Back to Journals » OncoTargets and Therapy » Volume 13
TAGLN and High-mobility Group AT-Hook 2 (HMGA2) Complex Regulates TGF-β-induced Colorectal Cancer Metastasis
Authors Zhou H, Li L, Xie W, Wu L, Lin Y, He X
Received 7 July 2020
Accepted for publication 12 September 2020
Published 15 October 2020 Volume 2020:13 Pages 10489—10498
DOI https://doi.org/10.2147/OTT.S263090
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Federico Perche
Huimin Zhou,1 Lan Li,1 Wenrui Xie,1 Lihao Wu,1 Ying Lin,2 Xingxiang He1
1Department of Gastroenterology and Hepatology, The First Affiliated Hospital, School of Clinical Medicine of Guangdong Pharmaceutical University, Guangzhou, People’s Republic of China; 2Department of Gastroenterology and Hepatology, The Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, Guangzhou, People’s Republic of China
Correspondence: Ying Lin; Xingxiang He Email [email protected]; [email protected]
Background: Colorectal cancer is one of the three most common cancers worldwide. Altered TGF-β signaling pathway in colorectal cancer is associated with metastasis and poor prognosis. It is also involved in epithelial-to-mesenchymal transition (EMT), which is essential in progression and metastasis. This study aims to investigate the role of transgelin (TAGLN) and high-mobility group AT-hook 2 (HMGA2) in the progression of colon cancer.
Methods: HT29 and HCT116 cells were treated with TGF-β, and the effects of inhibition of TAGLN and overexpression of HMGA2 on TGF-β treated cell on cell migration and invasion, expression of EMT markers, including E-cadherin, vimentin and fibronectin were detected as well as MMP2 and MMP9, which are critical in cancer cell metastasis. The interaction of TAGLN and HMGA2 was also investigated by using co-immunoprecipitation. The function of TAGLN in tumor metastasis and growth was investigated in vivo.
Results: We found that TGF-β could significantly promote the migration of HT29 and HCT116 cells, as well as TAGLN protein expression and nucleus translocation, while inhibition of TAGLN could effectively reverse the effects of TGF-β on HT29 and HCT116 cells, which was observed in terms of decreased cell migration and invasion. Knockdown of TAGLN could also rescue TGF-β-induced loss of E-cadherin, and decreased TGF-β-induced vimentin and fibronectin expression; the elevation of MMP9 and MMP2 was also reversed by inhibition of TAGLN. Further investigation confirmed the interaction of HMGA2 and TAGLN, as overexpression of HMGA2 restores the effects of TGF-β on HT29 cells, which were attenuated by TAGLN inhibition both in vitro and in vivo.
Conclusion: Overall, our study revealed that interaction between TAGLN and HMGA2 was involved in TGF-β-induced cell migration and promotion of colon cancer cells, suggesting that HMGA2 and TAGLN are potential molecular targets to prevent colon cancer progression.
Keywords: HMGA2, colorectal cancer, TGF-β, TAGLN
Introduction
Colorectal cancer is one of the most common cancers in adults in Western world, of which 90% is adenocarcinoma. The incidence rate of colorectal cancer keeps increasing worldwide, while improvements are achieved by thorough understanding of the mechanisms involved in colorectal cancer and therapy. However, advanced colorectal cancer has been associated with higher mortality rates. It should be noted that compared with the patients with nonmetastatic tumor sites whose five-year survival rate is approximately 80%, patients with metastatic disease have a significantly poorer five-year survival rate (20%).1 Thus, studies on the mechanism of metastasis in colorectal cancer are of value for the discovery of new therapies.
TGF-β is a critical cytokine which is involved in tumorigenesis.2 Altered TGF-β signaling has been reported in malignant gastrointestinal cancers, such as colorectal cancer, esophageal cancer, gastric cancer, liver cancer, and pancreatic cancer,2 and increased TGF-β level is associated with poor prognosis in patients with advanced cancers.3 Dual effects of TGF-β signaling were found in different stages of tumor development in which, during the early phase, TGF-β acts as a tumor suppressor to trigger cell cycle arrest and apoptosis,3 while promoting epithelial-mesenchymal transition (EMT), which is a critical pathological process in the development of colorectal cancer,4 cell proliferation, fibrosis and angiogenesis during tumor progression.5
Transgelin (TAGLN, also named SM22) is an actin-binding protein that belongs to the calponin family and was first identified in 1987.6 TAGLN is localized in vascular and visceral smooth muscle, and has been found to be a marker for smooth muscle differentiation.7 TAGLN is also expressed in fibroblasts, and some epithelium, including intestinal epithelial cells,8 glomerular epithelial cells,9 breast ductal epithelium8,10 and prostate epithelium in which TAGLN expression is induced by TGF-β. TGF-β plays key roles in regulation of TAGLN expression,11 cooperating as a TAGLN promoter, in which there are motifs of TGF-β control element (TCE), CArG boxes, and Smad-binding element (SBE). By interacting with these motifs, TGF-β promotes TAGLN expression.12 Recent evidence suggests that TAGLN has a role in tumor development and migration, as TAGLN regulates the level of metallomatrix protease-9 (MMP-9) in prostate, breast, and colon cancers.13
High-mobility group AT-hook 2 (HMGA2) is a member of HMGA family (HMGA1a, HMGA1b, and HMGA2) which contains three AT-hook basic domains binding the A/T-rich sequences at DNA minor groove to assemble transcriptional or enhancer complexes on chromatin.14 TGF-β/Smad signaling pathway has been shown to act on HMGA2 promoter in epithelial cells, which results in activation of some key regulators of EMT that plays a critical role in tumor metastasis.15 It has been reported that HMGA2 is involved in development of colorectal cancer, as increased expression of HMGA2 was found in colon carcinoma tissues,16 and progressively increased with the Dukes’ staging system for colorectal cancer.17 In invasive epithelial cells with EMT, overexpression of HMGA2 was observed, and loss of E-cadherin in a metastatic site was found.17 Overall, these results indicated that HMGA2 overexpression might play an important role in tumor metastasis and survival of colorectal cancer patients.18
These studies elucidate the role of TGF-β pathways in the pathogenesis of colorectal cancer, and further investigation on the underlying mechanism would benefit in developing new treatment strategies. Considering the interaction between TGF-β pathways and TAGLN and HMGA2, in this study, we examined the potential roles of TAGLN and HMGA2 and their interaction in TGF-β pathways in colorectal cancer. It would be of promising clinical value to provide new insight for the development of colon cancer therapy.
