Back to Journals » Cancer Management and Research » Volume 11
T cells targeting multiple tumor-associated antigens as a postremission treatment to prevent or delay relapse in acute myeloid leukemia
Authors Xue L, Hu Y, Wang J, Liu X, Wang X
Received 14 February 2019
Accepted for publication 12 June 2019
Published 16 July 2019 Volume 2019:11 Pages 6467—6476
DOI https://doi.org/10.2147/CMAR.S205296
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Eileen O'Reilly
Lei Xue,1 Yan Hu,1 Jian Wang,1 Xin Liu,1 Xingbing Wang1,2
1Department of Hematology, The First Affiliated Hospital of USTC, Division of Life Science and Medicine, University of Sciences and Technology of China, Hefei, Anhui 230001, People’s Republic of China; 2Department of Hematology, Anhui Provincial Hospital, Anhui Medical University, Hefei, Anhui, People’s Republic of China
Background: Relapse is a major problem in acute myeloid leukemia (AML) and adversely affects survival. Tumor-associated antigen-specific cytotoxic T lymphocyte (TAA-CTLs)-based therapy was introduced and increasingly used clinically to kill tumor cells via tumor antigen activation.
Method: In this study, we expanded autologous lymphocytes reactive to five TAA (NY-ESO-1, MAGE-A3, WT1, Survivin, and PRAME) and evaluated its safety and efficacy in 9 patients with AML at high risk of relapse.
Results: Before first TAA-CTL infusion, 5 patients were minimal residual disease (MRD) positive, whereas 4 were MRD negative. Patients received TAA-CTL infusion for 1–3 times. None of them had obvious adverse reactions during or post the infusion. Of the 4 MRD-negative patients who were infused with TAA-CTLs, one developed relapsed disease. Among 5 MRD+ patients, there was a demonstrable antileukemic effect of the TAA-CTLs alone without any concomitant chemotherapy in 2 patients, as demonstrated by the negative of MRD in bone marrow after TAA-CTL infusion.
Conclusions: In summary, we have observed preliminary indications of activity and safety after administration of autologous TAA-CTLs in patients with AML. The ultimate question of clinical efficacy, however, will need to be addressed in a larger trial with larger homogeneous patient population.
Keywords: tumor-associated antigen-specific T cells, immunotherapy, acute myeloid leukemia
Background
Acute myeloid leukemia (AML) is a malignant hematopoietic disorder characterized by proliferation of immature myeloid precursors with considerable impairment of the immune system. Chemotherapy was the primary therapeutic approach for AML. The complete remission rate of chemotherapy-treated AML was 60–80%; however many cases cannot attain continuous complete remission (CCR) for more than 5 years after the treatment. The failure is mainly due to residual leukemia relapse.1 Allogeneic hematopoietic stem cell transplantation (HSCT) is a potentially curative option for AML, while only a limited number of patients qualify. Considering the shortcomings of current AML therapy and the stagnation of treatment advances in the past few decades, new therapies are desperately needed.
