapeutic profiles of existing antibiotics.27 Nanoparticles can improve antibiotic efficacy, avoiding antimicrobial detection and deactivation in the biofilm matrix, providing targeted delivery to the cells and increasing the local concentration around the bacterial cells. In this framework, a noteworthy model system is based on the use of porous inorganic nanoparticles, for instance, mesoporous silica nanoparticles (MSNs). They are considered a promising delivery system for antimicrobial agents owing to their stable structure, flexible surface chemistry, good biocompatibility and high drug loading capacity.28,30 It has also been shown that the MSNs can protect the loaded antimicrobial agent from deactivation within the biofilm matrix and also allows for a better distribution through the biofilm when compared to free antibiotics.31,32 Previous studies, which have followed similar experimental designs involving the loading of an antimicrobial agent (antibiotic, enzyme, etc.) to the nanoparticles, have demonstrated some success.33,37 However, there is a lack of investigations into the fundamental interactions which govern the fate of nanoparticles when they come into contact with a biofilm. This information is vital when trying to identify specific physicochemical properties which influences nanoparticle – biofilm interactions.38
An essential part in the development of biofilm eradication systems is the investigation of the biofilm matrix and the influence of the EPS components on nanoparticle entrapment and penetration into the biofilm along with the interaction of the nanoparticles with bacterial cells. Biofilm diffusion resistance mechanisms towards antimicrobial agents are based in steric hindrance and repulsive electrostatic interactions, since bacterial cell walls and EPS components are (typically) negatively charged. The co-administration of penetration enhancers, such as electrolytes could be employed to improve the penetration of nanoparticles. For instance, the EPS resistance towards negatively charged nanoparticles could be reduced through electrostatic repulsion by the co-administration of cationic electrolytes.39
Herein, engineered MSNs with different surface chemistry were evaluated with regards to their entrapment in the biofilm of two S. aureus isolates, whose biofilm matrices differ significantly in composition. The role of the EPS components in nanoparticle-biofilm interactions was examined. Considering the importance of charge in these interactions, electrolyte screening using anionic (PBS) and a cationic (Tris) buffers were investigated as a tool for the enhanced administration of MSNs to bacterial biofilms. The impact of changing MSN surface functionalization with regards to vancomycin encapsulation, and the effect on cell viability was investigated. This systematic study aims to shine a light on the important parameters regarding nanoparticle-biofilm interactions and offer guidance to the design of biofilm eradication strategies, in order to improve antimicrobial efficacy and fight growing antimicrobial resistance trends.
Materials and Methods
Materials
Hexadecyltrimethylammonium bromide (CTAB, 98%), tetraethyl orthosilicate (TEOS, 99%), (3-aminopropyl)triethoxysilane (APTES, 99%), rhodamine B isothiocyanate (RITC, mixed isomers), triethanolamine (TEA, 99%), acetic acid, N1-(3-trimethoxysilylpropyl) diethylenetriamine (DETA), dimethylformamide (DMF), N-hydroxysuccinimide (NHS, 98%), N-(3-Dimethylaminopropyl)-N′-ethyl carbodiimide hydrochloride (EDC, 98%), glycerol, tryptic soy agar and tryptic soy broth (TSB), cell proliferation kit 1 (MTT), NaCl, glucose, tris(hydroxymethyl)aminomethane (Tris), PBS, dimethyl sulfoxide (DMSO) and sodium deuteroxide (NaOD) were purchased from Sigma-Aldrich. Ethanol (99.5%) and hydrochloric acid (HCl, 37%) were obtained from Merck. Succinic anhydride was acquired from Santa Cruz Biotechnology. Benzoic acid was purchased from Analar (BDH Chemicals Ltd). Crystal Violet (CV) was purchased from Honeywell Riedel-de-Haën™. All reagents were used as received without further purification. Clear 96-well plates were purchased from SARSTEDT, Germany. Costar® Black 96-well plates were purchased from Corning, UK. Grade 1 pure water, referred to as MilliQ water was obtained from an Elga Process Water System (Biopure 15 and Pureflex 2, Veolia, Ireland).
Synthesis and Functionalization of MSNs
The synthesis of MSNs was performed using an adapted Stöber method as previously described.14 The functionalization of MSNs was adapted from Gounani et al (2018);40 50 mg of bare-MSNs (MSN-B) were dispersed in 4.6 mL of absolute ethanol in an ultrasonic bath, then 200 µL of deionized water, 100 µL of acetic acid and 100 µL of DETA were added, and the reaction mixture stirred at room temperature for 1 h. The obtained aminated product (MSN-D) was washed three times with ethanol by centrifugation (9,000 rpm, 15 min). In order to obtain the carboxyl functionalized MSNs, a suspension of dried MSN-D in DMF (6 mL of 2 mg mL−1) was added to a solution of succinic anhydride in DMF (50 mL of 10 mg mL−1). The mixture was vigorously stirred overnight at room temperature. The carboxyl product (MSN-C) was washed three times with ethanol by centrifugation (9,000 rpm, 15 min). For the aromatic nanoparticles, a suspension of MSN-D in water (9 mL of 3.34 mg mL−1) was mixed with the aqueous solutions of benzoic acid (20 mL of 4.71 mg mL−1), NHS (2 mL of 114.7 mg mL−1) and EDC (2 mL of 150.8 mg mL−1). The mixture was kept stirring at room temperature overnight. The product MSN-A was recovered by extensive washing with ethanol by centrifugation (9,000 rpm, 15 min).
MSNs Characterization
Zeta potential of nanoparticles suspension in water (1 mg mL−1) was measured in a folded capillary zeta cell using a Zetasizer Nano-ZS apparatus (Malvern Instruments). All measurements were done in triplicates with 10 scans each. TEM analysis was performed using a FEI Tecnai G2 on samples dispersed in ethanol and deposited on carbon-coated copper grids. Average size and size distribution of nanoparticles were analyzed by Fiji software41 using TEM images, analyzing a minimum of 50 nanoparticles per sample. FT-IR was performed in a Bruker Vertex 70 spectrophotometer. Samples in ethanol dispersion were deposited onto NaCl FT-IR cards and dried at 70 °C. Spectra were obtained using a resolution of 4 cm−1, 64 scans and a spectral window from 4000 to 400 cm−1. Dissolution 1H NMR was used to dissolve the MSNs and obtain a clear spectrum of the surface groups, based on a previously reported method.42 Approximately 10–15 mg of MSNs were dissolved in 662 µL of D2O and 38 µL of NaOD. The mixture was incubated overnight at 37 °C under stirring. The spectra were obtained in a Varian Inova 300 MHz Spectrometer using 128 scans per sample.