Materials and Methods
Cell Culture
Human CRC cell lines HCT116, and HT29 were purchased from American Type Culture Collection. Cells were cultured in medium (RPMI-1640 (Gibco), 10% (v/v) FBS (Gibco), 1% streptomycin-penicillin (Invitrogen, cat lot.: 15640055), 1% L-glutamine (Thermo Fisher Scientific, cat lot.: 21051024) in an incubator with atmosphere containing 5% CO2 at 37°C. The transfections of overexpression vectors were performed by using lipofectamine 2000 (Invitrogen, cat lot.: 11668027) according to the manufacturer’s instructions.
Western Blotting Assay
Plasma and nucleus protein were extracted by using NE-PER™ nuclear and cytoplasmic extraction reagents (Thermo Scientific, cat lot.: 78835) lysing cells reported in previous studies.19,20 Briefly, protein quantitation was conducted by using the Pierce™ BCA protein assay kit (Thermo Fisher Scientific, cat lot.: 23225), and 10% SDS-polyacrylamide gels was used for separate the protein and then the protein was transferred onto PVDF membranes. 10% skimmed milk was used as blocking buffer, and then membranes were probed with primary antibodies against E-cadherin (Thermo Fisher Scientific, cat lot.:13–1700), vimentin (Thermo Fisher Scientific, cat lot.: MA5-11883), fibronectin (Thermo Fisher Scientific, cat lot.: MA5-11981), MMP2 (Thermo Fisher Scientific, cat lot.: MA5-14228), MMP9 (Thermo Fisher Scientific, cat lot.: MA5-14186), and GAPDH (Cell Signaling Technology, Danvers, TX) overnight at 4°C. Then the membrane was incubated with HRP-conjugated secondary antibody (Thermo Fisher Scientific, cat lot.: A-11059). Signals were visualized using enhanced chemiluminescence detection (GE Healthcare).
qPCR
Cells were collected and the RNAs from different groups were extracted by using TRIzol reagents (Invitrogen, USA). cDNA was synthesized by using PrimeScript™ RT reagent Kit with gDNA Eraser (Takara Bio, Japan) according to manufacturer’s instructions. qPCR was performed by using ABI 7300 real-time PCR system (Applied Biosystems, USA). The primer sequences were used as follows: TAGLN forward: 5ʹ-GTGTGATTCTGAGCAAATTGGTG-3ʹ, TAGLN reverse: 5ʹ-ACTGCTGCCATATCCTTACCTT-3ʹ; β-actin forward: 5ʹ-CATTGCTGACAGGATGCAGA-3ʹ, β-actin reverse: 5ʹ-CTGCTGGAAGGTGGACAGTGA-3ʹ. All samples were analyzed in triplicate and the 2−ΔΔCt method was used to calculate the gene expression levels.
Immunofluorescence Assay
In each well of a six-well plates, 1×105 cells/well were seeded, and following removal of the medium, cells were fixed with 4% paraformaldehyde solution for 30 min at room temperature. Immunofluorescence staining was performed by using primary antibody anti-TGF-β (Thermo Fisher Scientific, cat lot.: PA5-32631) (1:100) and incubated at 4°C overnight. Then, washed three times with phosphate buffer, cells were incubated with fluorescein isothiocyanate (FITC)-conjugated anti-rabbit immunoglobulin G (1:200; Beyotime Institute of Biotechnology, Haimen, China) in the dark for two hours at 37°C.
Cell Migration Assay
Cells (1×105per well) were re-suspended by RMPI-1640 medium and planted into the upper chambers of 24-well transwell plates (Corning, USA), and lower chamber was filled with medium. After 18 h incubation at 37°C, cells in the lower chamber were fixed with 4% paraformaldehyde and then stained with crystal violet. The cells remained in the upper chamber were removed. Migrated cells were counted in 10 random high-power fields. All experiments were performed in triplicate.