Recently, novel therapeutic approach focused on the manipulation of immune activity, including specific and nonspecific immunity, stimulation such as immunoregulatory antibodies, vaccines, and adoptive cell infusion.2 The direct clinical evidence of antileukemia-specific cytotoxic T lymphocytes (CTLs) originated from the relapse of chronic myeloid leukemia (CML) after donor lymphocyte infusion (DLI) treatment by allogeneic HSCT, followed by selective component T-cell infusion.3,4 Dendritic cell (DC)-based vaccines have been proven to be effective in AML and been used clinically.5,6 Weber G et al have shown that, in healthy donors, it is possible to induce T cells specifically targeting multiple leukemia-associated antigens (LAAs) which can recognize and kill AML cells.7 Infusion of donor multi-LAA-specific T cells to allogeneic HSCT-treated AML patients proved to be safe and associate with clinical benefit in individuals with highly treatment-resistant leukemia.8 By using of CTLs targeting multiple tumor-associated antigens (TAAs) (PRAME, SSX2, MAGEA4, NY-ESO-1, and Survivin), Ann Leen et al treated 18 lymphoma patients with relapsed refractory or high risk of recurrence. The proteins that investigators are targeting in this study are specific to the cancer cell. They either do not show or show up in low quantities on normal human cells. The results showed that infusion of autologous tumor-associated antigen-specific cytotoxic T lymphocytes (TAA-CTLs) is safe and can provide clinical benefits to patients with lymphomas.9 Furthermore, we also have shown that TAA-CTLs can be successfully induced and generated ex vivo from the peripheral blood of healthy volunteers, and the TAA-CTLs demonstrated specific killing activities.10 Based on the trial of Ann Leen,9 we selected five common TAA (NY-ESO-1, MAGE-A3, WT1, Survivin, and PRAME) to induce TAA-CTLs and after expansion, and evaluated its safety and efficacy in AML patients in this study.
Methods
Patients
The clinical trial protocol was approved by the ethics committee of the First Affiliated Hospital of USTC (Anhui Provincial Hospital), Hefei, China (Approval ID #2014–43). Patients were informed of the investigative nature of this study, and written consent in accordance with institutional regulations was obtained prior to study entry.
Nine patients with AML were enrolled in this study. Enrollment criteria for the study were adult patients with AML (between 18 and 70 years old, except acute promyelocytic leukemia), diagnosed according to World Health Organization criteria, at high risk of relapse as defined by: age ≥60 years or if <60 years without matched sibling donor for allo-HSCT, hyperleukocytosis at presentation (WBC ˃100×109/L), poor-risk cytogenetic or molecular markers, previous relapse, and/or minimal residual disease (MRD) positive.
Generation of DCs
Approximately, 100 mL of peripheral blood was obtained from each patient and mononuclear cells were isolated by using Ficoll–Hypaque (Invitrogen, Carlsbad, CA, USA) density-gradient centrifugation. Monocyte-derived DCs were generated from peripheral blood mononuclear cell (PBMC) as described previously [10]. DCs were prepared in a Good Manufacturing Practice (GMP)-compliant facility at the First Affiliated Hospital of USTC (Hefei, China). Briefly, PBMC concentration was adjusted to 4×106/mL with serum-free medium (SFM, Gibco, Carlsbad, CA, USA) containing 5% human AB serum and the suspended cells were removed after 2–4 hrs culture. The nonadherent cells were cryopreserved and subsequently used for specific T-cell stimulation. The adherent PBMCs were cultured for 5–6 days with the above-mentioned medium in the presence of IL-4 (1,000 U/mL, PeproTech, Rochy Hill, NJ, USA) and granulocyte macrophage colony-stimulating factor (GM-CSF; 800 U/mL, Xiamen Tebao, China) in a humidified 37 °C, 5% CO2 incubator. The medium was changed every 2–3 days. On the 5th or 6th day, immature DCs were matured in DC media with a cytokine cocktail consisting of IL-4 (1,000 U/mL, PeproTech), GM-CSF (800 U/mL, Xiamen Tebao, Xiamen, China), IL-6 (10 ng/mL, PeproTech), TNF-α (10 ng/mL, PeproTech), IL-1β (10 ng/mL, PeproTech), and PGE (1 mg/mL, Sigma-Aldrich), and were harvested after 48 hrs of maturation. The mature DCs were loaded with 15mer peptide libraries of five LAA peptides (WT-1, PRAME, NY-ESO-1, MAGEA3, and Survivin; all from JPT Peptide Technologies, Berlin, Germany) during days 7–8. The loaded DCs were co-cultured with autologous PBMC to prepare the leukemia-specific T cells from the 8th day to 25th day.
