")
Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 14
Tackling Drug Resistance in EGFR Exon 20 Insertion Mutant Lung Cancer
Authors Pacini L, Jenks AD, Vyse S, Wilding CP, Arthur A, Huang PH
Received 12 December 2020
Accepted for publication 22 February 2021
Published 9 March 2021 Volume 2021:14 Pages 301—317
DOI https://doi.org/10.2147/PGPM.S242045
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Martin H Bluth
Laura Pacini,* Andrew D Jenks,* Simon Vyse,* Christopher P Wilding, Amani Arthur, Paul H Huang
Division of Molecular Pathology, The Institute of Cancer Research, London, UK
*These authors contributed equally to this work
Correspondence: Paul H Huang
Division of Molecular Pathology, The Institute of Cancer Research, 237 Fulham Road, London, SW3 6JB, UK
Tel +44 207 153 5554
Email [email protected]
Abstract: Insertion mutations in exon 20 (Ex20ins) of the epidermal growth factor receptor (EGFR) gene are the largest class of EGFR mutations in non-small cell lung cancer (NSCLC) for which there are currently no approved targeted therapies. NSCLC patients with these mutations do not respond to clinically approved EGFR tyrosine kinase inhibitors (TKIs) and have poor outcomes. A number of early phase clinical trials are currently underway to evaluate the efficacy of a new generation of TKIs that are capable of binding to and blocking Ex20ins. Although these agents have shown some clinical activity, patient responses have been restricted by dose-limiting toxicity or rapid acquisition of resistance after a short response. Here we review the current understanding of the mechanisms of resistance to these compounds, which include on-target EGFR secondary mutations, compensatory bypass pathway activation and acquisition of an EMT phenotype. Taking lessons from conventional EGFR inhibitor therapy in NSCLC, we also consider other potential sources of resistance including the presence of drug-tolerant persister cells. We will discuss therapeutic strategies which have the potential to overcome different forms of drug resistance. We conclude by evaluating recent technological developments in drug discovery such as PROTACs as a means to better tackle TKI resistance in NSCLC harbouring Ex20ins mutations.
Keywords: EGFR, exon 20 insertions, poziotinib, lung cancer, drug resistance, PROTACs
Introduction
Lung cancer accounts for around 13% of all cancer diagnoses and is one of the leading causes of cancer mortality.1 Non-small cell lung cancer (NSCLC) comprises the vast majority of lung cancer cases (~85%)2 and activating mutations in the epidermal growth factor receptor (EGFR) gene are the second most prevalent oncogenic driver present in ~15–20% of NSCLC patients.3,4 There are a wide array of different EGFR mutations including the two most frequent mutations, L858R and Exon 19 deletions (Ex19del) which are often referred to as classical or sensitizing EGFR mutations. The third most common class of EGFR mutations are exon 20 insertions (Ex20ins) which account for ~4–10% of all EGFR mutations in NSCLC.5–7 EGFR Ex20ins are a class of mutations which are heterogeneous both in terms of size and location within the EGFR gene. They can be grouped together as insertions or duplications of 1–7 amino acids found between the α-C helix and following loop (762–774 amino acid sequence) of EGFR.5–8 The most frequently identified EGFR Ex20ins variants are V769_D770ins and D770_N771ins, which together account for half of all NSCLC cases that harbour Ex20ins.8 Activating Ex20ins have also been observed in the human epidermal growth factor receptor 2 (HER2) gene, another member of the EGFR family of receptor tyrosine kinases (RTK). Although HER2 mutations are present in only ~2% of NSCLC patients, Ex20ins are the most common HER2 mutation in lung cancer and occur between the α-C helix and following loop (767–783 amino acid) of the protein in a similar fashion to EGFR.9 Beyond NSCLC, EGFR Ex20ins have recently been described in 68% of sinonasal squamous cell carcinomas,10 a rare form of head and neck cancer, and both EGFR Ex20ins and HER2 Ex20ins were found in 18% and 3.6% of urothelial cancers, respectively.11 These data suggest that development of targeted therapies against Ex20ins may have therapeutic implications for other cancer types.
In lung cancer tumors with EGFR or HER2 mutations, blockade of EGFR or HER2 activity with targeted inhibitors can trigger rapid apoptosis in a manner consistent with the “oncogene addiction” model, in which cells are dependent on persistent kinase signalling for survival.12 As a kinase which is readily druggable with selective small molecule inhibitors, EGFR presents an attractive therapeutic target, and the success of EGFR inhibitors in NSCLC has paved the way for realising the potential of targeted therapy in oncology. However, EGFR Ex20ins represent a clinical unmet need as they are associated with de novo resistance to clinically approved EGFR inhibitors, including the competitive, reversible first-generation tyrosine kinase inhibitors (TKIs) (erlotinib and gefitinib) and the irreversible second-generation (afatinib) and third-generation inhibitors (osimertinib).6,9,13 One of the main challenges of targeting EGFR Ex20ins is that unlike classical EGFR mutations, Ex20ins mutations can activate EGFR without diminishing ATP affinity versus the wild-type kinase,14 a feature which negates the advantage of ATP-competitive inhibitors to selectively target mutant over wild-type EGFR. Moreover, 3D modelling suggests that EGFR Ex20ins possess a rigid C-helix conformation that creates a compact drug binding site, further blocking drug accessibility.9 Together, these features create an extremely narrow therapeutic window that prohibits clinically approved EGFR inhibitors from reaching therapeutic doses that can selectively target EGFR Ex20Ins mutants over wild-type EGFR without significant toxicity in patients. There is however one exception, the insertion mutant A763_Y764FQEA has a high affinity for first-generation EGFR inhibitors and there are multiple case studies that report responses to erlotinib in patients with this specific mutation.13,15 Beyond this exception, EGFR inhibitors are not currently used to treat EGFR Ex20Ins NSCLC patients. Instead, although the survival benefit is minimal, the current standard of care for the majority of EGFR Ex20ins patients remains cytotoxic chemotherapy comprising a platinum based agent such as cisplatin or carboplatin combined with a taxane or pemetrexed.16–18
EGFR inhibitors with the capacity to bind to and inactivate the compact ATP-binding site of Ex20ins (Ex20ins TKI; Table 1) include the covalent, irreversible EGFR inhibitors poziotinib (formerly HM781-36B), mobocertinib (TAK-788), and TAS6417 (CLN-081).9,19,20 Therapeutics which target the Ex20ins receptor but do not block the ATP-binding site include the EGFR and the hepatocyte growth factor receptor (HGFR or MET) dual targeting antibody amivantamab and the heat shock protein 90 (Hsp90) inhibitor luminespib (NVP-AUY922) (Figure 1).21,22 Pre-clinical studies and several ongoing clinical trials are currently evaluating these experimental therapeutics in NSCLC patients with EGFR and HER2 Ex20ins mutations.23–25 However, the limited clinical efficacy of these drugs reported to date highlights the challenges associated with Ex20ins mutant selectivity and drug resistance. In this review, we will outline the currently known resistance mechanisms identified for investigational agents that target Ex20ins and also describe candidate mechanisms based on the extensive clinical experience with first and third generation EGFR inhibitors in the context of classical EGFR mutations.
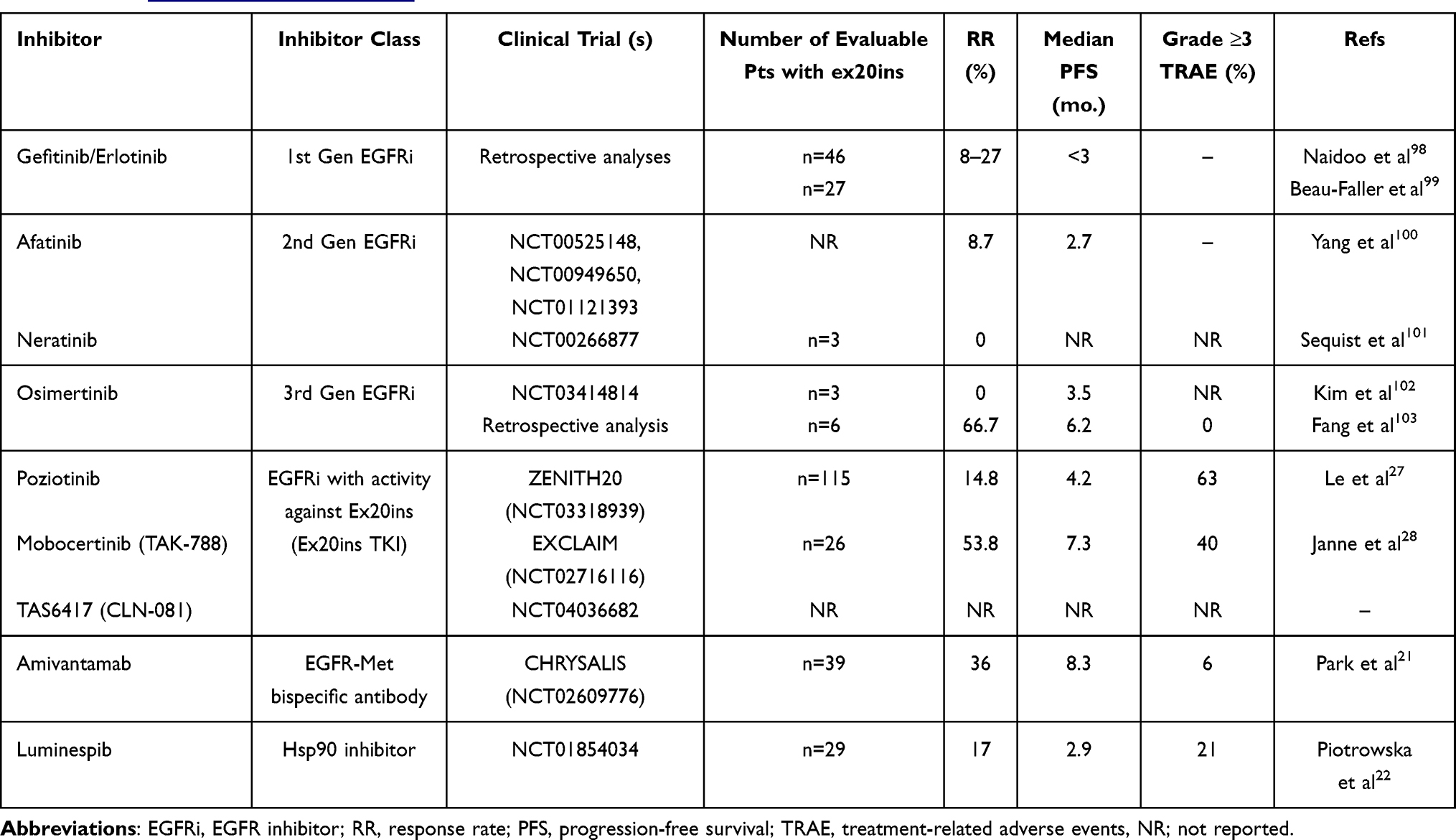 |
Table 1 Key Clinical Trial Results for NSCLC Harbouring EGFR Exon 20 Insertions. Details for Trials with NCT Numbers Can Be Accessed on Https://Clinicaltrials.gov/ |
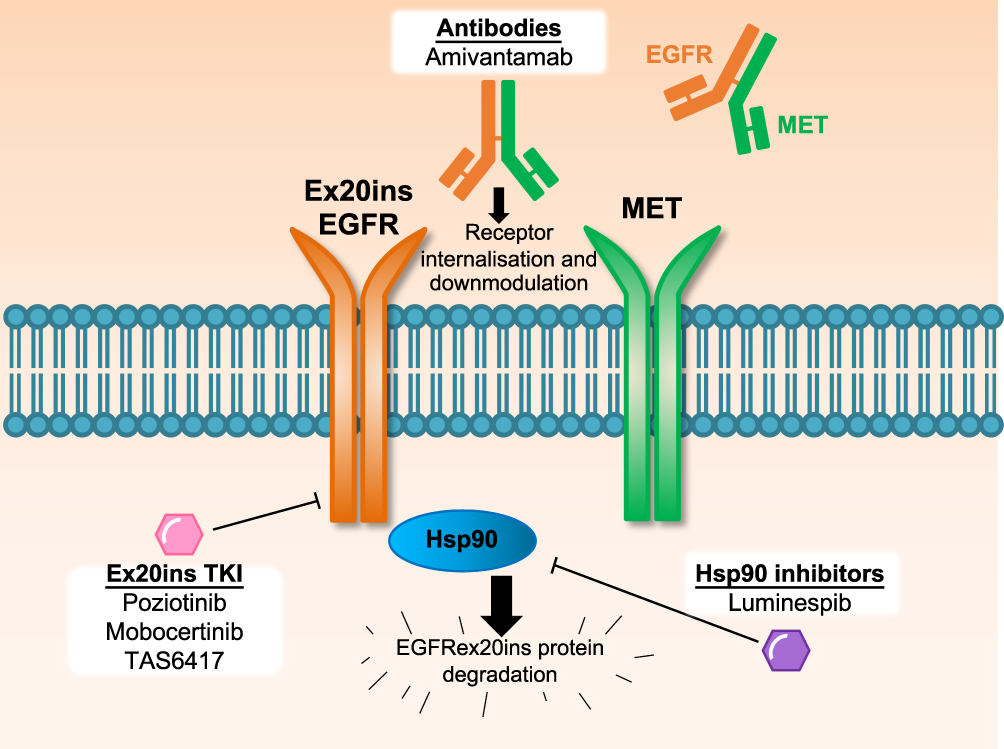 |