Cell Invasion Assay
Bio-coat matrigel invasion assay system (BD, USA) was used to evaluate cell invasion following the manufacturer’s protocol. Cells were re-suspended in RPMI-1640 medium and seeded into the upper chambers of 24-well transwell plates, and bottom chambers was filled RPMI-1640 medium with FBS (10%). After 24 h incubation, the supernatant cells were removed, while the cells on the bottom were fixed with 4% paraformaldehyde, stained with crystal violet, and counted. All experiments were performed in triplicate.
Co-immunoprecipitation
Cells were lysed at 4°C in ice-cold RIPA lysis buffer, and the lysates were centrifuged (12,000 g, 10 min) and supernatant were collected. Concentrations of proteins in the supernatant were determined by using Pierce™ BCA protein assay kit (Thermo Fisher Scientific, cat lot.: 23225). Co-immunoprecipitation was performed by using Pierce® co-immunoprecipitation kit (Pierce, cat lot.: 26149) following the manufacturer’s protocol. Samples were boiled and eluted by boiled in 2×SDS sample buffer for subsequent SDS-PAGE electrophoresis.
Tumor Formation Assay in vivo
BALA/c mice (six-to-eight weeks old, weight 20–25 g) were purchased from Vital River, Beijing, China. The xenograft model was generated by using BALB/c mice which were maintained at 20–26°C, 40–70% relative humidity, and 12 h/12 h light/dark condition. All experimental procedures were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The animal protocols were approved by the Institutional Animal Care and Use Committee of The First Affiliated Hospital, School of Clinical Medicine of Guangdong Pharmaceutical University. HT29 cells treated with TGF-β, TGF-β+si-TAGLN or control were collected and re-suspended in medium at a concentration of 2×106 cells/mL, and then 200 μL of cell suspension was injected into the right flank of nude mice. Tumor growth was detected at 1, 4, 8, 12, 16, 20, 24, 28, and 32 days after injection, and the volume of tumors were examined. All the mice were euthanized in an automated CO2 delivery system (LC500, Yuyan Instruments Co. Ltd, China) at 32 days after injection.
Statistical Analysis
All statistical analyses were performed by using SPSS software (Version 20; IBM, USA). Two-sided t tests and one-way ANOVA followed Tukey’s post hoc test was used to analyze the data, with a significance level of P<0.05.
Results
TGF-β-Induced TAGLN Expression Increased Migration and Invasion of CRC Cells
To preliminarily investigate the effects of TGF-β on TAGLN expression, cell migration and invasion, and screen cell line used for subsequent experiments, human colorectal cancer cell lines HT29 cells and HCT116 cells were treated with TGF-β at concentration of 10 nM. As shown in Figure 1A and B, TGF-β treatment induced significant increase of TAGLN protein expression in both of HT29 cells and HCT116 cells; enhanced cell migration and invasion were also found in HCT116 cells (Figure 1C) and HT29 cells (Figure 1D) treated with TGF-β. Furthermore, compared with HCT116 cells, relatively higher response in TAGLN expression, cell migration and invasion to TGF-β was found in HT29 cells.
 |
Figure 1 Increased cell migration and invasion and TAGLN nucleus translocation was induced by TGF-β. (A) Levels of TAGLN in HT29 and HT116 cells. (B) Representative images of Western blotting assay of TAGLN in HT29 and HT116 cells. (C) Cell migration and invasion in HCT116 cells (left panel) and representative images of crystal violet staining (right panel). (D) Cell migration and invasion in HT29 cells (left panel) and representative images of crystal violet staining (right panel). (E) Representative images of immunofluorescence of TAGLN. 200× magnification. *P <0.01, vs blank group. One-way ANOVA, Tukey’s post hoc test. |
Along with the increased expression of TAGLN in CRC cells, TGF-β also induced translocation of TAGLN to the nucleus. In Figure 1E, after treated with TGF-β for 24 hours, obvious nucleus localization of TAGLN could be observed. These results indicated altered TAGLN expression and nucleus translocation might involve in TGF-β induced HT29 and HCT116 cell migration and invasion.
Knockdown of TAGLN Inhibits TGF-β-Induced CRC Cell Migration and Invasion
To further study the role of TAGLN in TGF-β-induced HCT116 and HT29 cell migration and invasion, siRNA that targeted TAGLN (si-TAGLN) was constructed and transfected into HCT116 and HT29 cells. Inhibition of TAGLN by si-TAGLN could reverse TGF-β-induced migration and invasion in HCT116 cell (Figure 2A). We performed Western blot on the proteins involved in cell migration and EMT, including E-cadherin, vimentin, fibronectin, MMP2, and MMP9. During EMT, the epithelial cell marker E-cadherin is downregulated, while mesenchymal proteins vimentin, and fibronectin are often increased. As shown in Figure 2C, loss of E-cadherin and increased vimentin and fibronectin which are often observed in EMT are seen, while inhibition of TAGLN reversed the alternations of E-cadherin, vimentin and fibronectin induced by TGF-β. Studies in HCT116 cells further confirmed the findings in HT29 cells that knockdown of TAGLN inhibited TGF-β-induced cell migration and invasion (Figure 2B and C).