Vancomycin Loading in MSNs
For vancomycin loading into nanoparticles, 8 mg of antibiotic was added to 1 mL of MSNs suspensions (25 mg mL−1) in deionized water. The mixture was stirred at room temperature for 48 h and dried at 50 °C to facilitate drug encapsulation. The vancomycin loaded MSNs were washed twice with water by centrifugation (12000 rpm, 15 min) to remove unloaded antibiotic. The washing solutions were collected and evaluated regarding vancomycin concentration. The UV-visible spectrum was recorded in a Plate reader (SpectraMax iD3, Molecular devices). The loading content was calculated using a calibration curve (R2 = 0.99).43 To confirm vancomycin loading into MSNs, the carbon content of dried loaded and unloaded MSNs was measured (Exeter Analytical CE 440 elemental analyzer).
Bacterial Culture
The bacterial strains used in this study were methicillin-resistant S. aureus BH1CC (MRSA) and methicillin-sensitive S. aureus 8325–4 (MSSA). Bacterial stock cultures were stored in 25% (v/v) glycerol at −80°C. Aliquots were streaked on Tryptic soy agar plates and incubated at 37 °C for 24 h. A single bacterial colony was used to inoculate 50 mL of sterile TSB in an Erlenmeyer flask and incubated at 37°C with shaking at 200 rpm overnight. For biofilm formation, the overnight culture was diluted to a final optical density (OD600) of 0.001.
Biofilm Formation
Biofilm formation on the bottom of a multiwell well plate may hinder the assessment of nanoparticle entrapment, as nanoparticle sedimentation may influence fluorescent intensity measurements used to quantify biofilm entrapment. To overcome this, metallic pegs (3mm x 10mm, nickel-plated neodymium magnet, Supermagnete) were utilized in a method similar to the Calgary device (Figure S1). The pegs were suspended in a liquid culture medium which allowed for biofilm formation on the surface of the peg. The peg was then removed and used for the MSN entrapment assay. The MRSA strain BH1CC was cultured in TSB supplemented with 2% glucose to promote the formation of biofilm mediated by the fibronectin-binding proteins, and eDNA.44 MSSA biofilm culture media was supplemented with 4% NaCl, which activates expression of the ica operon-encoded enzymes used to synthesize PIA/PNAG, a major polysaccharide component of the MSSA EPS matrix.45 Biofilms were grown at 37 °C for 48 h under static conditions, before being washed three times in water to remove planktonic cells.
MSN Entrapment
To assess MSN entrapment, S. aureus biofilms were first incubated with buffers (Tris or PBS) or water (control) for 30 min. The pegs were then transferred into a sterile black 96-well plate, and each magnet was secured into position against the upper wall of the well using a small magnet (5mm x 5mm, nickel-plated neodymium magnet, Supermagnete). The biofilms were then exposed to various nanoparticles (MSN-B, MSN-D, MSN-C and MSN-A) at a concentration of 1 mg mL−1 for 3 h with fluorescent intensity readings taken every 10 mins to assess entrapment in a method similar to Fulaz et al, 201914 and Devlin et al.46 Briefly, the reduction in fluorescent intensity from MSNs exposed to biofilm coated pegs compared to that of a control was used to calculate the entrapment of MSNs. Assays were carried out in triplicate, with a minimum of 12 individual replicates per run.
MSN Binding Assay
To evaluate the adhesion of the MSNs to the bacteria, planktonic cells of MSSA and MRSA were incubated with nanoparticles (1 mg mL−1) for 1 h at 37°C, before being washed 5 times with water to remove unbound MSNs. The cells were then fixed with 5% glutaraldehyde in PBS (pH 7.4) for 30 min, washed twice with PBS, incubated with 1% osmium tetroxide prepared in PBS for 1 h, washed twice with PBS and dehydrated through the ethanol and hexamethyldisilazane (HMDS) sequence (ethanol: 30%, 50%, 75%, 95%, 100% and HMDS: ethanol 1:2, 2:1 and 100% HMDS). The samples were dried in the fume-hood overnight, deposited on carbon conductive adhesive tape and analyzed by scanning electron microscopy (SEM, Quanta 3D FEG Dual Beam, FEI).
Exposure of S. aureus Biofilms to Loaded MSNs
Before examining the activity of the vancomycin loaded MSNs against biofilm samples, the minimum inhibitory concentration (MIC) and minimum biofilm inhibitory concentration (MBIC) were identified for planktonic bacteria. Overnight cultures of MRSA and MSSA were exposed to free vancomycin (0.5–248 µg mL−1) and to the vancomycin loaded MSNs. The bacteria were incubated for 24 h at 37 °C under static conditions. Bacterial growth was assessed with absorbance readings at 600 nm to identify the MIC of the free vancomycin and the loaded MSNs against MRSA and MSSA cells. Following MIC identification, the spent culture media was removed, and crystal violet staining was used to identify the MBIC. To assess the antibiofilm effects of the loaded MSNs, biofilms were cultured for 24 h at 37 °C under static conditions. Following formation, biofilms were washed three times with MilliQ water to remove planktonic cells, before being treated for 24 h with vancomycin-loaded MSNs (0.25 and 1 mg mL−1), or free vancomycin (control). Following exposure, the nanoparticles solution was removed, and the biofilms were washed three times with MilliQ water to remove residual MSNs. An MTT assay was utilized to assess the bacterial cell viability following MSN exposure. Briefly, following the removal of planktonic cells and unbound MSNs, 150 µL of PBS and 50 µL of the MTT solution was added to each well and the plate was incubated for 2 h at 37 °C with shaking at 100 rpm. Following incubation, the MTT solution was removed, and 200 µL of dimethyl sulfoxide was added to solubilize the MTT solution, which was metabolized by live cells into an insoluble purple compound (formazan). The plate was incubated for 15 min at 37 °C with shaking at 100 rpm before absorbance readings were taken at 550 nm, using a plate reader (SpectraMax iD3, Molecular devices) to determine cell viability within the biofilm samples.47,48 Experiments were carried out in triplicate, with a minimum of 20 independent replicates per run.
Statistical Analysis
Statistical significance was determined using a Student’s t-test (two-tailed) for all experiments. A value of P < 0.05 was considered significant. Error bars were identified using standard error of the mean. All statistical analyses were performed on Excel (Microsoft Office).
Results and Discussion
Preparation and Characterization of Engineered Mesoporous Silica Nanoparticles (MSNs)
Different surface engineered mesoporous silica nanoparticles (MSNs) were prepared to investigate the role of MSNs surface composition in their diffusion and penetration into the biofilm matrix and cell targeting capability. Four families of nanoparticles were prepared: as-synthesized bare particles MSN-B, amino-functionalized and positively charged MSN-D, carboxy-functionalized and negatively charged MSN-C, benzene-functionalized (aromatic) and positively charged MSN-A. Representative TEM images of the synthesized nanoparticles (MSN-B, MSN-D, MSN-C and MSN-A) are represented in Figure 1, whereas the measured size, hydrodynamic diameter and surface charge are reported in Table 1.