Figure 1 Therapeutic approaches to target EGFR Ex20ins NSCLC in clinical trials. Several approaches with distinct mechanisms are being assessed in clinical trials to target EGFR Ex20ins NSCLC, which are refractory to current clinically approved EGFR inhibitors. Small molecule tyrosine kinase inhibitors with the capacity to target the EGFR Ex20ins (Ex20ins TKI) can inhibit kinase catalytic activity. The bispecific EGFR-MET antibody amivantamab binds to both receptor tyrosine kinases which can result in receptor internalisation and downmodulation of oncogene expression on the cell surface. The Hsp90 inhibitor luminespib can inhibit the Hsp90 chaperone system which is co-opted by mutant EGFR Ex20ins to prevent ubiquitin-mediated protein degradation. |
Clinical Trial Evaluation of Ex20ins Targeting Agents
Due to the lack of efficacy of approved EGFR inhibitors in EGFR Ex20ins NSCLC, targeted therapy is not normally considered and the standard of care for this subset of patients is chemotherapy.26 Retrospective analysis of 165 EGFR Ex20ins NSCLC patients found a significantly longer median progression-free survival (PFS) for patients treated with platinum-based chemotherapy (6.4 months) compared with all approved EGFR inhibitors (2.9 months).18 However, there are now several new targeted agents under clinical investigation with the potential to change the standard of care in these patients.
The most advanced candidate poziotinib is currently being assessed in a number of phase II clinical trials including an open label, single arm study (NCT03066206) to assess the safety and efficacy of poziotinib in advanced or relapsed NSCLC patients with confirmed EGFR or HER2 mutations. Early data of 40 patients with Ex20ins mutations from this trial demonstrated a promising 58% objective response rate (ORR) after 8 weeks treatment.25 There was no restriction in the number or type of prior systemic therapies received, and responses were observed in 8 of 13 patients (62%) previously treated with an alternative TKI, indicating the potential role of poziotinib in heavily pre-treated patients. However, poziotinib is also a potent inhibitor of wild-type EGFR, and this trial reported that 60% of patients experienced grade 3 or greater adverse events, most commonly rash and diarrhoea. Furthermore, 45% of patients required a dose reduction from the starting dose of 16 mg poziotinib daily to 12 mg daily, and 17.5% of patients required a dose reduction to 8 mg daily.
Poziotinib has also been investigated in the phase II ZENITH20 trial (NCT03318939), an open-label, multi-cohort, multi-centre study which includes a cohort of pre-treated patients with a proven EGFR or HER2 Ex20ins mutation.27 This cohort of 115 patients had a median of 2 prior lines of therapy, and treatment with poziotinib 16 mg once daily conferred an ORR of 14.8% falling below the pre-specified primary endpoint of an ORR of 17%. 65% of patients had an observed reduction in tumor size with a disease control rate of 68.7% and a median PFS of 4.2 months. Toxicity remained a concern with 63% of patients reporting grade 3–4 treatment related adverse events. As a result, 68% of patients required dose reductions to subtherapeutic doses; 16% requiring a one-step reduction to 14 mg, 30% requiring a 2-step reduction to 12 mg, 22% requiring a dose of 10 mg or less and 10% of the study population permanently discontinued treatment. Adverse events were consistent with those previously reported for irreversible second-generation EGFR TKIs, most commonly diarrhoea and skin rash. This large multi-centre trial is ongoing with a cohort of treatment-naïve Ex20ins NSCLC patients and a split daily dosing regimen of poziotinib to determine if this regimen reduces incidence of adverse events and the requirement for dose reductions.
Mobocertinib is another covalent, irreversible inhibitor that selectively targets EGFR and HER2 Ex20ins.28 It is being assessed in the ongoing phase I/II EXCLAIM trial (NCT02716116) to determine the safety of administering mobocertinib as a single agent or in combination with pemetrexed or carboplatin. Preliminary results have been presented, with 28 patients with locally advanced or metastatic NSCLC harbouring EGFR or HER2 Ex20ins treated with 160 mg mobocertinib once daily included for analysis.28 Of these 28 patients, 26 were evaluable for treatment response with 14 having a partial response, giving an ORR of 53.8%, and 23 (88.5%) achieved disease control. In terms of toxicity, 21.7% of patients required a dose reduction due to treatment related adverse events with 10.9% discontinuing treatment as a result. The most common side effects included diarrhoea, rash and nausea. Based on this data, the FDA granted mobocertinib breakthrough designation status, however it remains to be determined whether toxicity will be present as an issue moving forwards into its phase III trial, EXCLAIM-2, which is now enrolling treatment-naïve NSCLC patients with EGFR Ex20ins (NCT04129502).29
TAS6417 is a covalent, irreversible EGFR inhibitor specifically designed to target the ATP binding site of the EGFR Ex20ins kinase domain.30 Promising pre-clinical work suggests that TAS6417 has a wide therapeutic window to target EGFR Ex20ins mutants over wild-type EGFR in cell line models.20 Clinical data for TAS6417 has yet to be reported, however a phase 1/2a clinical trial (NCT04036682) is ongoing to establish the maximum tolerated dose for NSCLC patients with EGFR Ex20ins.
An EGFR and MET-targeted bispecific antibody, amivantamab, has shown promising efficacy against EGFR Ex20ins NSCLC in engineered mouse models with a reduction in tumor volume, as well as a reduction in total and phospho EGFR and MET and the inhibition of downstream signaling pathways protein kinase B (AKT) and extracellular signal-regulated kinase (ERK). Amivantamab has also shown superior efficacy to poziotinib in tackling EGFR Ex20ins NSCLC with lower skin toxicity and loss of body weight in mice.31 Promising clinical activity has been observed in the phase 1 first-in-human study CHRYSALIS (NCT02609776) involving 50 NSCLC patients with 13 distinct EGFR Ex20ins mutations, of which 39 patients were evaluable for response. After a median follow-up of 4 months, ORR for amivantamab was 36% across the 39 patients, with 8.3 months median PFS.21 The safety profile for amivantamab was manageable, with 36% of patients experiencing grade >3 adverse events, of which 6% were treatment related. Based on this data, the FDA granted breakthrough designation status for amivantamab in March 2020 to accelerate the clinical investigation of this antibody in EGFR Ex20ins NSCLC. A phase 3 clinical trial, the PAPILLON study, is currently underway to investigate the potential of a combination of amivantamab with carboplatin-pemetrexed chemotherapy compared to chemotherapy alone in NSCLC patients with EGFR Ex20ins (NCT04538664).32
The Hsp90 inhibitor luminespib is generally well-tolerated, though reversible low-grade ocular-toxicity is common.22 In a phase II study (NCT01124864) involving patients with advanced NSCLC with several molecularly-defined subtypes, luminespib showed an ORR of ~17% among EGFR-mutant NSCLC. Notably, one patient with an Ex20ins mutation responded to luminespib.33 Based on further pre-clinical evidence that Hsp90 inhibition is effective in models that harbour EGFR Ex20ins,34 a phase II clinical trial for luminespib in NSCLC patients that specifically harbour EGFR Ex20ins (NCT01854034) found a 17% ORR in 29 patients and a median PFS of 2.9 months.22 The study met its primary endpoint for ORR, indicating that Hsp90 inhibitors could potentially be used as a therapeutic strategy in patients with EGFR Ex20ins. It should be noted however, that a high degree of luminespib-related toxicities reported in clinical trials reflects the general challenge of using Hsp90 inhibitors in patients and may ultimately be a limiting factor for further clinical development.