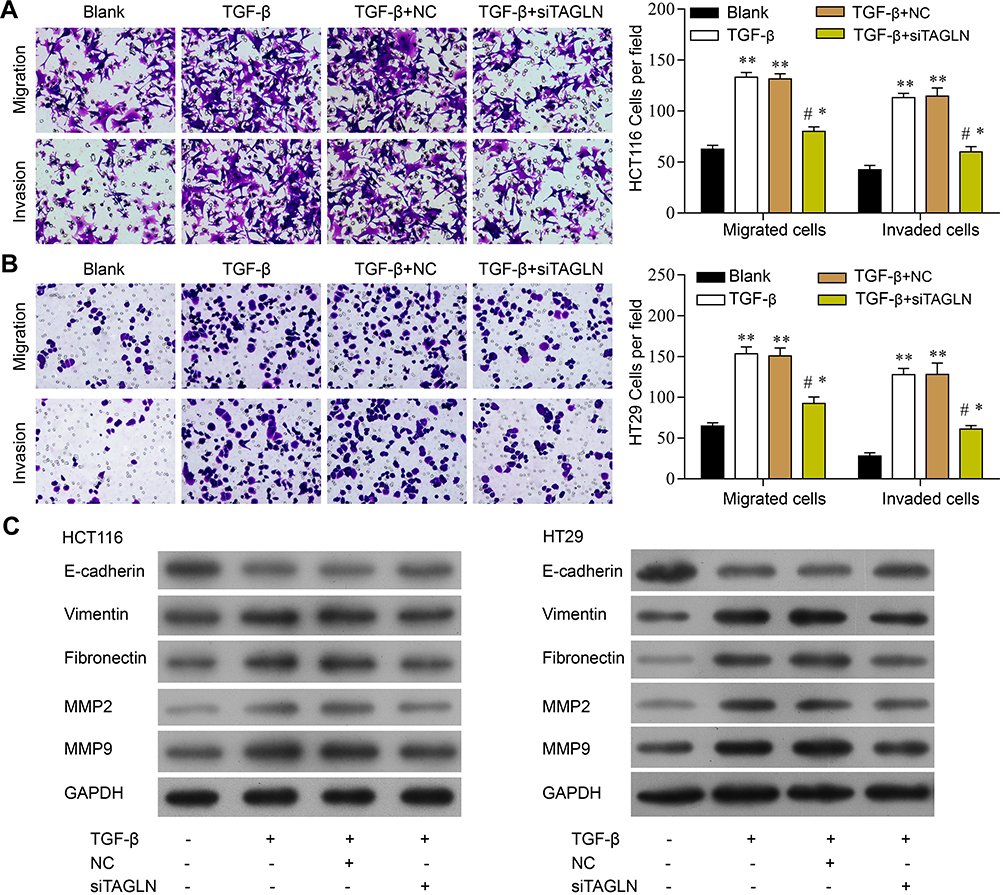 |
Figure 2 TGF-β-induced cell migration and invasion was reversed by knockdown of TAGLN. (A) Cell migration and invasion in HCT116 cells (left panel) and representative images of crystal violet staining (right panel). (B) Cell migration and invasion in HT29 cells (left panel) and representative images of crystal violet staining (right panel). (C) Representative images of Western blotting assay of E-cadherin, vimentin, fibronectin, MMP2, MMP9, and GADPH. 200 x magnification. *P<0.05, **P<0.01, ***P<0.05. One-way ANOVA, Tukey’s post hoc test. |
Nucleus translocation of TAGLN was investigated, and decreased plasma TAGLN protein expression in western-blotting assay (Figure 3A) and nucleus localization in immunofluorescence assay (Figure 3C) were found in si-TAGLN+TGF-β group compared with those in TGF-β group or si-TAGLN NC+TGF-βgroup. HMGA2 was found to be regulated by the TGF-β1/Smad3 signaling pathway,21 thus we investigated the interaction between TAGLN and HMGA2 by using co-immunoprecipitation assay. Results revealed an interaction between TAGLN and HMGA2 in both HCT116 and HT29 cells (Figure 3B). In addition, knockdown of TAGLN led to less HMGA2 pulldown (Figure 3B).
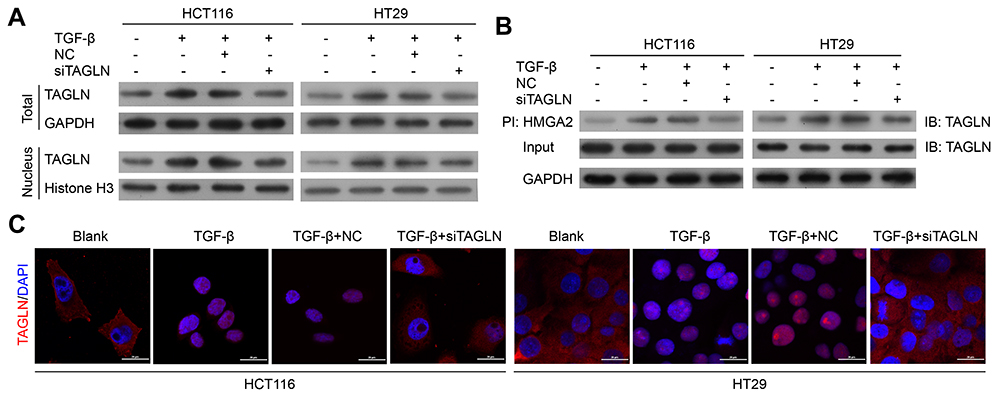 |
Figure 3 TGF-β-induced TAGLN nucleus translocation in CRC cells. (A) Representative images of Western blotting assay of nucleus and total TAGLN levels. (B) Representative images of immunoprecipitation of TAGLN and HMGA2. (C) Representative images of immunofluorescence of TAGLN. 200 x magnification. |
These results confirm that the TGF-β-TAGLN signaling pathway affects cell migration-related malignant behavior, and HMGA2 might also be involved.