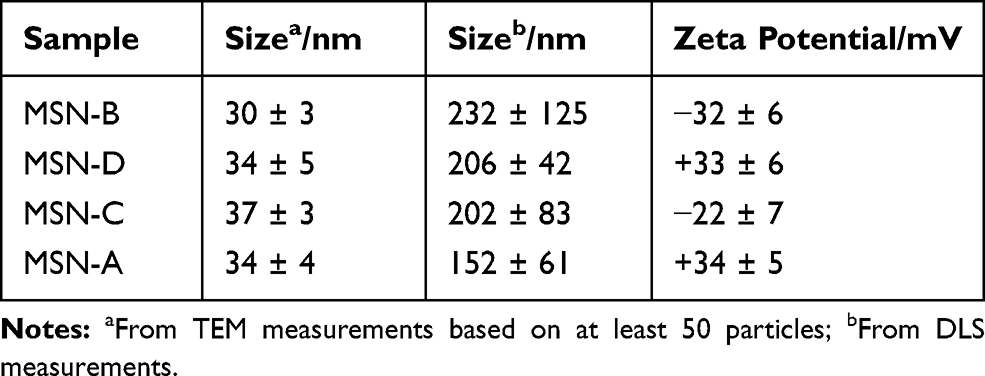 |
Table 1 Physicochemical Characterization of Functionalized MSNs (Bare (B), Amine (D), Carboxyl (C) and Aromatic (A)) |
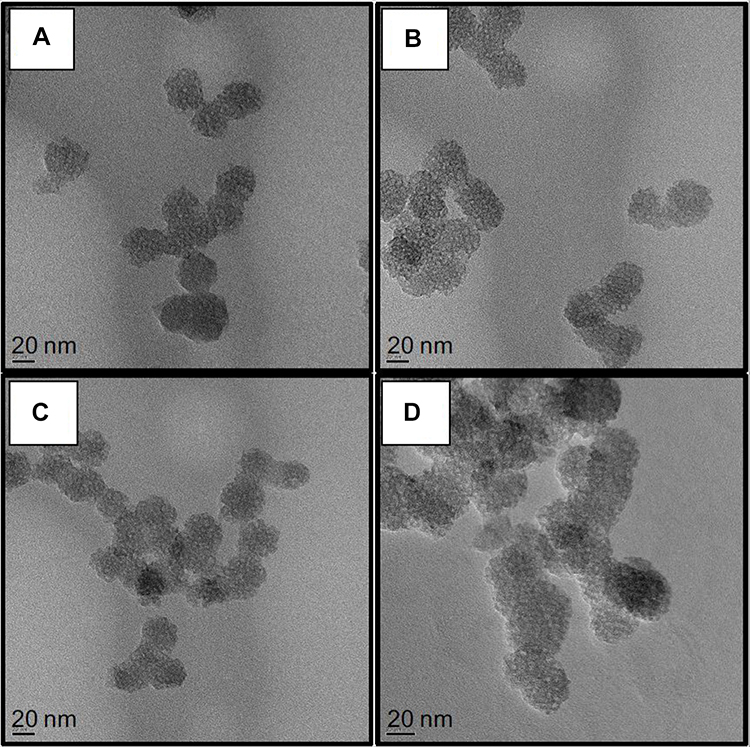 |
Figure 1 Representative TEM images of A) bare (MSN-B), B) amine (MSN-D), C) carboxyl (MSN-C) and D) aromatic (MSN-A) functionalized MSNs. Bars represent 20 nm. |
The obtained nanoparticles are spherical with a clear porous structure and a diameter of approximately 30 nm. As expected, the surface functionalization did not significantly impact the nanoparticle size. The hydrodynamic size of the nanoparticles obtained by DLS, as expected, shows a significant overestimation of their size when compared to TEM measurements, owing mainly to the influence of surface coating and hydration layer on the diffusion of the nanoparticles.49,50
In the FT-IR spectra (Figure S2) it is possible to observe the main bands related to the Si-O symmetrical (812 cm−1) and asymmetrical (1092 cm−1) stretching.51 With the addition of amine-containing silane (red) new bands arise, such as C-N stretching (1232 cm−1) and N-H bending (1403 cm−1) of primary amines, N-H stretching (3671 cm−1) of secondary amines or amide and alkane stretching bands (2979, 2902 cm−1).51,52 After the carboxyl functionalization (blue) we can observe the rise of carbonyl stretching mode (1698 cm−1). For the aromatic nanoparticles (green), the C=C stretching band at 1638 cm−153 can be observed.
To further confirm the surface functionalization dissolution 1H NMR was carried out (Figure 2). Instead of using a suspension of the MSNs, they were dissolved with NaOD, leaving in solution only the surface groups previously grafted onto the nanoparticle surface. The signals related to DETA functionalization (red) could be assigned to the protons in the molecule. These peaks are still present after the carboxyl functionalization (blue) followed by the emergence of α-carbonyl protons. With the addition of the aromatic group (green), new signals for the protons in the aromatic ring were observed.
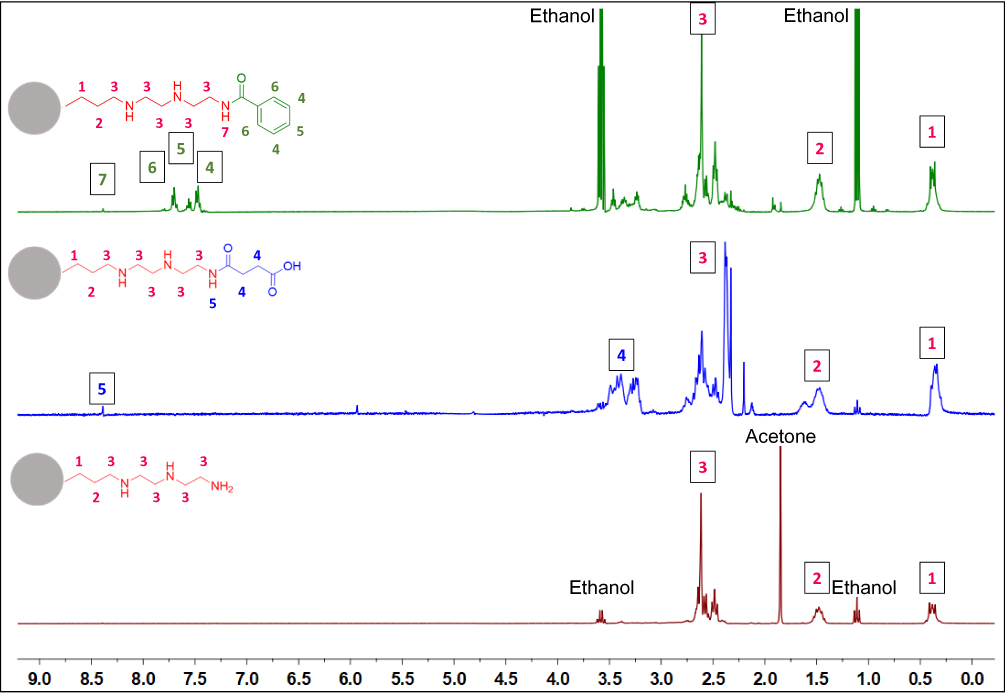 |
Figure 2 1H NMR spectra of functionalized MSNs aromatic (MSN-A)-green, carboxyl (MSN-C)-blue, and amine (MSN-D)-red. |
MSN Interaction with S. aureus Biofilms
The four families of nanoparticles were used to systematically study the effect of MSN surface chemistry on their interaction with S. aureus biofilms, especially in terms of entrapment in the biofilm, which was evaluated as a function of MSN charge and surface composition.