The clinical data to date highlight the challenges of targeting EGFR Ex20ins without significant toxicity due to wild-type EGFR inhibition. The ORR for these new agents remains low compared to approved EGFR inhibitors in the context of NSCLC bearing L858R and Ex19del (ORR >60%).35–37 The design of EGFR inhibitors with a greater therapeutic index may result in higher response rates and better drug tolerability. However, an outstanding question is whether intrinsic and acquired drug resistance will be a major limiting factor to the clinical efficacy of these agents that target EGFR Ex20ins. The short median PFS of 2.9 months for luminespib, 4.2 months reported for poziotinib27 and 7.3 months for mobocertinib23 contrasts with 10.1 months for osimertinib in L858R and Ex19del NSCLC in the second-line setting.38 While insufficient dosing due to toxicity may contribute to short-term responses in patients, early data from the use of poziotinib in EGFR Ex20ins patients suggests rapid acquisition of drug resistance, and the specific mechanisms of resistance have some overlap with those observed to arise in classical mutant EGFR NSCLC treated with approved EGFR inhibitors.39 Therefore, it is important to consider and anticipate the potential routes of drug resistance in order to achieve durable responses in patients with Ex20ins mutations.
Known Mechanisms of Ex20ins TKI Resistance
Despite having only recently been evaluated in clinical trials, clinical mechanisms of resistance have already been reported for some of the aforementioned Ex20ins TKIs.39 Here we will outline the currently known resistance mechanisms to poziotinib and other Ex20ins TKIs focusing on on-target mechanisms and compensatory bypass mechanisms of resistance described in the literature (Figure 2). An overview of genomic alterations, mutations, amplifications and copy number losses which are present at baseline or occur at relapse post-TKI treatment in Ex20ins patients are summarized in Table 2.39–41 We also outline potential therapeutics and druggable targets, which could be utilised to overcome TKI resistance.
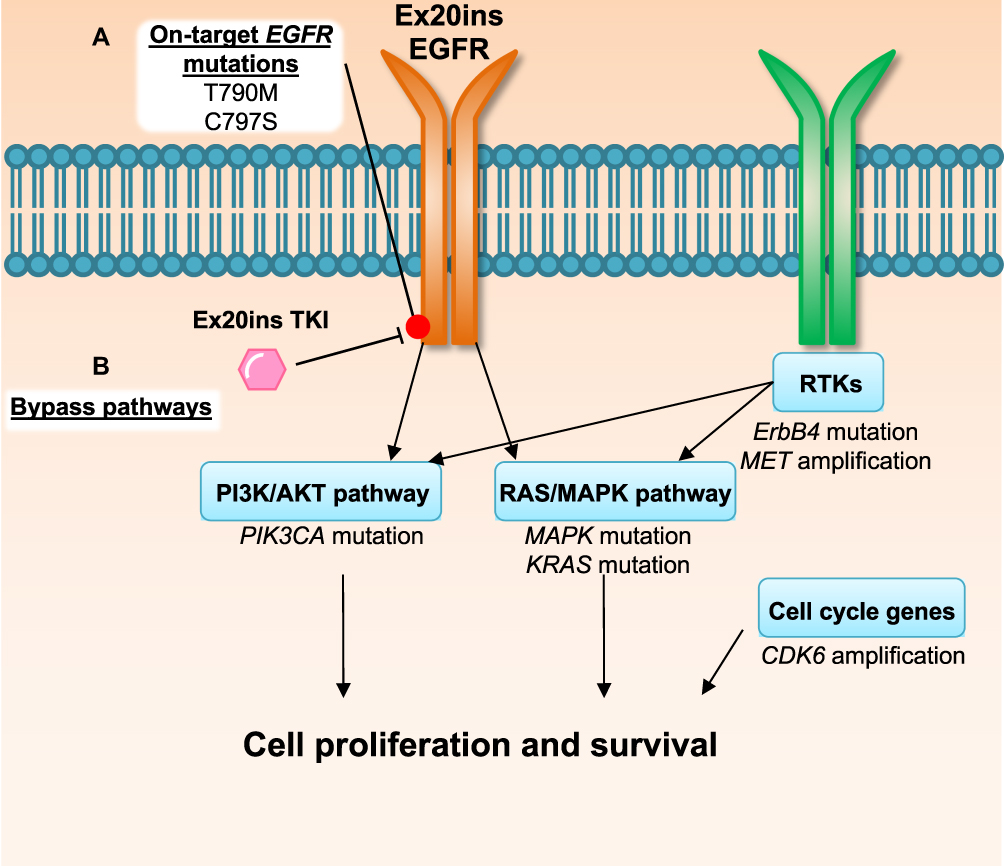 |
Figure 2 Mechanisms of EGFRex20ins TKI resistance. Evidence from the use of poziotinib in patients and in pre-clinical models39 suggests drug resistance can be driven by (A) acquisition of secondary on-target mutations in EGFR or (B) mutations or amplification in other oncogenic pathway proteins that result in activation of compensatory bypass pathways including the PI3K/AKT pathway, RAS/MAPK pathway, alternative RTKs and cell cycle genes. |
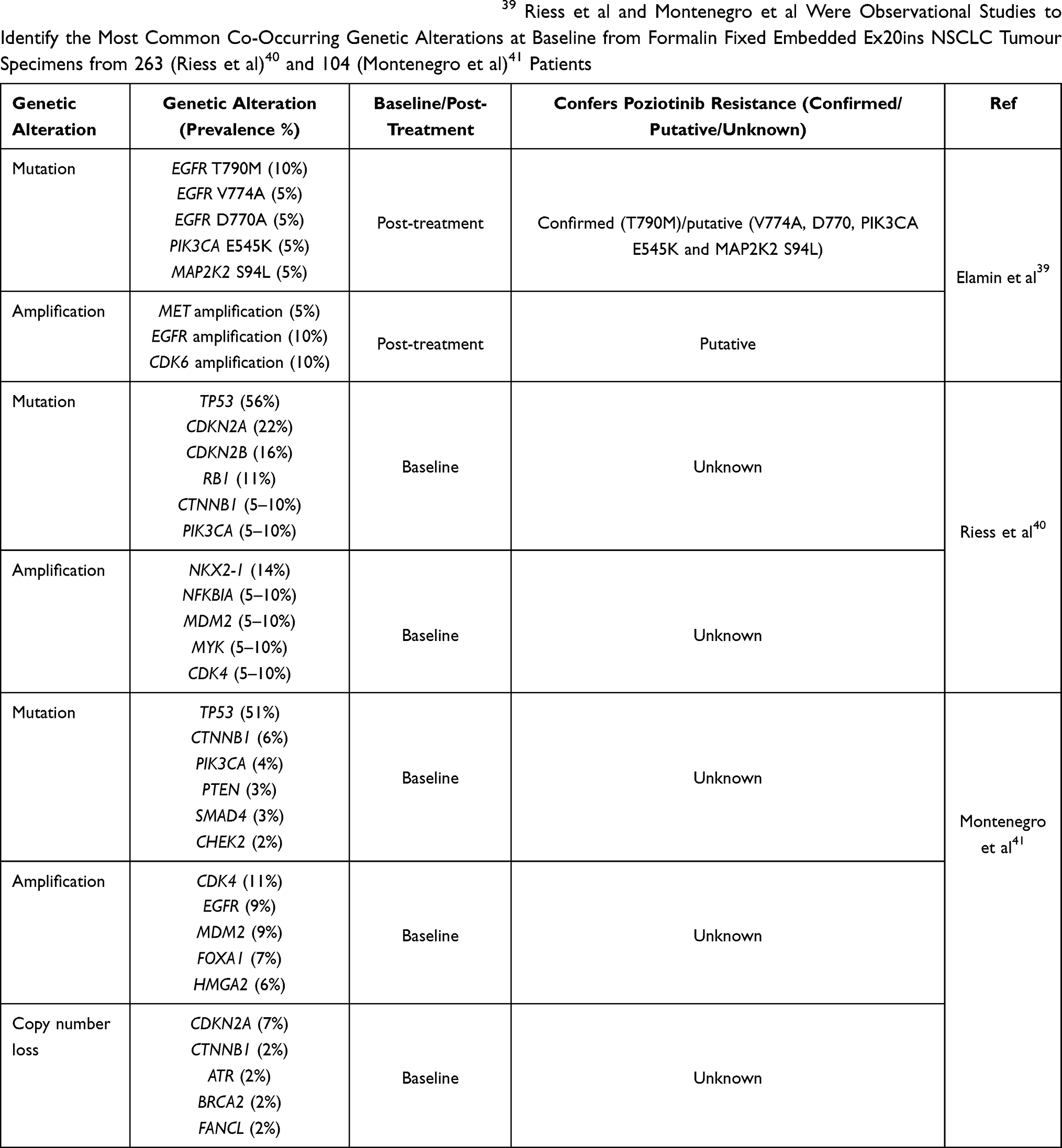 |
Table 2 The Prevalence of Baseline or Post-Treatment Resistance Associated Genetic Alterations in EGFR Ex20ins Patients. Studies Presented in the Table Utilized Different Sample Collection Methods, Elamin et al Evaluated Tumour Specimens Pre-Poziotinib and on Progression from 20 Patients Who Responded to Poziotinib.39 Riess et al and Montenegro et al Were Observational Studies to Identify the Most Common Co-Occurring Genetic Alterations at Baseline from Formalin Fixed Embedded Ex20ins NSCLC Tumour Specimens from 263 (Riess et al)40 and 104 (Montenegro et al)41 Patients |
On-Target Mechanisms of Resistance
A well-established mechanism of resistance to clinically approved EGFR inhibitors is the acquisition of on-target secondary mutations in EGFR, including the T790M gatekeeper and C797S point mutation. T790M is located in the ATP binding pocket and confers resistance to competitive first generation inhibitors by sterically hindering drug binding and increasing the affinity of mutant EGFR for ATP, thus decreasing the affinity and binding of reversible TKIs.42 This mutation can be effectively overcome with the irreversible inhibitor osimertinib, which covalently binds to the C797 residue of EGFR and shows greater selectivity for EGFR T790M mutations over wild-type EGFR.43,44 However 7% of NSCLC patients with classical EGFR mutations that are treated with osimertinib as a first line therapy develop the C797S mutation, the second most common mechanism of acquired resistance observed after MET amplification (15%).45 The C797S mutation renders osimertinib ineffective by preventing the formation of the key covalent bond between this irreversible inhibitor and the thiol group (-SH) of cysteine in the EGFR 797 residue.46–48 This second critical point mutation is a common mechanism of resistance to irreversible inhibitors which prevents permanent inactivation of the kinase.