Overexpression of HMGA2 Reversed the Effects of TAGLN Inhibition on CRC Cell Migration and Invasion
To investigate the interaction of HMGA2 of TAGLN and its role in TGF-β signaling pathway in CRC cells, we overexpressed HMGA2 (empty vector as the control) in HT29 and HCT116 cells that are treated with TGF-β and si-TAGLN or its control siRNA, as a blank group, TGF-β group, TGF-β+si-TAGLN NC group, TGF-β+si-TAGLN group, TGF-β+si-TAGLN+Vector group, TGF-β+si-TAGLN+HMGA2 group. As expected, overexpression of HMGA2 significantly rescued si-TAGLN induced decreases of cell migration and invasion in TGF-β treated HT29 cells (Figure 4A), and TGF-β triggered unclear translocation of TAGLN was also significantly restored (Figure 4C), which a higher TAGLN protein level was found in unclear than cytoplasm in the TGF-β+si-TAGLN+HMGA2 group. Western blotting assay revealed that overexpression of HMGA2 could restore TGF-β induced alternation, including cell migration, TAGLN expression, and protein expression of inhibition of TAGLN induced E-cadherin, vimentin, fibronectin, MMP9 and MMP2, which were attenuated by TAGLN knockdown (Figure 4D). These results indicated that interaction between TAGLN and HMGA2 are involved in TGF-β signaling pathway. Studies in HCT116 cells further confirmed the findings in HT29 cells that knockdown of TAGLN inhibited TGF-β-induced cell migration and invasion (Figure 4B–D).
 |
Figure 4 Overexpression of HMGA2 restored altered CRC cell migration and invasion induced by inhibition of TAGLN. (A) Cell migration and invasion in HCT116 cells (left panel) and representative images of crystal violet staining (right panel). (B) Cell migration and invasion in HT29 cells (left panel) and representative images of crystal violet staining (right panel). (C) Representative images of Western blotting assay of nucleus and total TAGLN levels. (D) Representative images of Western blotting assay of E-cadherin, vimentin, fibronectin, MMP2, MMP9, and GADPH. 200 x magnification. *P<0.05, **P<0.01, vs Blank group; ***P<0.05, vs si-TAGLN NC+TGF-β; ****P<0.05, vs si-TAGLN +Vector. One-way ANOVA, Tukey’s post hoc test. |
TGF-β-Induced TAGLN Expression Increased Migration and Invasion of Tumor Promoting
Tumor inoculation of HT29 cells in nude mouse was used as animal model to evaluate the effect of si-TAGLN on cancer cell growth, in which mice were implanted with untreated HT29 cells (blank) or HT29 cells that pretreated with TGF-β and si-TAGLN (TGF-β+si-TAGLN group) or TGF-β control siRNA (NC group). As shown in Figure 5A and B, in the si-TAGLN group, compared with the blank group and control group, the growth of the tumor was significantly inhibited. Western blotting assay of the tumor tissues in nude mouse confirmed the findings, as decreased protein level of TAGLN and HMGA2 in the TGF-β+si-TAGLN group; Western blotting assay revealed that TAGLN knockdown attenuated TGF-β induced alternation in HT29 cells, including cell migration, TAGLN expression, and protein expression of inhibition of TAGLN induced E-cadherin, vimentin, fibronectin, MMP9 and MMP2 (Figure 5C). The interaction between TAGLN and HMGA2 was further confirmed, as shown in Figure 5D, and weakened HMGA2 expression was also observed after inhibition of TAGLN compared with the control group (Figure 5E).