It has been previously reported that specific nanoparticle surface functionalization has a significant effect on interactions with bacterial biofilms.54 An important consideration when investigating NP-biofilm interactions is the role of the biofilm EPS matrix.33 EPS composition and charge can influence the interactions which occur when NPs come into contact with a biofilm. Here we used two S. aureus strains, one MRSA and one MSSA, which form biofilms with protein/eDNA or polysaccharide matrices, respectively (Table S1), in order to investigate the role which these components have on nanoparticle entrapment (Figure 3).
 |
Figure 3 Biofilm entrapment by MRSA (Black) and MSSA (Grey) of bare (MSN-B), amine (MSN-D), carboxyl (MSN-C) and aromatic (MSN-A) nanoparticles after 180 min exposure in water. Error bars represent standard error of the mean (n = 3). |
The entrapment results show that both the overall charge and specific surface functionalization have a crucial role in determining the interaction between nanoparticles and bacterial biofilms. Regardless of the biofilm matrix, the MSN-D shows the highest entrapment. Given that many of the components which make up the biofilm matrix (proteins, polysaccharides and eDNA) along with the bacterial cell envelope have an overall negative charge (Table S1), strong electrostatic attractive forces with the amine group on the NP surface are expected and explain the higher entrapment of MSN-D. The entrapment was seen to be lower for MSN-A despite having a similar overall positive charge. This could be due to the aromatic ring, which confers a more hydrophobic behavior compared to the free amine group; therefore, weaker interactions are expected to occur, leading to a lower entrapment.
Significant differences between the entrapment of negatively charged MSNs in MRSA and MSSA biofilms can be observed. It is clear from Figure 3 that a significantly higher number of negatively charged MSNs (MSN-B and MSN-C) were entrapped in MSSA biofilms when compared to MRSA biofilms. More information on the cause of this higher entrapment can be found when looking at the different components which make up the matrix of each biofilm. MSSA biofilms are made up primarily of polysaccharides, in this specific case PNAG.55 PNAG has an overall net positive charge,56 and so electrostatic attractive forces are expected to occur between the OH and COOH groups on the surface of the MSN-B and MSN-C particles. These interactions are hypothesized to be influencing the differences in nanoparticle entrapment between MRSA and MSSA biofilms and highlights the complicated situation in predicting the interaction between nanoparticles and biofilms. It indicates that not only are there a range of nanoparticle characteristics (size, charge, surface functionalization, etc.) which can affect these interactions, but also the components of the EPS matrix need to be considered.
Another important aspect is the fact that bacterial biofilms are known to hinder the penetration of antimicrobial agents via different mechanisms, such as electrostatic interactions and steric hindrance.57 The administration of penetration enhancers can be used to modify the interactions between biofilm and nanomaterial.39 Given that the bacterial cell surface and EPS components are negatively charged (Table S1), cationic penetration enhancers could potentially shield the biofilm charge and as a consequence, increase nanoparticles penetration. Furthermore, the addition of an electrolyte would decrease the Debye length of the EPS functional groups,58 thus increasing the matrix effective pore size.59 Previous studies suggested the electrolyte screening interactions are relevant for nanoparticles larger than 10 nm.57,60 For the reasons mentioned above, the entrapment of different surface-functionalized nanoparticles was investigated in the presence of a negatively charged PBS buffer and a positively charged Tris buffer (Figure 4).
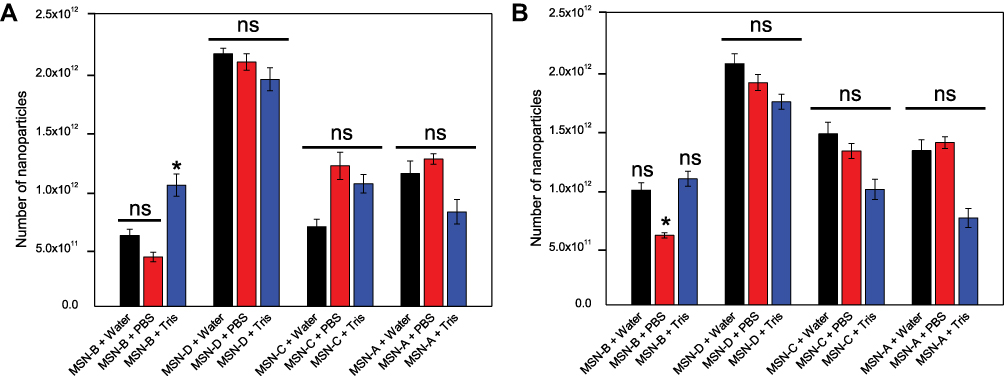 |
Figure 4 MSNs entrapment in S. aureus biofilms A) MRSA and B) MSSA in the presence of water (Black), PBS (Red) and Tris (Blue) after 180 min exposure. Error bars represent standard error of the mean (n = 3). *represents a significant difference in MSN entrapment between test conditions, p < 0.05; ns represents no significant difference in MSN entrapment between test conditions, p > 0.05. |
It can be noted that in the case of MSNs, the administration of PBS (negatively charged) or Tris (positively charged) buffers did not significantly alter the entrapment of nanoparticles in MRSA and MSSA biofilms. It is worth mentioning that in the case of MSN-B, the use of Tris buffer significantly increased the capture of nanoparticles in MRSA biofilms and increased slightly for MSSA biofilms, while the addition of PBS is seen to reduce the entrapment of MSN-B in MSSA biofilms. The larger entrapment of MSN-B in MRSA biofilm samples exposed to the positively charged buffer compared to the control could be a result of the composition of the EPS matrix. As the matrix of MRSA biofilms is primarily composed of negatively charged proteins, with the addition of a cationic penetration enhancer (Tris), the overall negative charge could be shielded, allowing for a higher entrapment. Such an increase in entrapment is not observed in the MSSA biofilm samples as the matrix is primarily composed of PNAG, a polysaccharide with an overall positive charge and so the effects of the buffer are not as significant while by adding an anionic penetration enhancer (PBS) hinders the entrapment of MSN-B. For the MSN-A, positively charged and with a more hydrophobic surface, the administration of PBS buffer led to a slight increase in their penetration in both biofilms.
The myriad of factors influencing nanoparticle-biofilm interactions contributes to the difficulty in finding novel treatment strategies to overcome the rising trend of antimicrobial resistance. For novel treatments involving NPs as carriers of antimicrobial agents to be effective against biofilm forming bacteria, it is vital to ensure that the NPs are localized close to the bacterial cells to deliver a high local concentration of the antimicrobial agent. As such, methods need to be identified, which will lead to an increased local concentration of the antimicrobial agent not only within the EPS matrix but also around the bacterial cells. Tailoring the surface functionalization of the nanoparticle carriers could lead to promising results. To investigate if the MSNs used would interact and bind to the cell wall of the bacteria, binding assays were carried out with planktonic S. aureus cells and MSNs. Planktonic cells were incubated with MSNs for one hour, following which the unbound MSNs were removed through washing steps, and the cells were imaged using SEM (Figure 5).