On-target secondary mutations in EGFR have also been observed to confer resistance to Ex20ins targeting TKIs. A recent study from Elamin et al has shown that resistance to poziotinib can occur through the acquisition of T790M in pre-clinical models and EGFR Ex20ins NSCLC patients.39 The study found that co-expression of an Ex20ins (S768supSVD) and T790M in engineered Ba/F3 cells caused resistance to poziotinib. This study also analysed blood samples and biopsies collected at baseline and upon disease progression from 50 NSCLC Ex20ins patients enrolled in a poziotinib phase II clinical trial (NCT03066206). Patient samples were analysed using next generation sequencing. Of the 20 patients who went on to have disease progression, a number of on-target secondary mutations were observed. These mutations included T790M (n=2), V774A (n=1) and D770A (n=1).39
To determine whether poziotinib binds to EGFR via the C797 residue, Robichaux et al generated Ba/F3 cells engineered with the EGFR C797S point mutation.9 At the time of this study, the C797S mutation had only been observed in response to osimertinib in patients with classical EGFR mutations and T790M. The addition of C797S to classical EGFR mutants co-expressing T790M was found to confer poziotinib resistance (the half maximal inhibitory concentration IC50 >10 μM), indicating poziotinib does bind to the C797 residue.9
Mobocertinib has also been shown to inhibit EGFR and HER2 via covalent modification of EGFR C797 and its equivalent residue C805 in HER2, indicating point mutations in these amino acids may confer resistance to multiple Ex20ins TKIs.49 Koga et al demonstrated that the C805S secondary resistance mutation can confer poziotinib resistance in a Ba/F3 model with HER2 Ex20ins mutations (A775_G776insYVMA and G776delinsVC). Using N-ethyl-N-nitrosourea (ENU), a mutagen known to cause random mutations, poziotinib resistant clones were generated by growing ENU mutagenized Ba/F3 HER2 Ex20ins cells in the presence of poziotinib. All clones were sequenced to identify on-target secondary HER2 mutations and notably, C805S accounted for resistance in 31% of the resistant clones and was the only on-target secondary mutation identified. The acquisition of the C805S mutation was found to confer a 100-fold increased resistance to poziotinib. The authors demonstrated that luminespib was able to reduce the viability of HER2 Ex20ins mutant expressing cells regardless of the presence of the C805S on-target mutation.50 The mechanism through which the Hsp90 inhibitor overcomes this resistance mechanism was not explored by Koga et al, however the Hsp90 chaperone complex has been shown to protect cellular proteins from ubiquitin degradation. EGFR and HER2 Ex20ins mutants are reliant upon Hsp90 for maintaining mutant receptor protein levels51 and Hsp90 inhibitors have been shown to reduce expression of mutant EGFR and HER2 proteins.52
Compensatory Bypass Pathways
Co-occurring mutations and gene amplifications in alternative oncogenic drivers are also putative resistance mechanisms in cancers with EGFR activating mutations. Elamin et al identified that co-occurring mutations in the Kirsten rat sarcoma 2 viral oncogene homolog (KRAS) and the Erb-B2 receptor tyrosine kinase 4 (ErbB4) were observed in genetically engineered mouse models (GEMM) harbouring tumors expressing EGFR Ex20ins (D770insNPG), following treatment with poziotinib.39 Additionally, activation of the mitogen-activated protein kinase (MAPK), the mitogen-activated protein kinase kinase (MEK), AKT and ERK was elevated in GEMM tumors which progressed on poziotinib treatment compared to sensitive tumors, suggesting that acquired resistance to poziotinib is associated with the reactivation of MAPK and phosphatidylinositol 3-kinase (PI3K) pathways.39 The same study utilized Ex20ins NSCLC patient biopsies from a poziotinib phase II clinical trial which were taken prior to treatment and upon disease progression. 1/20 patients with Ex20ins NSCLC had an E545K mutation in phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) following progression on poziotinib. Mitogen-activated protein kinase 2 (MAPK2) S94L mutation (1 patient), MET amplification (1 patient) and cyclin dependent kinase 6 (CDK6) amplification (2 patients) were also identified.39 Notably, MET and CDK6 amplifications have been previously described as mechanisms of acquired resistance to approved EGFR TKIs and are potential druggable targets to overcome resistance to TKIs that target Ex20ins.53,54
Putative Mechanisms of Ex20ins TKI Resistance
Ex20ins TKIs are still undergoing the initial stages of clinical development and our understanding of their resistance mechanisms is limited. However, based on recent studies it appears that these compounds may share overlapping acquired resistance mechanisms to first- and third-generation EGFR inhibitors, including the acquisition of the point mutations T790M and C797S described above. These findings, together with the extensive knowledge gleaned from over a decade of clinical use of approved EGFR TKIs in NSCLC bearing classical mutations allows us to make informed predictions about additional potential acquired resistance mechanisms to TKIs that target Ex20ins. This is key to anticipating and forecasting effective therapeutic strategies to overcome drug resistance in this patient group. Here we will discuss two mechanisms that may play a role in the acquisition of resistance in the context of EGFR Ex20ins based on pre-clinical data from cellular models of classical EGFR mutations treated with first and third generation EGFR inhibitors, namely epithelial to mesenchymal transition (EMT) and drug tolerance. The cell line models and experimental design employed in these studies, and their EGFR mutational status, genomic alterations associated with resistance and drug dosing regimens are summarized in Table 3.
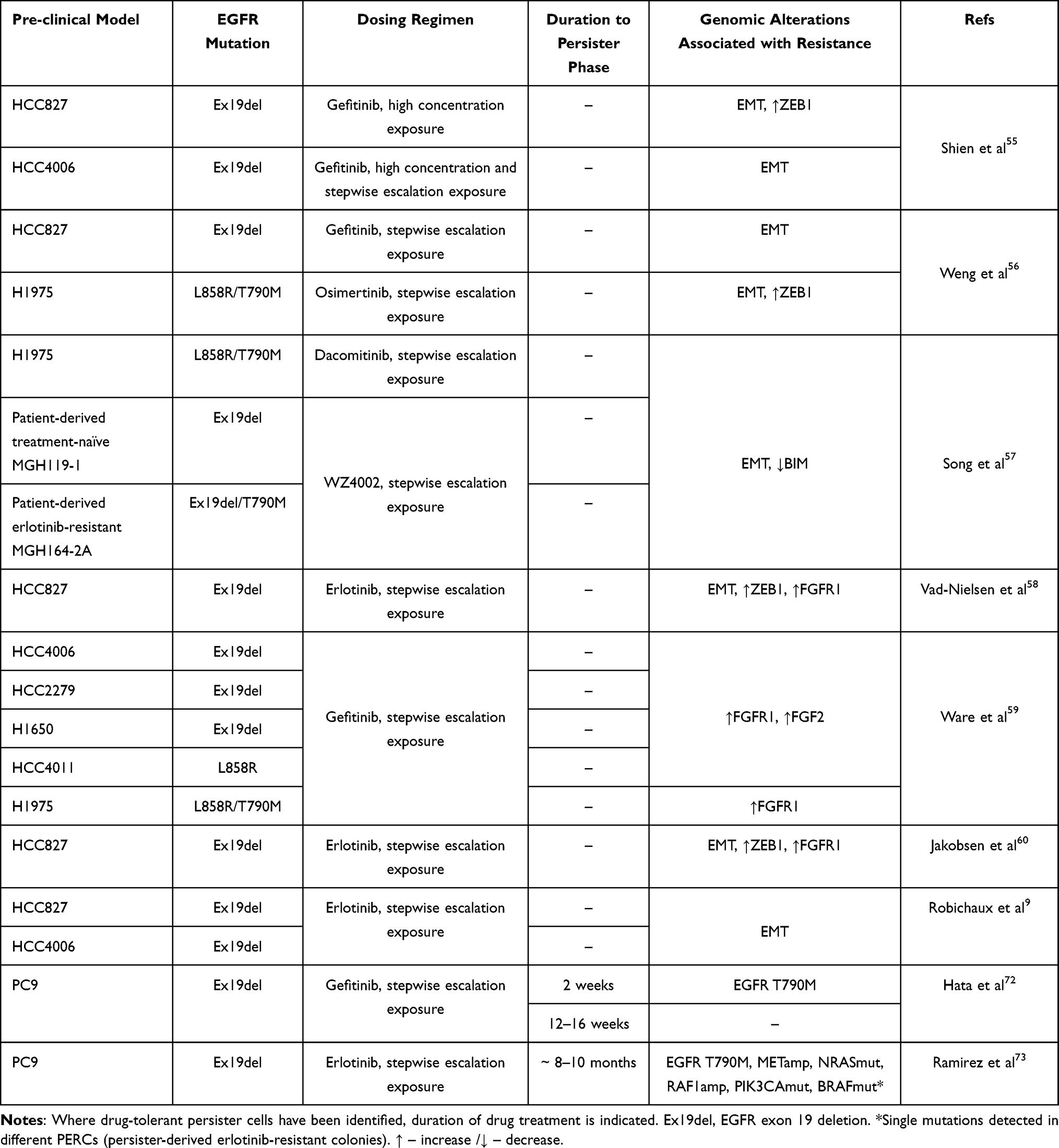 |
Table 3 A Summary of the Characteristics of EGFR Mutant Cell Line Models Employed in Studies to Investigate EGFR Inhibitor Resistance and the Reported Genomics Alterations Associated with Drug Resistance |
Epithelial to Mesenchymal Transition
EMT has been shown to confer resistance to clinically approved EGFR inhibitors in classical mutant EGFR NSCLC which lack EGFR on-target mutations or compensatory bypass mechanisms (Table 3).55 The acquisition of an EMT phenotype in response to gefitinib treatment has been observed both in vitro and in patients with a decrease in expression of the epithelial marker E-cadherin.56 EMT protects against EGFR-mediated TKI cell death through increased expression of the mesenchymal transcription factor zinc finger E-box binding homeobox 1 (ZEB1) which in turn inhibits the expression of the Bcl-2-like protein 11 (BIM). BIM is a pro-apoptotic protein required for EGFR TKI-induced apoptosis, therefore lower levels of BIM in cells that undergo EMT protect against EGFR TKI induced cell death.57 ZEB1 has also been linked to increased expression of the fibroblast growth factor receptor 1 (FGFR1) which is associated with resistance to EGFR inhibitors and often concomitant with EMT.58 Activation of an autocrine fibroblast growth factor 2 (FGF2)-FGFR1 growth loop drives resistance to EGFR TKIs through FGFR1-mediated activation of PI3K/AKT and MEK/ERK pathways.59 Moreover, FGFR1 inhibition has been shown to restore sensitivity to EGFR TKIs in acquired resistant cell models with an EMT phenotype.59,60 EMT has also been indirectly identified as a potential mechanism by which NSCLC cells can become resistant to poziotinib. NSCLC cell lines with classical EGFR mutations (HCC4006 (Ex19del) and HCC827 (Ex19del)) which underwent EMT in response to escalating concentrations of erlotinib were also resistant to poziotinib.9 Further preclinical and translational studies are required to determine if EMT is a bona fide mechanism of resistance in EGFR Ex20ins patients.