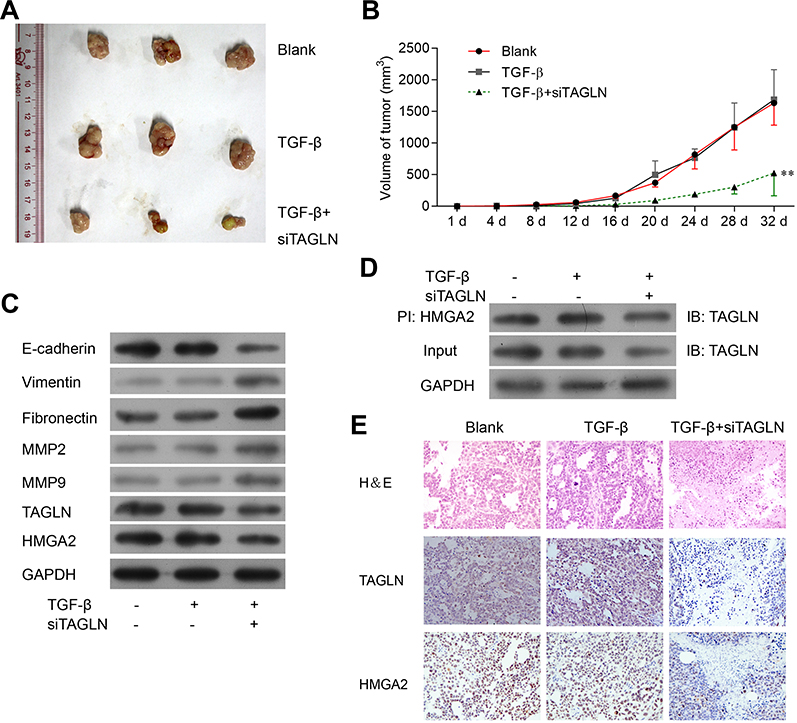 |
Figure 5 TGF-β-induced TAGLN expression increased migration and invasion of tumor promoting. (A) Image of tumors in nude mice inoculated with HT29 cells (Blank) and HT29 cells pretreated with TGF-β and si-TAGLN (TGF-β+si-TAGLN group). (B) Tumor volume was measured every four days. Tumor volume=1/2*L*W*W. *P<0.01, vs TGF-β group. (C) Representative images of Western blotting of metastasis markers in tumor tissues. (D) Co-IP assay was performed to detect the interaction of TAGLN and HMGA2 in tumor tissues. (E) Representative images of H&E, TAGLN and HMGA2 staining. 200 x magnification. |
Discussion
In this study, we investigated the role of TAGLN and HMGA2 in TGF-β-induced increase in cell migration of colorectal cancer cell lines HT29 and HCT116. We found that TGF-β could significantly promote the migration of CRC cells probably through induction of TAGLN protein expression and nucleus translocation. Knockdown of TAGLN could also rescue TGF-β-induced loss of metastasis markers’ expression; the elevation of MMP9 and MMP2 were also reversed by inhibition of TAGLN. Further investigation confirmed the interaction of HMGA2 and TAGLN, as overexpression of HMGA2 restored effects of TGF-β on HT29 and HCT116 cells that were attenuated by TAGLN inhibition.
Colorectal cancer is one of the three most common cancers worldwide.1 Altered TGF-β signaling pathway was reported in colorectal cancer, as increased TGF-β levels were detected in primary tumor and blood samples, and were found to correlate with metastasis and poor prognosis.22,23 TGF-β was reported to induce cell growth in colon carcinomas and promote cell proliferation in aggressive cancer cells,24 while inhibition of TGF-β prevents CRC metastasis by suppressing immune response to cancer cells by immune system.25 EMT is considered a key process that confers cancer cells with migration and invasion abilities.26 During tumor progression, TGF-β signaling was also found to be involved in the EMT that is critical in colorectal cancer, in which TGF-β activates RhoA and rho-associated protein kinase to induce actin polymerization.15,21,27 In our study, we found TGF-β-dependent activation of TAGLN expression is accompanied with decreased expression of E-cadherin, and increased levels of vimentin, fibronectin. It has been shown that TGF-β induced activation of TAGLN expression in tumor cells, especially in epithelial cells, can lead to a process of EMT, and TGF-β stimulated fibroblasts can lead to their transformation into myofibroblasts, which is important in tumor progression.
TGF-β is one of the most important signaling pathways in TAGLN regulation, in which TGF-β activates TAGLN expression,12,28 as our finding shows decreased TAGLN expression and altered nucleus translocation was found in TGF-β-treated CRC cells. We detected the function of TGF-β in vitro and in vivo. Our data show that it promoted tumor metastasis and progression accompanied with the activation of TAGLN. However, the relationship of TAGLN expression and metastases remains unclear. A few studies have been published where increased TAGLN expression is reported in connection with metastasis in colorectal cancer.29,30 TAGLN is a cytoplasmic protein that was first identified in smooth muscle,6 TAGLN highly expresses in smooth muscle cells as well as all other types of cells.31 TAGLN primarily participates in actin cytoskeleton remodeling, which is critical in cell morphogenesis, cell migration, differentiation, proliferation and apoptosis. In recent years, number of studies reported altered expression of TAGLN in many kinds of tumors, and dysfunction of TAGLN are also found in colorectal cancers.8,32–36 In cancer samples in comparison with healthy controls increased level of TAGLN was observed patients with advanced disease,32 in which invasion of tumor cells into the muscle layer was found. These results further confirmed in our study that si-TAGLN significantly decreased the cell invasion and migration.