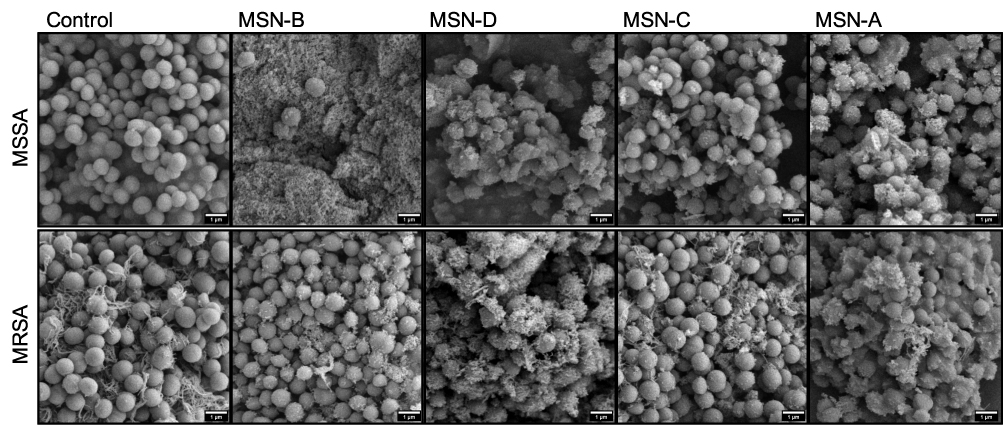 |
Figure 5 Representative SEM images of S. aureus planktonic cells incubated with MSNs. |
It was found that positively charged MSNs (MSN-D and MSN-A) primarily localized around the bacterial cells of both S. aureus strains. This localization is believed to be a result of strong electrostatic attraction forces between the negatively charged peptidoglycan in the cell wall61 and the positively charged functional groups on the MSNs surface. On the other hand, negatively charged MSNs (MSN-B and MSN-C) seems to interact more with the EPS surrounding the cells, than attaching to the bacterial cells, particularly in the case for MSSA.
Increased interaction with the cell wall can, in principle, allow for the delivery of a higher local concentration of an antimicrobial agent directly to the bacterial cells. To test this hypothesis, vancomycin was loaded onto the MSNs, to assess if enhanced bacterial cell-binding could increase the antibacterial effect of the antimicrobial agent. The loading (Table 2) showed that twice the concentration of vancomycin was loaded onto negatively charged MSNs (MSN-B and MSN-C). Planktonic bacteria were cultured in the presence of the loaded MSNs and free vancomycin to identify the minimum inhibitory concentration (MIC) and the minimum biofilm inhibitory concentration (MBIC). The loading of vancomycin did not significantly affect the MIC or the MBIC compared to free vancomycin (Table S2). As the planktonic cells do not have the protection of the EPS matrix against antimicrobial agents the free vancomycin can easily access the bacterial cell, and so the specific binding of MSNs to the bacterial cell does not produce a significant decrease in the MIC or MBIC. To observe the effects of vancomycin loading against biofilms, samples were treated with the loaded MSNs for 24 hours, which was followed by cell viability (MTT assay of metabolic activity) as shown in Figures 6 and 7.
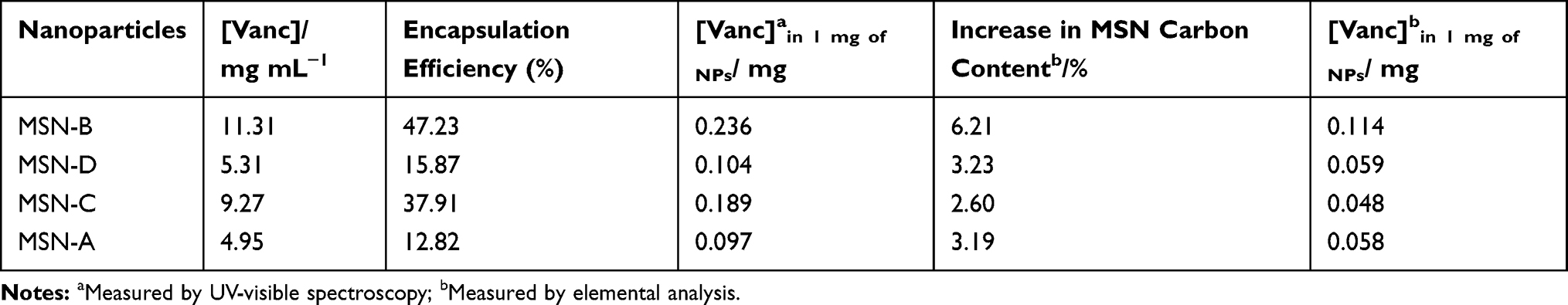 |
Table 2 Vancomycin Loading on the Different MSNs |
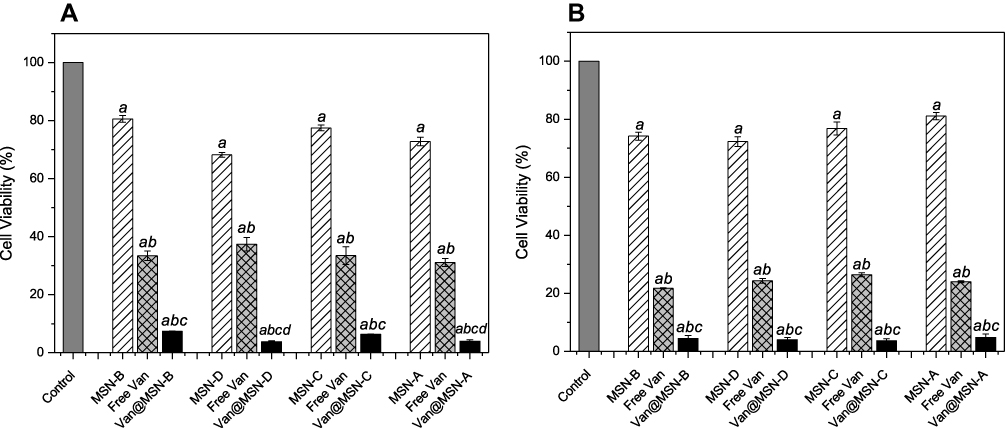 |
Figure 6 Viability of A) MRSA and B) MSSA biofilm cells after exposure to unloaded MSNs (1 mg mL-1), vancomycin-loaded MSNs (1 mg mL-1), free vancomycin or water for 24 h, as determined by MTT assay. Error bars represent standard error of the mean (n = 3). Letters (a, b, c, d) represent a significant difference in antibacterial activity between test conditions, p < 0.05. |
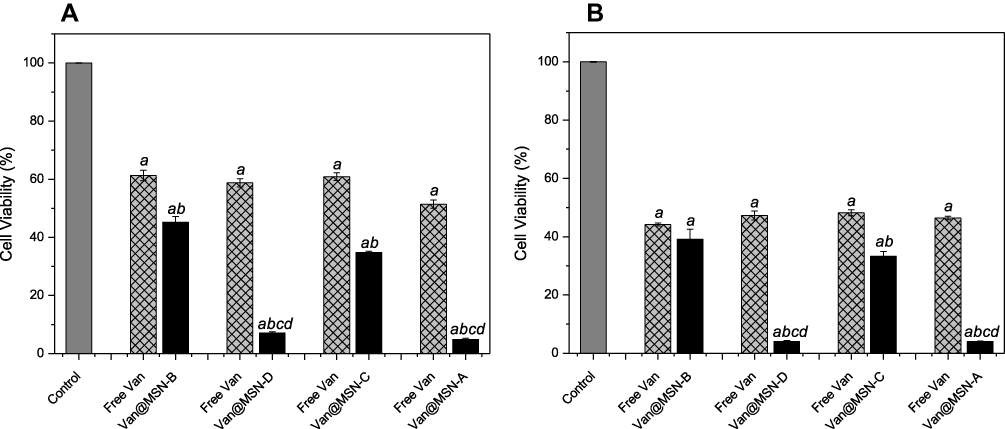 |
Figure 7 Viability of A) MRSA and B) MSSA biofilm cells after exposure to vancomycin-loaded MSNs (0.25 mg mL-1), free vancomycin or water for 24 h as determined by MTT assay. Error bars represent standard error of the mean (n = 3). Letters (a, b, c, d) represent a significant difference in antibacterial activity between test conditions, p < 0.05. |
When 1 mg mL−1 of nanoparticles solution was used in the biofilm treatment, all vancomycin loaded MSNs had similar effects on MRSA and MSSA biofilm cell viability, although positively charged MSNs had a lower overall antimicrobial loading. The increased activity of positively charged MSNs is attributed to the higher binding of the MSNs to the cell wall of the bacteria (Figure 5) increasing the delivery directly to the cells. Furthermore, it was identified that the loaded MSNs had a much more significant effect on cell viability as compared to free vancomycin administered at the same concentration. For the MRSA, a 3-6-fold decrease in cell viability was observed when vancomycin was loaded inside MSNs, compared to the same concentration of free vancomycin. In the case of MSSA, a reduction of 10–42% in the cell viability was observed when loaded nanoparticles were used. This is a result of MSSA bacteria being more susceptible to vancomycin compared to MRSA (Table S2), and therefore treatment with free vancomycin produces a more significant reduction in cell viability in MSSA biofilm samples as compared with MRSA biofilms. The loading of the antimicrobial agent inside the structure of the nanoparticles offers protection to the drug from interactions in the EPS matrix which can inhibit its penetration, while also increasing its concentration into the deepest regions of the biofilm.62