Drug Tolerance
It is now well established that the emergence of minimal residual disease can be attributed to a subpopulation of drug tolerant persister (DTP) cells.61,62 DTP cells are defined as the small subpopulation of cells that remain viable in the presence of anti-cancer treatments, despite not harboring classic genetic mutations commonly associated with drug resistance. They undergo a drug tolerant reversible state which has been observed in numerous cancer models in vitro and in vivo in response to drug pressure, suggesting a general phenomenon.63–68 Despite no evidence of on-target resistance mutations, drug sensitivity can be >100-fold less in DTP cells when compared to the bulk tumor cells. Although the properties of DTP cells have not been fully characterized, it has been demonstrated that these cells harbor specific epigenetic modifications and a reversible drug tolerant slow-growing phenotype.62,69 Experiments in several cell line models suggest that the ability of these DTP cells to maintain viability following drug exposure to both targeted therapy and chemotherapy involves a transient chromatin state dependent on insulin-like growth factor 1 receptor (IGF-1R) signaling, histone demethylase KDM5A and KDM6B activity and decreased histone acetylation.62,69 This reversible DTP state could also account for the re-sensitization of patient tumors to TKIs after the interruption of treatment for an extended period of time (drug holiday). For example, some NSCLC patients with classical EGFR mutations who respond well to treatment with gefitinib and later experience therapy failure, showed a second response to the same EGFR TKI after a drug holiday.70,71
DTP cells in NSCLC have been studied in vitro using the PC9 cell line (Ex19del mutation). Hata et al showed that acquired resistance to gefitinib can occur as a result of either pre-existing EGFR T790M containing cellular subpopulations or from initially T790M-negative DTP cells.72 These DTP cells provide a reservoir of cells that can then acquire de novo T790M or other resistance-associated mutations after prolonged exposure to gefitinib. The cells also showed diminished apoptosis after exposure to osimertinib, indicating they may be less responsive to third-generation EGFR inhibitors.72 A second study explored the evolution of PC9 DTP cells derived DTP cells from a single clonal population after prolonged exposure to erlotinib.73 Different DTP cells derived from the same clonal population were found to acquire a diverse set of resistance mechanisms, including those most commonly observed in NSCLC patients in the clinic such as EGFR T790M mutation and MET amplification. These data suggest that different genetic and epigenetic drug resistance mechanisms can arise independently within the same initial cell population passing through the persister bottleneck, thereby complicating strategies to overcome resistance.73
Given that DTP cells have been observed in response to clinically approved EGFR inhibitors, it is tempting to speculate that a similar phenomenon may be seen in EGFR Ex20ins tumors. Upon treatment with Ex20ins-targeted TKIs, a small subpopulation of clones may enter a resistant slow-growing state facilitating escape from drug pressure. Multiple de novo resistance mechanisms can then arise in these DTP clones which will allow them to revert to a fast-growing state, eventually becoming the dominant population in a relapsed tumor.73 Understanding the biological mechanisms driving the evolution of DTP cells will undoubtedly help in the design of more effective upfront therapeutic strategies for EGFR Ex20ins patients.
Future Perspectives
Given the dose limiting toxicities in the current generation of EGFR Ex20ins TKIs, there is an urgent need for new compounds with a wider therapeutic index which are both effective and safe for use in Ex20ins patients. Furthermore, it is also essential to identify innovative approaches to overcome key resistance mechanisms anticipated with the current generation of Ex20ins TKIs. In this section we describe new methods to discover next-generation compounds which may be more effective in the treatment of Ex20ins patients including proteolysis targeting chimeras (PROTACs) and the mammalian membrane two-hybrid drug screen (MaMTH-DS) methodology. We also outline recent advances in monoclonal antibodies (mAb) combinations targeting on-target EGFR resistance mutants and explore new therapeutic opportunities in overcoming DTP tumor cells in patients.
PROTACs
PROTACs are valuable tools for the discovery of EGFR Ex20ins targeting agents. PROTACs consist of a bifunctional molecule containing a target protein binding ligand and an E3 ligase ligand which are bridged by a crosslinker. After the formation of a ternary complex composed of the protein target, PROTAC and E3 ligase, the ubiquitin proteasome system is recruited to degrade the protein of interest (in this case a transmembrane RTK). After degradation, the bifunctional PROTAC molecule is released and can enter the next degradation cycle, allowing a sustained reduction in receptor signaling and providing potential for PROTAC activity at lower concentrations than comparable TKIs.74–76 Promising PROTACs have been reported for mutant EGFR in various cellular models.77,78 Burslem et al described the development of a PROTAC for RTKs based on the reversible EGFR/HER2 inhibitor lapatinib by using a ligand that binds to the E3 ligase, VHL (von Hippel-Lindau).77 Interestingly, this lapatinib-based compound was also shown to be capable of degrading EGFR Ex20ins protein (ASV duplication) in engineered HeLa cells.77 By virtue of the ability of PROTACs to degrade EGFR rather than just inhibit its kinase activity, the authors showed that PROTACs offered several advantages over conventional TKIs. This included marked improvement in potency in preclinical models as well as sustained inactivation of downstream effector signaling compared to kinase inhibition by TKIs. These effects minimize compensatory pathway activation and could circumvent kinome rewiring which is a frequently observed resistance mechanism in response to TKIs (Figure 3).77 However, phase I clinical trials of PROTACs have yet to report on the safety profile of these compounds. PROTACs have the potential to cause adverse clinical effects due to prolonged on-target and off-target protein degradation.79–82 For example, proteins that are part of the same complex or in close proximity with the target protein can be degraded even if not directly bound to a PROTAC.83 In addition disruption of cellular proteostasis can occur, through either competition with endogenous E3 binding substrates or accumulation of ubiquitinated proteins which can saturate the proteolysis machinery.84 Finally, some proteins are refractory to PROTAC-mediated degradation, which may limit the suitability of this therapeutic strategy for targeting certain oncogenes.85,86
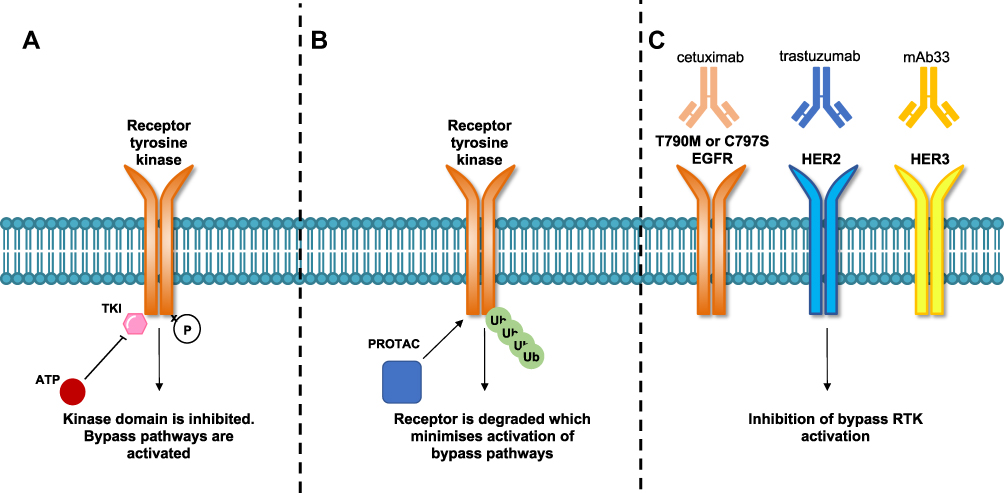 |
Figure 3 Comparison of tyrosine kinase inhibitor, PROTAC and therapeutic monoclonal antibodies mechanism of action. (A) TKIs bind to the kinase domain of the receptor which inhibits receptor phosphorylation and activation. Upon acquisition of drug resistance either develops on-target mutations or activate compensatory bypass pathways. (B) PROTACs degrade tyrosine kinase receptors through protein ubiquitination and receptor degradation. The degradation of the receptor is thought to minimize the activation of compensatory bypass pathways. (C) A combination of three monoclonal antibodies can target T790M and C797S mutant EGFR tumors. Cetuximab (EGFR), trastuzumab (HER2) and mAb33 (HER3) when used together were shown to suppress HER2, HER3, MET and AXL compensatory bypass pathway activation.91 |
MaMTH-DS
MaMTH-DS is a split-ubiquitin-based-technology which has recently been used to identify new EGFR targeting agents. It involves a high-throughput screening methodology that is based on targeting functional RTK protein-protein interactions.87 Rather than relying on classical in vitro kinase methods, this drug discovery platform utilizes full-length integral membrane proteins in their natural membrane context in live mammalian cells. In this assay, cells are transfected to stably express a bait RTK which is fused to the C-terminus of ubiquitin and an artificial transcription factor. In addition, the Src homology 2 domain-containing adaptor protein 1 (Shc1) is fused to the N-terminus of ubiquitin and expressed as the prey due to its ability to interact with a wide variety of phosphorylated RTKs. Upon activation of the bait RTK, proteolytic cleavage and release of the transcription factor leads to the activation of a luciferase reporter system. This methodology provides a useful strategy to identify inhibitors that block RTK phosphorylation resulting in a reduction in the luciferase readout. As proof of principle, Saraon et al used this platform to screen a EGFR inhibitor resistant Exon19del/T790M/C797S triple mutant NSCLC model against a library of 2960 small molecules.88 They identified 4 new compounds that inhibit this triple mutant which is resistant to irreversible EGFR inhibitors including poziotinib. Importantly, two of these compounds, AZD7762 and EMI1, would not have been identified using in vitro kinase assays. For instance, the specificity of the checkpoint kinase (Chk) inhibitor AZD7762 for mutant EGFR depends on additional factors only present in the live-cell format while the mechanisms of action of the small molecule EMI1 is reliant on direct inhibition of microtubule polymerization, which indirectly affects mutant but not wild-type EGFR signaling and trafficking. This work demonstrates the utility and potential of MaMTH-DS as a screening platform that could be used to identify new candidate drugs for Ex20ins and associated on-target resistance mutations.
Therapeutic Monoclonal Antibodies
MAbs represent an important component in the arsenal of targeted cancer therapy for NSCLC treatment. MAbs that bind to the extracellular domain of EGFR are not affected by the acquisition of common on-target resistance mechanisms (eg T790M or C797S) that are found in the intracellular domain of the receptor. Cetuximab is a mAb that binds to the extracellular domain of EGFR, preventing ligand binding and blocking receptor activation.89 Experimental strategies able to overcome EGFR T790M or C797S resistant mutants have exploited the combinatorial use of MAbs, such as cetuximab, trastuzumab (anti-HER2 mAb) and mAb33 (anti-HER3 mAb).90 In particular, it has been shown that a triple combination of mAbs (3xmAbs) that simultaneously target EGFR, HER2 and HER3 inhibited tumor growth with low toxicity in a xenograft NSCLC model with classical EGFR mutations in combination with T790M.91 In tumors which had acquired T790M, the 3xmAbs combination was shown to inhibit tumor growth in a similar fashion to osimertinib, but through a mechanism of cell senescence rather than apoptosis. This mAb combination overcame resistance to osimertinib in tumors that either expressed C797S or upregulated HER2 and HER3 as compensatory bypass mechanisms (Figure 3).91 In another study from the same group, the combination of the TKI osimertinib and mAbs cetuximab and trastuzumab had a long-lasting effect in preventing onset of resistance to osimertinib by suppressing signaling from compensatory RTKs, such as HER2, HER3, MET and AXL.92 These findings suggest that the combinatorial mAbs strategy may offer a feasible pharmacological option for treating Ex20ins lung cancer patients that develop both on-target and bypass resistance mechanisms to TKIs such as poziotinib. Limited clinical evidence for the efficacy of afatinib in combination with cetuximab has been reported in patients with EGFR Ex20ins,93 however the 3xmAbs combination has yet to be assessed in this context.