In addition, we found that HMGA2 are downregulated by TGF-β, and overexpression of HMGA2 reversed the function of TAGLN inhibition, indicating that HMGA2 exerted direct effects on cell migration. The role of HMGA proteins in colorectal carcinomas has been widely evaluated.37,38 Previous studies reported that HMGA2 mRNA in tumor tissues in patients with adenocarcinoma was significantly upregulated compared with the normal tissues.39–42 TGF-β/Smad signaling pathway is critical in HMGA2 expression, of which the activation is a key regulator of EMT that is known to play a key role in cancer metastasis.15 Loss of epithelial markers, such as E-cadherin and increase of mesenchymal markers, such as vimentin and fibronectin, are typical characteristics of EMT in cancer development. In summary, TGF-β treatment promoted CRC tumor metastasis and malignancy through upregulation of TAGLN. Mechanically, the translocation of TAGLN and the interaction of TAGLN and HMGA2 mediated EMT in CRC.
Acknowledgments
This work was supported by the National Natural Science Foundation of China for Young Scientists (No. 81602110).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Haggar FA, Boushey RP. Colorectal cancer epidemiology: incidence, mortality, survival, and risk factors. Clin Colon Rectal Surg. 2009;22(4):191–197. doi:10.1055/s-0029-1242458
2. Massague J. TGFbeta signalling in context. Nat Rev Mol Cell Biol. 2012;13(10):616–630.
3. Neuzillet C, Tijeras-Raballand A, Cohen R, et al. Targeting the TGFbeta pathway for cancer therapy. Pharmacol Ther. 2015;147:22–31.
4. Massague J. TGFbeta in Cancer. Cell. 2008;134(2):215–230.
5. Colak S, Ten Dijke P. Targeting TGF-beta Signaling in Cancer. Trends in Cancer. 2017;3(1):56–71.
6. Lees-Miller JP, Heeley DH, Smillie LB, Kay CM. Isolation and characterization of an abundant and novel 22-kDa protein (SM22) from chicken gizzard smooth muscle. J Biol Chem. 1987;262(7):2988–2993.
7. Lawson D, Harrison M, Shapland C. Fibroblast transgelin and smooth muscle SM22alpha are the same protein, the expression of which is down-regulated in many cell lines. Cell Motil Cytoskeleton. 1997;38(3):250–257.
8. Shields JM, Rogers-Graham K, Der CJ. Loss of transgelin in breast and colon tumors and in RIE-1 cells by Ras deregulation of gene expression through Raf-independent pathways. J Biol Chem. 2002;277(12):9790–9799.
9. Ogawa A, Sakatsume M, Wang X, et al. SM22alpha: the novel phenotype marker of injured glomerular epithelial cells in anti-glomerular basement membrane nephritis. (1660-2129 (Electronic)). Nephron Exp Nephrology. 2007;106(3):e77–e87
10. Wulfkuhle JD, Sgroi D, Krutzsch H, et al. Proteomics of human breast ductal carcinoma in situ. (0008-5472 (Print)). Cancer res. 2002;62(22):6740–6749
11. Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84(3):767–801.
12. Qiu P, Feng XH, Li L. Interaction of Smad3 and SRF-associated complex mediates TGF-beta1 signals to regulate SM22 transcription during myofibroblast differentiation. J Mol Cell Cardiol. 2003;35(12):1407–1420.
13. Nair RR, Solway J, Boyd DD. Expression cloning identifies transgelin (SM22) as a novel repressor of 92-kDa type IV collagenase (MMP-9) expression. J Biol Chem. 2006;281(36):26424–26436. doi:10.1074/jbc.M602703200
14. Reeves R, Nissen MS. The A.T-DNA-binding domain of mammalian high mobility group I chromosomal proteins. A novel peptide motif for recognizing DNA structure. J Biol Chem. 1990;265(15):8573–8582.
15. Thuault S, Valcourt U, Petersen M, Manfioletti G, Heldin CH, Moustakas A. Transforming growth factor-beta employs HMGA2 to elicit epithelial-mesenchymal transition. J Cell Biol. 2006;174(2):175–183.
16. Helmke BM, Markowski DN, Meyer A, Bullerdiek J. The expression of HMGA2 varies strongly among colon carcinomas. Anticancer Res. 2012;32(5):1589–1593.
17. Li Y, Zhao Z, Xu C, Zhou Z, Zhu Z, You T. HMGA2 induces transcription factor Slug expression to promote epithelial-to-mesenchymal transition and contributes to colon cancer progression. Cancer Lett. 2014;355(1):130–140. doi:10.1016/j.canlet.2014.09.007
18. Wang X, Liu X, Li AY, et al. Overexpression of HMGA2 promotes metastasis and impacts survival of colorectal cancers. Clin Cancer Res. 2011;17(8):2570–2580. doi:10.1158/1078-0432.CCR-10-2542
19. Hung S-C, Pochampally RR, Chen S-C, Hsu S-C, Prockop DJ. Angiogenic effects of human multipotent stromal cell conditioned medium activate the PI3K-Akt pathway in hypoxic endothelial cells to inhibit apoptosis, increase survival, and stimulate angiogenesis. Stem Cells. 2007;25(9):2363–2370. doi:10.1634/stemcells.2006-0686
20. Giannoni E, Guignard L, Knaup Reymond M, et al. Estradiol and progesterone strongly inhibit the innate immune response of mononuclear cells in newborns. Infect Immun. 2011;79(7):2690–2698. doi:10.1128/IAI.00076-11
21. Song X, Liu W, Xie S, et al. All-transretinoic acid ameliorates bleomycin-induced lung fibrosis by downregulating the TGF-beta1/Smad3 signaling pathway in rats. Lab Invest. 2013;93(11):1219–1231.