To assess if the preferential binding of the positively charged MSNs to the bacterial cell membrane would improve treatment of S. aureus infections, a lower nanoparticle concentration solution was used (0.25 mg mL−1). The positively charged MSNs significantly outperformed the negatively charged MSNs with a 5-8-fold decrease in viable cells despite having a lower antimicrobial loading (P < 0.005), indicating that specific binding to the bacterial cell membrane is critical in improving the delivery of antimicrobial agents into biofilms. When compared to free vancomycin at the same concentration, it is clear both positively and negatively charged MSNs show considerably higher bacterial cell killing activity (Figure 7).
Another critical factor to take into account for future research when considering the treatment of biofilm infections is the dispersal of the EPS matrix, which represents an ideal environment for future bacterial colonization63 and so must be removed to ensure total elimination of the infection. Coupling EPS targeting agents with antibiotic-loaded MSNs could lead to promising new techniques to target biofilm infections. The results presented show that by tailoring the nanoparticle surface chemistry, lower concentrations of antimicrobial agents can be used to target bacterial biofilms helping the development of novel treatment strategies.
Conclusions
Infections caused by the formation of antimicrobial tolerant biofilms constitute a significant healthcare issue. New approaches are needed to develop in order to overcome this problem. Enhancing antimicrobial efficacy through encapsulation in nanomaterials is a promising area of research. However, the complex interactions which govern nanoparticle-biofilm phenomena are not yet fully understood. This systematic study has investigated how NP characteristics and biofilm matrix composition impact entrapment and binding to bacterial cells in order to enhance dispersal and eradication of biofilms. Positively charged MSNs had a higher entrapment in both protein/eDNA and polysaccharide S. aureus biofilm matrices and were shown to be localized near the bacterial cell surface. Although, negatively charged MSNs were more efficiently loaded with vancomycin, positively charged vancomycin-loaded MSNs more efficiently localized to the surface of biofilm cells and were more active in reducing biofilm cell viability. These results demonstrate that tailoring nanoparticle surface chemistry offers the potential to improve the treatment options for chronic infections involving bacterial biofilms. These technologies can increase the delivery of antimicrobial agents directly to the bacterial cells within a biofilm while also protecting the loaded antimicrobial from deactivation in the biofilm matrix. Future studies will need to investigate how nanocarriers are affected by physiological fluids, and a deeper understand regarding the implication of ECM proteins and how they would interact with different nanoparticles is essential. Furthermore, how the improved dispersal of the biofilm matrix could enhance the treatment of biofilm infections needs further investigation. Combining NP-encapsulated therapeutic agents to target both the bacterial cells and remove the protein/polysaccharides within the biofilm matrix may represent an alternative strategy for the treatment of S. aureus biofilm-associated infections.
Acknowledgments
We thank Science Foundation Ireland (SFI) for the support of this research under grant number 15/IA/3008. We also would like to thank Prof. Dr Kenneth Dawson from the Centre for BioNano Interactions (CBNI) UCD for the Zetasizer equipment.
Author Contributions
Henry Devlin and Stephanie Fulaz contributed equally to this work. All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Golkar Z, Bagasra O, Pace DG. Bacteriophage therapy: a potential solution for the antibiotic resistance crisis. J Infect Dev Ctries. 2014;8(2):129–136. doi:10.3855/jidc.3573
2. World Health Organization. Antibiotic resistance. Available from: https://www.who.int/en/news-room/fact-sheets/detail/antibiotic-resistance.
3. World Health Organization. New report calls for urgent action to avert antimicrobial resistance crisis. Published 2019. Available from: https://www.who.int/news-room/detail/29-04-2019-new-report-calls-for-urgent-action-to-avert-antimicrobial-resistance-crisis.