Therapeutic Targeting of DTP Cells
In order to fully tackle the challenge of drug resistance and tumor relapse, it will be necessary to identify ways to effectively overcome DTP cells and residual disease following EGFR TKI treatment. The DTP cell state is reliant upon specific signaling pathways and epigenetic alterations, which present a therapeutic opportunity for drugs that can target these dependencies. A study from Rusan et al showed that the DTP cellular state is transcriptionally addicted to specific genes and pathways in a variety of cancer models.94 In the PC9 cell line, the authors found that DTP cells arising from erlotinib treatment could be targeted by combining erlotinib with THZ1, which is a CDK7/12 inhibitor that blocks the transcriptional response in DTP cells (Figure 4). A genome-wide CRISPR/Cas9 screen performed in PC9 treated with erlotinib in combination with THZ1 demonstrated that suppression of genes associated with transcriptional complexes (such as EP300 or CREBBP) enhanced the THZ1/erlotinib therapeutic synergy. In addition, a new drug tolerant pathway associated with the dysregulation of UFMylation protein response and endoplasmic reticulum (ER) stress was characterized using this approach.95 Components of the post-translational UFMylation pathway have only recently been characterized. They play an important role in cell survival as regulators of ER homeostasis and are linked to several types of cancer including lung cancer.96,97 Suppressing expression of genes involved in the UFMylation pathway protects DTP cells against THZ1 and erlotinib combination treatment by promoting a protective unfolded protein response (UPR) associated with the stimulator of interferon response CGAMP interactor 1 (STING) upregulation. This triggers pro-tumorigenic inflammatory signaling and dependency on the apoptotic repressor B-cell lymphoma-extra large (Bcl-xL).94 The dysregulation of the UFMylation pathway and ER stress response is a key TKI drug tolerance pathway that activates survival signaling which could be therapeutically exploited, however further work is required to identify whether similar DTP cellular pathways are present in Ex20ins tumors.
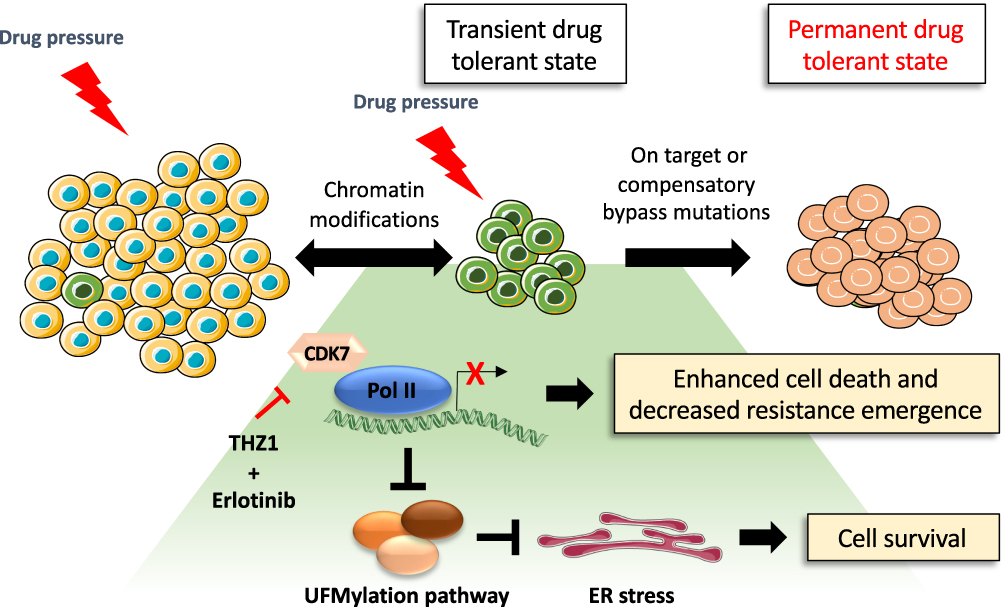 |
Figure 4 Proposed model of drug tolerant persister cell evolution. Under drug pressure a subpopulation of transient drug tolerant persister cells can emerge through epigenetic mechanisms. This transient DTP population can acquire permanent genetic modifications which allows for the emergence of a drug tolerant population. The transcriptionally dependent state of persister cells induced by targeted therapy can be exploited by the treatment with THZ1 which blocks transcriptional responses, promoting cancer cell death.94 However, THZ1 treatment in combination with erlotinib suppresses the expression of the UFMylation pathway components which can trigger a protective unfolded protein response associated with tolerable levels of ER stress and cell survival.94 |
Conclusion
The current generation of TKIs capable of targeting Ex20ins has shown preclinical promise in the treatment of this rare group of NSCLC patients. However, early clinical data finds that this strategy suffers from a poor therapeutic index and inevitable primary and acquired drug resistance. Recent pre-clinical and clinical studies indicate that resistance may be acquired through the acquisition of EGFR on-target mutations or the activation of compensatory bypass pathways.39 In some cases, resistance mechanisms that mirror what has been observed with the common classical EGFR activating mutants in response to clinically approved EGFR inhibitors are applicable to this current generation of Ex20ins TKIs. But there is still a large gap in our knowledge of the myriad ways in which these tumors evolve when subjected to drug selection. In addressing this class of mutations, there is clearly a twin challenge of not only identifying a new generation of drugs with a better therapeutic index but also developing an in-depth understanding of the spectrum of biological mechanisms of drug resistance. The advent of new drug discovery tools, such as MaMTH-DS and PROTACs technology, should facilitate the rapid identification of new therapeutics that might ultimately be useful as first-line or salvage therapy; while a better understanding of mechanisms of resistance arising from residual DTP cells may hold the key to achieving durable responses in this NSCLC patient group of unmet need.
Acknowledgments
This work was supported by grants from the Institute of Cancer Research (ICR) and Cancer Research UK (C36478/A19281). Laura Pacini, Andrew Jenks, and Simon Vyse are co-first authors for this work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. doi:10.3322/caac.21262
2. Duma N, Santana-Davila R, Molina JR. Non–small cell lung cancer: epidemiology, screening, diagnosis, and treatment. Mayo Clin Proc. 2019;94(8):1623–1640. doi:10.1016/j.mayocp.2019.01.013
3. Pakkala S, Ramalingam SS. Personalized therapy for lung cancer: striking a moving target. JCI Insight. 2018;3:15. doi:10.1172/jci.insight.120858
4. D’Angelo SP, Pietanza MC, Johnson ML, et al. Incidence of EGFR exon 19 deletions and L858R in tumor specimens from men and cigarette smokers with lung adenocarcinomas. J Clin Oncol. 2011;29(15):2066–2070. doi:10.1200/JCO.2010.32.6181
5. Oxnard GR, Lo PC, Nishino M, et al. Natural history and molecular characteristics of lung cancers harboring EGFR exon 20 insertions. J Thorac Oncol. 2013;8(2):179–184. doi:10.1097/JTO.0b013e3182779d18
6. Yasuda H, Kobayashi S, Costa DB. EGFR exon 20 insertion mutations in non-small-cell lung cancer: preclinical data and clinical implications. Lancet Oncol. 2012;13(1):23–31. doi:10.1016/S1470-2045(11)70129-2
7. Harrison PT, Vyse S, Huang PH. Rare epidermal growth factor receptor (EGFR) mutations in non-small cell lung cancer. Semin Cancer Biol. 2020;61:167–179. doi:10.1016/j.semcancer.2019.09.015
8. Vyse S, Huang PH. Targeting EGFR exon 20 insertion mutations in non-small cell lung cancer. Signal Transduct Target Ther. 2019;4(1):5. doi:10.1038/s41392-019-0038-9
9. Robichaux JP, Elamin YY, Tan Z, et al. Mechanisms and clinical activity of an EGFR and HER2 exon 20-selective kinase inhibitor in non-small cell lung cancer. Nat Med. 2018;24(5):638–646. doi:10.1038/s41591-018-0007-9
10. Udager AM, Rolland DCM, McHugh JB, et al. High-frequency targetable EGFR mutations in sinonasal squamous cell carcinomas arising from inverted sinonasal papilloma. Cancer Res. 2015;75(13):2600–2606. doi:10.1158/0008-5472.CAN-15-0340
11. Madison RW, Gupta SV, Elamin YY, et al. Urothelial cancer harbours EGFR and HER2 amplifications and exon 20 insertions. BJU Int. 2020;125(5):739–746. doi:10.1111/bju.15006
12. Weinstein IB, Joe A. Oncogene addiction. Cancer Res. 2008;68(9):3077–3080. doi:10.1158/0008-5472.CAN-07-3293
13. Arcila ME, Nafa K, Chaft JE, et al. EGFR exon 20 insertion mutations in lung adenocarcinomas: prevalence, molecular heterogeneity, and clinicopathologic characteristics. Mol Cancer Ther. 2013;12(2):220–229. doi:10.1158/1535-7163.MCT-12-0620
14. Yasuda H, Park E, Yun CH, et al. Structural, biochemical, and clinical characterization of epidermal growth factor receptor (EGFR) exon 20 insertion mutations in lung cancer. Sci Transl Med. 2013;5(216):216ra177. doi:10.1126/scitranslmed.3007205
15. Voon PJ, Tsui DWY, Rosenfeld N, Chin TM. Letter to Editor: EGFR Exon 20 Insertion A763-Y764insFQEA and response to Erlotinib. Mol Cancer Ther. 2013;12:20–25. doi:10.1158/1535-7163.MCT-13-0192
16. Schiller JH, Harrington D, Belani CP, et al. Comparison of four chemotherapy regimens for advanced non–small-cell lung cancer. N Engl J Med. 2002;346(2):92–98. doi:10.1056/nejmoa011954
17. Noronha V, Choughule A, Patil VM, et al. Epidermal growth factor receptor exon 20 mutation in lung cancer: types, incidence, clinical features and impact on treatment. Onco Targets Ther. 2017;10:2903–2908. doi:10.2147/OTT.S133245
18. Yang G, Li J, Xu H, et al. EGFR exon 20 insertion mutations in Chinese advanced non-small cell lung cancer patients: molecular heterogeneity and treatment outcome from nationwide real-world study. Lung Cancer. 2020;145:186–194. doi:10.1016/j.lungcan.2020.03.014
19. Doebele RC, Riely GJ, Spira AI, et al. First report of safety, PK, and preliminary antitumor activity of the oral EGFR/HER2 exon 20 inhibitor TAK-788 (AP32788) in non–small cell lung cancer (NSCLC). J Clin Oncol. 2018;36(suppl):abstr 9015.