22. Picon A, Gold LI, Wang J, Cohen A, Friedman E. A subset of metastatic human colon cancers expresses elevated levels of transforming growth factor beta1. Cancer Epidemiol Biomarkers Prevent. 1998;7(6):497–504.
23. Tsushima H, Kawata S, Tamura S, et al. High levels of transforming growth factor beta 1 in patients with colorectal cancer: association with disease progression. Gastroenterology. 1996;110(2):375–382. doi:10.1053/gast.1996.v110.pm8566583
24. Schroy P, Rifkin J, Coffey RJ, Winawer S, Friedman E. Role of transforming growth factor beta 1 in induction of colon carcinoma differentiation by hexamethylene bisacetamide. Cancer Res. 1990;50(2):261–265.
25. Tauriello DVF, Palomo-Ponce S, Stork D, et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. 2018;554(7693):538–543. doi:10.1038/nature25492
26. Scheel C, Weinberg RA. Cancer stem cells and epithelial–mesenchymal transition: concepts and molecular links. Semin Cancer Biol. 2012;22(5–6):396–403. doi:10.1016/j.semcancer.2012.04.001
27. Kim I-G, Lee J-H, Kim S-Y, Hwang H-M, Kim T-R, Cho E-W. Hypoxia-inducible transgelin 2 selects epithelial-to-mesenchymal transition and γ-radiation-resistant subtypes by focal adhesion kinase-associated insulin-like growth factor 1 receptor activation in non-small-cell lung cancer cells. Cancer Sci. 2018;109(11):3519–3531. doi:10.1111/cas.13791
28. Chen S, Kulik M, Lechleider RJ. Smad proteins regulate transcriptional induction of the SM22alpha gene by TGF-beta. Nucleic Acids Res. 2003;31(4):1302–1310.
29. Lin Y, Buckhaults PJ, Lee JR, et al. Association of the actin-binding protein transgelin with lymph node metastasis in human colorectal cancer. Neoplasia. 2009;11(9):864–873. doi:10.1593/neo.09542
30. Zhang J, Song MQ, Zhu JS, et al. Identification of differentially-expressed proteins between early submucosal non-invasive and invasive colorectal cancer using 2D-DIGE and mass spectrometry. Int J Immunopathol Pharmacol. 2011;24(4):849–859.
31. Shapland C, Hsuan JJ, Totty NF, Lawson D. Purification and properties of transgelin: a transformation and shape change sensitive actin-gelling protein. J Cell Biol. 1993;121(5):1065–1073.
32. Peng J, Zhang Q, Ma Y, et al. A rat-to-human search for proteomic alterations reveals transgelin as a biomarker relevant to colorectal carcinogenesis and liver metastasis. Electrophoresis. 2009;30(17):2976–2987.
33. Yeo M, Kim DK, Park HJ, et al. Loss of transgelin in repeated bouts of ulcerative colitis-induced colon carcinogenesis. Proteomics. 2006;6(4):1158–1165.
34. Zhao L, Wang H, Deng YJ, et al. Transgelin as a suppressor is associated with poor prognosis in colorectal carcinoma patients. Modern Pathol. 2009;22(6):786–796.
35. Yeo M, Park HJ, Kim DK, et al. Loss of SM22 is a characteristic signature of colon carcinogenesis and its restoration suppresses colon tumorigenicity in vivo and in vitro. Cancer. 2010;116(11):2581–2589.
36. Li SY, An P, Cai HY, et al. Proteomic analysis of differentially expressed proteins involving in liver metastasis of human colorectal carcinoma. Hepatobiliary Pancreatic Dis Int. 2010;9(2):149–153.
37. Chiappetta G, Manfioletti G, Pentimalli F, et al. High mobility group HMGI(Y) protein expression in human colorectal hyperplastic and neoplastic diseases. Int j Cancer. 2001;91(2):147–151.
38. Abe N, Watanabe T, Sugiyama M, et al. Determination of high mobility group I(Y) expression level in colorectal neoplasias: a potential diagnostic marker. Cancer Res. 1999;59(6):1169–1174.
39. Lin Y, Liu AY, Fan C, et al. MicroRNA-33b Inhibits Breast Cancer Metastasis by Targeting HMGA2, SALL4 and Twist1. Sci Rep. 2015;5:9995.
40. Sarhadi VK, Wikman H, Salmenkivi K, et al. Increased expression of high mobility group A proteins in lung cancer. J Pathol. 2006;209(2):206–212.
41. He QY, Wang GC, Zhang H, et al. miR-106a-5p Suppresses the Proliferation, Migration, and Invasion of Osteosarcoma Cells by Targeting HMGA2. DNA Cell Biol. 2016;35(9):506–520.
42. Aguirre-Gamboa R, Gomez-Rueda H, Martinez-Ledesma E, et al. SurvExpress: an online biomarker validation tool and database for cancer gene expression data using survival analysis. PLoS One. 2013;8(9):e74250.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.