4. Costerton JW, Stewart PS, Greenberg EP. Bacterial biofilms: a common cause of persistent infections. Science. 1999;284(5418):1318–1322. doi:10.1126/science.284.5418.1318
5. de la Fuente-nunez C, Reffuveille F, Fernandez L, Hancock REW. Bacterial biofilm development as a multicellular adaptation: antibiotic resistance and new therapeutic strategies. Curr Opin Microbiol. 2013;16(5):580–589. doi:10.1016/j.mib.2013.06.013
6. Davies D. Understanding biofilm resistance to antibacterial agents. Nat Rev Drug Discov. 2003;2(2):114–122. doi:10.1038/nrd1008
7. Hall-Stoodley L, Stoodley P, Kathju S, et al. Towards diagnostic guidelines for biofilm-associated infections. FEMS Immunol Med Microbiol. 2012;65(2):127–145. doi:10.1111/j.1574-695X.2012.00968.x
8. Fux CA, Costerton JW, Stewart PS, Stoodley P. Survival strategies of infectious biofilms. Trends Microbiol. 2005;13(1):34–40. doi:10.1016/j.tim.2004.11.010
9. Leid JG. Bacterial biofilms resist key host defenses. Microbe. 2009;4:66–70.
10. Mah TF. Biofilm-specific antibiotic resistance. Future Microbiol. 2012;7(9):1061–1072. doi:10.2217/fmb.12.76
11. Flemming HC, Wingender J, Szewzyk U, Steinberg P, Rice SA, Kjelleberg S. Biofilms: an emergent form of bacterial life. Nat Rev Microbiol. 2016;14(9):563–575. doi:10.1038/nrmicro.2016.94
12. Flemming HC, Wingender J. The biofilm matrix. Nat Rev Microbiol. 2010;8(9):623–633. doi:10.1038/nrmicro2415
13. Stewart PS. Mechanisms of antibiotic resistance in bacterial biofilms. Int J Med Microbiol. 2002;292(2):107–113. doi:10.1078/1438-4221-00196
14. Fulaz S, Hiebner D, Barros CHN, et al. Ratiometric imaging of the in situ pH distribution of biofilms by use of fluorescent mesoporous silica nanosensors. ACS Appl Mater Interfaces. 2019;11(36):32679–32688. doi:10.1021/acsami.9b09978
15. Singh S, Singh SK, Chowdhury I, Singh R. Understanding the mechanism of bacterial biofilms resistance to antimicrobial agents. Open Microbiol J. 2017;11:53–62. doi:10.2174/1874285801711010053
16. Rossolini GM, Arena F, Pecile P, Pollini S. Update on the antibiotic resistance crisis. Curr Opin Pharmacol. 2014;18:56–60. doi:10.1016/j.coph.2014.09.006
17. Center for Disease Control and Prevention. Biggest threats and data 2019 AR threats report. Published 2019. Available from: https://www.cdc.gov/drugresistance/biggest-threats.html?CDC_AA_refVal=https%3A%2F%2Fwww.cdc.gov%2Fdrugresistance%2Fbiggest_threats.html.
18. Ventola CL. The antibiotic resistance crisis: part 1: causes and threats. PT. 2015;40(4):277–283.
19. Gross M. Antibiotics in crisis. Curr Biol. 2013;23(24):R1063–R1065. doi:10.1016/j.cub.2013.11.057
20. McCarthy H, Rudkin JK, Black NS, Gallagher L, O’Neill E, O’Gara JP. Methicillin resistance and the biofilm phenotype in Staphylococcus aureus. Front Cell Infect Microbiol. 2015;5:1. doi:10.3389/fcimb.2015.00001
21. Joo HS, Otto M. Molecular basis of in vivo biofilm formation by bacterial pathogens. Chem Biol. 2012;19(12):1503–1513. doi:10.1016/j.chembiol.2012.10.022
22. O’Gara JP. ica and beyond: biofilm mechanisms and regulation in Staphylococcus epidermidis and Staphylococcus aureus. FEMS Microbiol Lett. 2007;270(2):179–188. doi:10.1111/j.1574-6968.2007.00688.x
23. Hibbitts A, O’Leary C. Emerging nanomedicine therapies to counter the rise of methicillin-resistant Staphylococcus aureus. Materials. 2018;11(2):321. doi:10.3390/ma11020321
24. Tamimi NAM, Ellis P. Drug development: from concept to marketing! Nephron Clin Pract. 2009;113(3):C125–C131. doi:10.1159/000232592
25. Chong CR, Sullivan DJ. New uses for old drugs. Nature. 2007;448(7154):645–646. doi:10.1038/448645a
26. Qayyum S, Khan AU. Nanoparticles vs. biofilms: a battle against another paradigm of antibiotic resistance. Medchemcomm. 2016;7(8):1479–1498. doi:10.1039/C6MD00124F
27. Gupta A, Landis RF, Rotello VM. Nanoparticle-based antimicrobials: surface functionality is critical. F1000Res. 2016;5:364. doi:10.12688/f1000research.7595.1
28. Xu C, Yu MH, Noonan O, et al. Core-cone structured monodispersed mesoporous silica nanoparticles with ultra-large cavity for protein delivery. Small. 2015;11(44):5949–5955. doi:10.1002/smll.201501449
29. Xu C, Niu YT, Popat A, Jambhrunkar S, Karmakar S, Yu CZ. Rod-like mesoporous silica nanoparticles with rough surfaces for enhanced cellular delivery. J Mater Chem B. 2014;2(3):253–256. doi:10.1039/C3TB21431A
30. Chen Y, Chen HR, Shi JL. In vivo bio-safety evaluations and diagnostic/therapeutic applications of chemically designed mesoporous silica nanoparticles. Adv Mater. 2013;25(23):3144–3176. doi:10.1002/adma.201205292
31. Rasamiravaka T, El Jaziri M. Quorum-sensing mechanisms and bacterial response to antibiotics in P. aeruginosa. Curr Microbiol. 2016;73(5):747–753. doi:10.1007/s00284-016-1101-1
32. Han CD, Romero N, Fischer S, Dookran J, Berger A, Doiron AL. Recent developments in the use of nanoparticles for treatment of biofilms. Nanotechnol Rev. 2017;6(5):383–404. doi:10.1515/ntrev-2016-0054
33. Xu C, He Y, Li ZH, Nor YA, Ye QS. Nanoengineered hollow mesoporous silica nanoparticles for the delivery of antimicrobial proteins into biofilms. J Mater Chem B. 2018;6(13):1899–1902. doi:10.1039/C7TB03201C
34. Tan YL, Ma S, Liu CG, Yu WG, Han F. Enhancing the stability and antibiofilm activity of DspB by immobilization on carboxymethyl chitosan nanoparticles. Microbiol Res. 2015;178:35–41. doi:10.1016/j.micres.2015.06.001
35. Baelo A, Levato R, Julian E, et al. Disassembling bacterial extracellular matrix with DNase-coated nanoparticles to enhance antibiotic delivery in biofilm infections. J Control Release. 2015;209:150–158. doi:10.1016/j.jconrel.2015.04.028
36. Mu HB, Tang JJ, Liu QJ, Sun CL, Wang TT, Duan JY. Potent antibacterial nanoparticles against biofilm and intracellular bacteria. Sci Rep. 2016;6:18877. doi:10.1038/srep18877