20. Udagawa H, Hasako S, Ohashi A, et al. TAS6417/CLN-081 Is a pan-mutation-selective EGFR tyrosine kinase inhibitor with a broad spectrum of preclinical activity against clinically relevant EGFR mutations. Mol Cancer Res. 2019;17(11):2233–2243. doi:10.1158/1541-7786.MCR-19-0419
21. Park K, John T, Kim S-W, et al. Amivantamab (JNJ-61186372), an anti-EGFR-MET bispecific antibody, in patients with EGFR exon 20 insertion (exon20ins)-mutated non-small cell lung cancer (NSCLC). J Clin Oncol. 2020;38(15_suppl):9512. doi:10.1200/jco.2020.38.15_suppl.9512
22. Piotrowska Z, Costa DB, Oxnard GR, et al. Activity of the Hsp90 inhibitor luminespib among non-small-cell lung cancers harboring EGFR exon 20 insertions. Ann Oncol. 2018;29(10):2092–2097. doi:10.1093/annonc/mdy336
23. Gonzalvez F. TAK-788: an EGFR inhibitor, currently in Phase II clinical trials, targeting lung cancers with Exon20 insertion mutations.
24. Elamin Y, Robichaux J, Heymach J. Preliminary results of a Phase II study of poziotinib in EGFR Exon 20 mutant advanced NSCLC. J Thorac Oncol. 2017;12(8):S1536. doi:10.1016/j.jtho.2017.06.020
25. Heymach J, Negrao M, Robichaux J, et al. OA02.06 A Phase II Trial of Poziotinib in EGFR and HER2 exon 20 mutant Non-Small Cell Lung Cancer (NSCLC). AACR. 2020;(10):S323–S324. doi:10.1016/j.jtho.2018.08.243
26. Leduc C, Merlio JP, Besse B, et al. Clinical and molecular characteristics of non-small-cell lung cancer (NSCLC) harboring EGFR mutation: results of the nationwide French Cooperative Thoracic Intergroup (IFCT) program. Ann Oncol. 2017;28(11):2715–2724. doi:10.1093/annonc/mdx404
27. Le X, Goldman JW, Clarke JM, et al. Poziotinib shows activity and durability of responses in subgroups of previously treated EGFR exon 20 NSCLC patients. J Clin Oncol. 2020;38(15_suppl):9514. doi:10.1200/jco.2020.38.15_suppl.9514
28. Janne PA, Neal JW, Camidge DR, et al. Antitumor activity of TAK-788 in NSCLC with EGFR exon 20 insertions. J Clin Oncol. 2019;37(15_suppl):9007. doi:10.1200/JCO.2019.37.15_suppl.9007
29. ClinicalTrials.gov. TAK-788 as first-line treatment versus platinum-based chemotherapy for Non-Small Cell Lung Cancer (NSCLC) With EGFR Exon 20 insertion mutations. Available from: https://clinicaltrials.gov/ct2/show/NCT04129502. Accessed Dec 15, 2020.
30. Hasako S, Terasaka M, Abe N, et al. TAS6417, A novel EGFR inhibitor targeting Exon 20 insertion mutations. Mol Cancer Ther. 2018;17(8):1648–1658. doi:10.1158/1535-7163.MCT-17-1206
31. Yun J, Lee S-H, Kim S-Y, et al. Antitumor activity of amivantamab (JNJ-61186372), an EGFR-cMet bispecific antibody, in diverse models of EGFR Exon 20 insertion-driven NSCLC. Cancer Discov. 2020. doi:10.1158/2159-8290.CD-20-0116
32. A study of combination amivantamab and carboplatin-pemetrexed therapy, compared with carboplatin-pemetrexed, in participants with advanced or metastatic non-small cell lung cancer characterized by Epidermal Growth Factor Receptor (EGFR) Exon 20 insertions - full text view - ClinicalTrials.gov. Availalble from: https://clinicaltrials.gov/ct2/show/NCT04538664?term=amivantamab&draw=2&rank=1.
33. Felip E, Barlesi F, Besse B, et al. Phase 2 study of the HSP-90 Inhibitor AUY922 in previously treated and molecularly defined patients with advanced non–small cell lung cancer. J Thorac Oncol. 2018;13(4):576–584. doi:10.1016/j.jtho.2017.11.131
34. Jorge SE, Lucena-Araujo AR, Yasuda H, et al. EGFR Exon 20 insertion mutations display sensitivity to Hsp90 inhibition in preclinical models and lung adenocarcinomas. Clin Cancer Res. 2018;24:6548–6555. doi:10.1158/1078-0432.CCR-18-1541
35. Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13(3):239–246. doi:10.1016/s1470-2045(11)70393-x
36. Mok TS, Wu Y-L, Thongprasert S, et al. Gefitinib or carboplatin–paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361(10):947–957. doi:10.1056/NEJMoa0810699
37. Soria J-C, Ohe Y, Vansteenkiste J, et al. Osimertinib in untreated EGFR -mutated advanced non–small-cell lung cancer. N Engl J Med. 2018;378(2):113–125. doi:10.1056/NEJMoa1713137
38. Mok TS, Wu Y-L, Ahn M-J, et al. Osimertinib or platinum–pemetrexed in EGFR T790M–positive lung cancer. N Engl J Med. 2017;376(7):629–640. doi:10.1056/nejmoa1612674
39. Elamin Y, Robichaux J, Carter B, et al. MA09.03 identification of mechanisms of acquired resistance to poziotinib in EGFR Exon 20 mutant Non-Small Cell Lung Cancer (NSCLC). J Thorac Oncol. 2019;14(10):S282–S283. doi:10.1016/j.jtho.2019.08.567
40. Riess JW, Gandara DR, Frampton GM, et al. Diverse EGFR Exon 20 insertions and co-occurring molecular alterations identified by comprehensive genomic profiling of NSCLC. J Thorac Oncol. 2018;13(10):1560–1568. doi:10.1016/j.jtho.2018.06.019
41. Montenegro GB, Nagasaka M, Ma P, et al. P2.01-100 spectrum of EGFR Exon 20 insertion mutations and co-occurring genetic alterations in patients with non-small-cell lung cancer. J Thorac Oncol. 2019;14(10):S680. doi:10.1016/j.jtho.2019.08.1443
42. Yun C-H, Mengwasser KE, Toms AV, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci. 2008;105(6):2070–2075. doi:10.1073/pnas.0709662105
43. Yosaatmadja Y, Silva S, Dickson JM, et al. Binding mode of the breakthrough inhibitor AZD9291 to epidermal growth factor receptor revealed. J Struct Biol. 2015;192(3):539–544. doi:10.1016/j.jsb.2015.10.018
44. Zhou W, Ercan D, Chen L, et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature. 2010;462(7276):1070–1074. doi:10.1038/nature08622
45. Ramalingam SS, Cheng Y, Zhou C, et al. Mechanisms of acquired resistance to first-line osimertinib: preliminary data from the phase III FLAURA study. Ann Oncol. 2018;29:viii740. doi:10.1093/annonc/mdy424.063
46. Butterworth S, Cross DAE, Finlay MRV, Ward RA, Waring MJ. The structure-guided discovery of osimertinib: the first U.S. FDA approved mutant selective inhibitor of EGFR T790M. Medchemcomm. 2017;8(5):820–822. doi:10.1039/c7md90012k
47. Thress KS, Paweletz CP, Felip E, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med. 2015;21(6):560–562. doi:10.1038/nm.3854
48. Ercan D, Choi HG, Yun C-H, et al. EGFR mutations and resistance to irreversible pyrimidine-based EGFR inhibitors. Clin Cancer Res. 2015;21(17):3913–3923. doi:10.1158/1078-0432.CCR-14-2789
49. Chouitar J, Vincent S, Brake R, Li S. P2.13-32 TAK-788 is a novel and potent tyrosine kinase inhibitor with selective activity against EGFR/HER2. J Thorac Oncol. 2018;13(10):S811. doi:10.1016/j.jtho.2018.08.1427
50. Koga T, Kobayashi Y, Tomizawa K, et al. Activity of a novel HER2 inhibitor, poziotinib, for HER2 exon 20 mutations in lung cancer and mechanism of acquired resistance: An in vitro study. Lung Cancer. 2018;126:72–79. doi:10.1016/j.lungcan.2018.10.019
51. Shimamura T, Shapiro GI. Heat shock protein 90 inhibition in lung cancer. J Thorac Oncol. 2008;3(6 SUPPL 2):152–159. doi:10.1097/JTO.0b013e318174ea3a
52. Xu W, Soga S, Beebe K, et al. Sensitivity of epidermal growth factor receptor and ErbB2 exon 20 insertion mutants to Hsp90 inhibition. Br J Cancer. 2007;97(6):741–744. doi:10.1038/sj.bjc.6603950
53. Qin Q, Li X, Liang X, et al. CDK46 inhibitor palbociclib overcomes acquired resistance to third-generation EGFR inhibitor osimertinib in non-small cell lung cancer (NSCLC). Thorac Cancer. 2020;11(9):2389–2397. doi:10.1111/1759-7714.13521
54. Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316(5827):1039–1043. doi:10.1126/science.1141478
55. Shien K, Toyooka S, Yamamoto H, et al. Acquired resistance to EGFR inhibitors is associated with a manifestation of stem cell-like properties in cancer cells. Cancer Res. 2015;73(10):3051–3061. doi:10.1158/0008-5472.CAN-12-4136
56. Weng CH, Chen LY, Lin YC, et al. Epithelial-mesenchymal transition (EMT) beyond EGFR mutations per se is a common mechanism for acquired resistance to EGFR TKI. Oncogene. 2019;38(4):455–468. doi:10.1038/s41388-018-0454-2
57. Song KA, Niederst MJ, Lochmann TL, et al. Epithelial-to-mesenchymal transition antagonizes response to targeted therapies in lung cancer by suppressing BIM. Clin Cancer Res. 2018;24(1):197–208. doi:10.1158/1078-0432.CCR-17-1577
58. Vad-Nielsen J, Gammelgaard KR, Daugaard TF, Nielsen AL. Cause-and-effect relationship between FGFR1 expression and epithelial-mesenchymal transition in EGFR-mutated non-small cell lung cancer cells. Lung Cancer. 2019;132:132–140. doi:10.1016/j.lungcan.2019.04.023
59. Ware KE, Hinz TK, Kleczko E, et al. A mechanism of resistance to gefitinib mediated by cellular reprogramming and the acquisition of an FGF2-FGFR1 autocrine growth loop. Oncogenesis. 2013;2(3):e39–e39. doi:10.1038/oncsis.2013.4
60. Jakobsen KR, Demuth C, Madsen AT, et al. MET amplification and epithelial-to-mesenchymal transition exist as parallel resistance mechanisms in erlotinib-resistant, EGFR-mutated, NSCLC HCC827 cells. Oncogenesis. 2017;6(4):e307. doi:10.1038/oncsis.2017.17
61. Bivona TG, Doebele RC. A framework for understanding and targeting residual disease in oncogene-driven solid cancers. Nat Med. 2016;22(5):472–478. doi:10.1038/nm.4091
62. Sharma SV, Lee DY, Li B, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141(1):69–80. doi:10.1016/j.cell.2010.02.027
63. Chen J, Li Y, Yu TS, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488(7412):522–526. doi:10.1038/nature11287
64. Fallahi‐Sichani M, Becker V, Izar B, et al. Adaptive resistance of melanoma cells to RAF inhibition via reversible induction of a slowly dividing de‐differentiated state. Mol Syst Biol. 2017;13(1):905. doi:10.15252/msb.20166796
65. Su Y, Wei W, Robert L, et al. Single-cell analysis resolves the cell state transition and signaling dynamics associated with melanoma drug-induced resistance. Proc Natl Acad Sci U S A. 2017;114(52):13679–13684. doi:10.1073/pnas.1712064115
66. Liau BB, Sievers C, Donohue LK, et al. Adaptive chromatin remodeling drives glioblastoma stem cell plasticity and drug tolerance. Cell Stem Cell. 2017;20(2):233–246.e7. doi:10.1016/j.stem.2016.11.003
67. Jordan NV, Bardia A, Wittner BS, et al. HER2 expression identifies dynamic functional states within circulating breast cancer cells. Nature. 2016;537(7618):102–106. doi:10.1038/nature19328
68. Roesch A, Vultur A, Bogeski I, et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1Bhigh cells. Cancer Cell. 2013;23(6):811–825. doi:10.1016/j.ccr.2013.05.003
69. Roesch A, Fukunaga-Kalabis M, Schmidt EC, et al. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell. 2010;141(4):583–594. doi:10.1016/j.cell.2010.04.020
70. Kurata T, Tamura K, Kaneda H, et al. Effect of re-treatment with gefitinib (“Iressa”, ZD1839) after acquisition of resistance [1]. Ann Oncol. 2004;15(1):173. doi:10.1093/annonc/mdh006
71. Oh IJ, Ban HJ, Kim KS, Kim YC. Retreatment of gefitinib in patients with non-small-cell lung cancer who previously controlled to gefitinib: a single-arm, open-label, phase II study. Lung Cancer. 2012;77(1):121–127. doi:10.1016/j.lungcan.2012.01.012
72. Hata AN, Niederst MJ, Archibald HL, et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat Med. 2016;22(3):262–269. doi:10.1038/nm.4040
73. Ramirez M, Rajaram S, Steininger RJ, et al. Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nat Commun. 2016;7:10690. doi:10.1038/ncomms10690
74. Burslem GM, Crews CM. Proteolysis-targeting chimeras as therapeutics and tools for biological discovery. Cell. 2020;181(1):102–114. doi:10.1016/j.cell.2019.11.031
75. Hu B, Zhou Y, Sun D, et al. PROTACs: new method to degrade transcription regulating proteins. Eur J Med Chem. 2020:207. doi:10.1016/j.ejmech.2020.112698.
76. An S, Fu L. Small-molecule PROTACs: an emerging and promising approach for the development of targeted therapy drugs. EBioMedicine. 2018;36:553–562. doi:10.1016/j.ebiom.2018.09.005
77. Burslem GM, Smith BE, Lai AC, Crew AP, Hines J, Crews Correspondence CM. The advantages of targeted protein degradation over inhibition: an RTK case study. Cell Chem Biol. 2018;25(67–77.e3):67–77.e3. doi:10.1016/j.chembiol.2017.09.009
78. Zhang H, Zhao HY, Xi XX, et al. Discovery of potent epidermal growth factor receptor (EGFR) degraders by proteolysis targeting chimera (PROTAC). Eur J Med Chem. 2020:189. doi:10.1016/j.ejmech.2020.112061.
79. Zorba A, Nguyen C, Xu Y, et al. Delineating the role of cooperativity in the design of potent PROTACs for BTK. Proc Natl Acad Sci U S A. 2018;115(31):E7285–E7292. doi:10.1073/pnas.1803662115
80. Matyskiela ME, Lu G, Ito T, et al. A novel cereblon modulator recruits GSPT1 to the CRL4 CRBN ubiquitin ligase. Nature. 2016;535(7611):252–257. doi:10.1038/nature18611
81. He Y, Khan S, Huo Z, et al. Proteolysis targeting chimeras (PROTACs) are emerging therapeutics for hematologic malignancies. J Hematol Oncol. 2020;13(1):1–24. doi:10.1186/s13045-020-00924-z
82. Bondeson DP, Smith BE, Burslem GM, et al. Lessons in PROTAC design from selective degradation with a promiscuous warhead. Cell Chem Biol. 2018;25(1):78–87.e5. doi:10.1016/j.chembiol.2017.09.010
83. Hsu JHR, Rasmusson T, Robinson J, et al. EED-targeted PROTACs degrade EED, EZH2, and SUZ12 in the PRC2 Complex. Cell Chem Biol. 2020;27(1):41–46.e17. doi:10.1016/j.chembiol.2019.11.004
84. Moreau K, Coen M, Zhang AX, et al. Proteolysis‐targeting chimeras in drug development: a safety perspective. Br J Pharmacol. 2020;177(8):1709–1718. doi:10.1111/bph.15014
85. Ishoey M, Chorn S, Singh N, et al. Translation termination factor GSPT1 is a phenotypically relevant off-target of heterobifunctional phthalimide degraders. ACS Chem Biol. 2018;13(3):553–560. doi:10.1021/acschembio.7b00969
86. Gasic I, Groendyke BJ, Nowak RP, et al. Tubulin resists degradation by cereblon-recruiting PROTACs. Cells. 2020;9:5. doi:10.3390/cells9051083
87. Petschnigg J, Groisman B, Kotlyar M, et al. The mammalian-membrane two-hybrid assay (MaMTH) for probing membrane-protein interactions in human cells. Nat Methods. 2014;11(5):585–592. doi:10.1038/nmeth.2895
88. Saraon P, Snider J, Kalaidzidis Y, et al. A drug discovery platform to identify compounds that inhibit EGFR triple mutants. Nat Chem Biol. 2020;16:577–586. doi:10.1038/s41589-020-0484-2
89. Li S, Schmitz KR, Jeffrey PD, Wiltzius JJW, Kussie P, Ferguson KM. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell. 2005;7(4):301–311. doi:10.1016/j.ccr.2005.03.003
90. Gaborit N, Abdul-Haia A, Mancini M, et al. Examination of HER3 targeting in cancer using monoclonal antibodies. Proc Natl Acad Sci U S A. 2015;112(3):839–844. doi:10.1073/pnas.1423645112
91. Mancini M, Gal H, Gaborit N, et al. An oligoclonal antibody durably overcomes resistance of lung cancer to third‐generation EGFR inhibitors. EMBO Mol Med. 2018;10(2):294–308. doi:10.15252/emmm.201708076
92. Romaniello D, Mazzeo L, Mancini M, et al. A combination of approved antibodies overcomes resistance of lung cancer to osimertinib by blocking bypass pathways. Clin Cancer Res. 2018;24(22):5610–5621. doi:10.1158/1078-0432.CCR-18-0450
93. van Veggel B, de Langen AJ, Hashemi SMS, et al. Afatinib and cetuximab in four patients with EGFR Exon 20 insertion–positive advanced NSCLC. J Thorac Oncol. 2018;13(8):1222–1226. doi:10.1016/j.jtho.2018.04.012
94. Rusan M, Li K, Li Y, et al. Suppression of adaptive responses to targeted cancer therapy by transcriptional repression. Cancer Discov. 2018;8(1):59–73. doi:10.1158/2159-8290.CD-17-0461
95. Terai H, Kitajima S, Potter DS, et al. ER stress signaling promotes the survival of cancer “Persister Cells” tolerant to EGFR tyrosine Kinase inhibitors. Cancer Res. 2018;78(4):1044–1057. doi:10.1158/0008-5472.CAN-17-1904
96. Tatsumi K, Sou YS, Tada N, et al. A novel type of E3 ligase for the Ufm1 conjugation system. J Biol Chem. 2010;285(8):5417–5427. doi:10.1074/jbc.M109.036814
97. Wei Y, Xu X. UFMylation: a Unique & Fashionable Modification for Life. Genomics, Proteomics Bioinforma. 2016;14(3):140–146. doi:10.1016/j.gpb.2016.04.001
98. Naidoo J, Sima CS, Rodriguez K, et al. Epidermal growth factor receptor exon 20 insertions in advanced lung adenocarcinomas: clinical outcomes and response to erlotinib. Cancer. 2015;121(18):3212–3220. doi:10.1002/cncr.29493
99. Beau-Faller M, Prim N, Ruppert A-M-M, et al. Rare EGFR exon 18 and exon 20 mutations in non-small-cell lung cancer on 10 117 patients: a multicentre observational study by the French ERMETIC-IFCT network. Ann Oncol. 2013;25(1):126–131. doi:10.1093/annonc/mdt418
100. Yang JCH, Wu YL, Schuler M, et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): Analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol. 2015;16(2):141-151. doi:10.1016/S1470-2045(14)71173-8
101. Sequist LV, Besse B, Lynch TJ, et al. Neratinib, an irreversible pan-ErbB receptor tyrosine kinase inhibitor: results of a phase II trial in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2010;28(18):3076–3083. doi:10.1200/JCO.2009.27.9414
102. Kim TM, Ock C-Y, Kim M, et al. Phase II study of osimertinib in NSCLC patients with EGFR exon 20 insertion mutation: a multicenter trial of the Korean Cancer Study Group (LU17-19). Ann Oncol. 2019;30:v628. doi:10.1093/annonc/mdz260.051
103. Fang W, Huang Y, Hong S, et al. EGFR exon 20 insertion mutations and response to osimertinib in non-small-cell lung cancer. BMC Cancer. 2019;19(1):1–9. doi:10.1186/s12885-019-5820-0
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.