37. Ramasamy M, Lee JH, Lee J. Direct one-pot synthesis of cinnamaldehyde immobilized on gold nanoparticles and their antibiofilm properties. Colloid Surf B. 2017;160:639–648. doi:10.1016/j.colsurfb.2017.10.018
38. Fulaz S, Vitale S, Quinn L, Casey E. Nanoparticle-biofilm interactions: the role of the EPS matrix. Trends Microbiol. 2019;27(11):915–926. doi:10.1016/j.tim.2019.07.004
39. Harper RA, Carpenter GH, Proctor GB, et al. Diminishing biofilm resistance to antimicrobial nanomaterials through electrolyte screening of electrostatic interactions. Colloid Surf B. 2019;173:392–399. doi:10.1016/j.colsurfb.2018.09.018
40. Gounani Z, Asadollahi MA, Meyer RL, Arpanaei A. Loading of polymyxin B onto anionic mesoporous silica nanoparticles retains antibacterial activity and enhances biocompatibility. Int J Pharm. 2018;537(1–2):148–161. doi:10.1016/j.ijpharm.2017.12.039
41. Schmid B, Schindelin J, Cardona A, Longair M, Heisenberg M. A high-level 3D visualization API for Java and ImageJ. BMC Bioinformatics. 2010;11:274. doi:10.1186/1471-2105-11-274
42. Hristov DR, Rocks L, Kelly PM, et al. Tuning of nanoparticle biological functionality through controlled surface chemistry and characterisation at the bioconjugated nanoparticle surface. Sci Rep. 2015;5:17040. doi:10.1038/srep17040
43. Zhou XJ, Weng WZ, Chen B, et al. Mesoporous silica nanoparticles/gelatin porous composite scaffolds with localized and sustained release of vancomycin for treatment of infected bone defects. J Mater Chem B. 2018;6(5):740–752. doi:10.1039/C7TB01246B
44. O’Neill E, Pozzi C, Houston P, et al. A novel Staphylococcus aureus biofilm phenotype mediated by the fibronectin-binding proteins, FnBPA and FnBPB. J Bacteriol. 2008;190(11):3835–3850. doi:10.1128/JB.00167-08
45. O’Neill E, Pozzi C, Houston P, et al. Association between methicillin susceptibility and biofilm regulation in Staphylococcus aureus isolates from device-related infections. J Clin Microbiol. 2007;45(5):1379–1388. doi:10.1128/JCM.02280-06
46. Devlin H, Hiebner D, Barros C, et al. A high throughput method to investigate nanoparticle entrapment efficiencies in biofilms. Colloids and surfaces. B, Biointerfaces. 2020;193:111123. doi: 10.1016/j.colsurfb.2020.111123
47. Wu YT, Zhu H, Willcox M, Stapleton F. Removal of biofilm from contact lens storage cases. Invest Ophthalmol Vis Sci. 2010;51(12):6329–6333. doi:10.1167/iovs.10-5796
48. Walencka E, Rozalska S, Sadowska B, Rozalska B. The influence of Lactobacillus acidophilus-derived surfactants on staphylococcal adhesion and biofilm formation. Folia Microbiol. 2008;53(1):61–66. doi:10.1007/s12223-008-0009-y
49. Kaasalainen M, Aseyev V, von Haartman E, et al. Size, stability, and porosity of mesoporous nanoparticles characterized with light scattering. Nanoscale Res Lett. 2017;12(1):74. doi:10.1186/s11671-017-1853-y
50. Sere S, De Roo B, Vervaele M, et al. Altering the biodegradation of mesoporous silica nanoparticles by means of experimental parameters and surface functionalization. J Nanomater. 2018;2018:7390618.
51. Ojea-Jimenez I, Urban P, Barahona F, et al. Highly flexible platform for tuning surface properties of silica nanoparticles and monitoring their biological interaction. ACS Appl Mater Interfaces. 2016;8(7):4838–4850. doi:10.1021/acsami.5b11216
52. Mittal N, Samanta A, Sarkar P, Gupta R. Postcombustion CO2 capture using N-(3-trimethoxysilylpropyl)diethylenetriamine-grafted solid adsorbent. Energy Sci Eng. 2015;3(3):207–220. doi:10.1002/ese3.64
53. Foschiera JL, Pizzolato TM, Benvenutti EV. FTIR thermal analysis on organofunctionalized silica gel. J Braz Chem Soc. 2001;12(2):159–164. doi:10.1590/S0103-50532001000200006
54. Li XN, Yeh YC, Giri K, et al. Control of nanoparticle penetration into biofilms through surface design. Chem Commun. 2015;51(2):282–285. doi:10.1039/C4CC07737G
55. Zapotoczna M, O’Neill E, O’Gara JP. Untangling the diverse and redundant mechanisms of Staphylococcus aureus biofilm formation. PLoS Pathog. 2016;12(7):6. doi:10.1371/journal.ppat.1005671
56. Arciola CR, Campoccia D, Ravaioli S, Montanaro L. Polysaccharide intercellular adhesin in biofilm: structural and regulatory aspects. Front Cell Infect Microbiol. 2015;5:7. doi:10.3389/fcimb.2015.00007
57. Besinis A, De Peralta T, Tredwin CJ, Handy RD. Review of nanomaterials in dentistry: interactions with the oral microenvironment, clinical applications, hazards, and benefits. ACS Nano. 2015;9(3):2255–2289. doi:10.1021/nn505015e
58. Harper RA, Saleh MM, Carpenter G, et al. Soft, adhesive (+) alpha tocopherol phosphate planar bilayers that control oral biofilm growth through a substantive antimicrobial effect. Nanomedicine. 2018;14(7):2307–2316. doi:10.1016/j.nano.2017.12.024
59. Zhang ZS, Nadezhina E, Wilkinson KJ. Quantifying diffusion in a biofilm of streptococcus mutans. Antimicrob Agents Chemother. 2011;55(3):1075–1081. doi:10.1128/AAC.01329-10
60. Golmohamadi M, Clark RJ, Veinot JGC, Wilkinson KJ. The role of charge on the diffusion of solutes and nanoparticles (silicon nanocrystals, nTiO2, nAu) in a biofilm. Environ Chem. 2013;10(1):34–41. doi:10.1071/EN12106
61. Monteiro JM, Covas G, Rausch D, et al. The pentaglycine bridges of Staphylococcus aureus peptidoglycan are essential for cell integrity. Sci Rep. 2019;9:5010. doi:10.1038/s41598-019-41461-1
62. Ramos M, Da Silva PB, Sposito L, et al. Nanotechnology-based drug delivery systems for control of microbial biofilms: a review. Int J Nanomedicine. 2018;13:1179–1213. doi:10.2147/IJN.S146195
63. Fleming D, Rumbaugh K. The consequences of biofilm dispersal on the host. Sci Rep. 2018;8:10738. doi:10.1038/s41598-018-29121-2
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.