Preparation and characterization of TAA-CTLs
TAA-CTLs were generated by stimulation of PBMC with peptide-pulsed DCs at an effector-to-target ratio of 10:1 in SFM (Gibco). The TAA-CTLs were manufactured under GMP conditions by the First Affiliated Hospital of USTC with standard operating procedures and predefined batch release criteria. For initial stimulation, a cytokine mix containing IL-7 (10 ng/mL), IL-12 (10 ng/mL), IL-15 (5 ng/mL), and IL-6 (100 ng/mL, all Peprotech, USA) was added at day 0. Medium was changed and continued to be cultured for 4–6 days if the cell number is less than 3×106/mL, otherwise cells were transfered to new culture well and cultured for another 4–6 days. At days 11–13, the cells were counted and then the loaded DCs, cytokines IL-7, IL-15 (all Peprotech, USA), and IL-2 (50 U/mL, Beijing Sihuan, Beijing, China), were added for the second stimulation, and cultured for 3–4 days continuously to amplify TAA-specific CTL. Contamination of fungi, bacteria, and endotoxin in all the cultured samples were detected during the course of cell culture. At the 21th–25th days, partial cells were frozen and the phenotype was tested and remaining was centrifuged. After removal of the supernatants, the cells were suspended in 100 mL of physiological saline containing 1% albumin and IL-2, then intravenously injected to the patient.
Treatment procedures
During this study, patients received TAA-CTLs (1–5×107 cells/m2 for each time) via intravenous infusion on day 1 followed by low-dose s.c. IL-2×14 days. Some of patients received decitabine (15 mg/m2) chemotherapy on day −6 to −2 before TAA-CTL infusion. Patients were monitored for toxicities based on Common Toxicity Criteria v4.0. The glucocorticoids should be avoided in this period. BM aspirates were obtained for analysis within 28 days of the TAA-CTL infusion.
Interferon gamma (IFN-γ) enzyme-linked immunospot assay
Enzyme-linked immunospot (ELISpot) assay was used to determine the frequency of T cells secreting IFN-γ in response to TAAs pepmixes according to the manufacturer’s instructions (Mabtech, Nacka Strand, Sweden). To reduce interassay variability, patient PBMC samples were cryopreserved and batched for ELISpot analysis. Briefly, recognition of the pooled TAAs (NY-ESO-1, MAGEA3, PRAME, Survivin, and WT1, all from JPT Peptide Technologies, Berlin, Germany) was tested as compared with no-peptide (media) control and positive control group in triplicate. PBMCs were washed and resuspended and stimulated with the pooled TAAs at a concentration of 1 µg/µL. The plates were incubated for 24 hrs at 37 °C. For development, plates were washed with PBS and incubated with biotinylated IFN-γ detection antibody for 2 hrs at 37 °C, followed by incubation with streptavidin-coupled alkaline phosphatase complex for 1 hr at room temperature and spots were then developed by incubation with 3-amino-9-ethylcarbazole substrate solution. The secreation of IFN-γ was quantified by calculation of spot-forming cells (SFCs) per 2×105 monocytes. The numbers of spots was observed and analyzed using ordinary light microscope.
Immunophenotyping
CTLs were stained with extracellular antibodies targetting CD3, CD4, CD8, CD19, CD45RA and RO, CD56, CD62L, and CCR7 (all BD Biosciences, San Diego, CA, USA). Control samples labeled with appropriate isotype-matched antibodies were included in each experiment. For each sample, 10,000 cells were analyzed by FACS Calibur using Cell Quest software (Becton Dickinson, San Jose, CA, USA).
Results
Patient characteristics and clinical results
Patients—who either were MRD positive after receiving standard therapy or were considered at high risk for relapse (Table 1)—received infusion of TAA-CTLs in the First Affiliated Hospital of USTC, where their vital signs were monitored before and immediately after infusion. A total of 9 patients (5 male, 4 female) were enrolled in the study and evaluable for responses (Table 1). The median age was 52 years (range, 29–66 years).
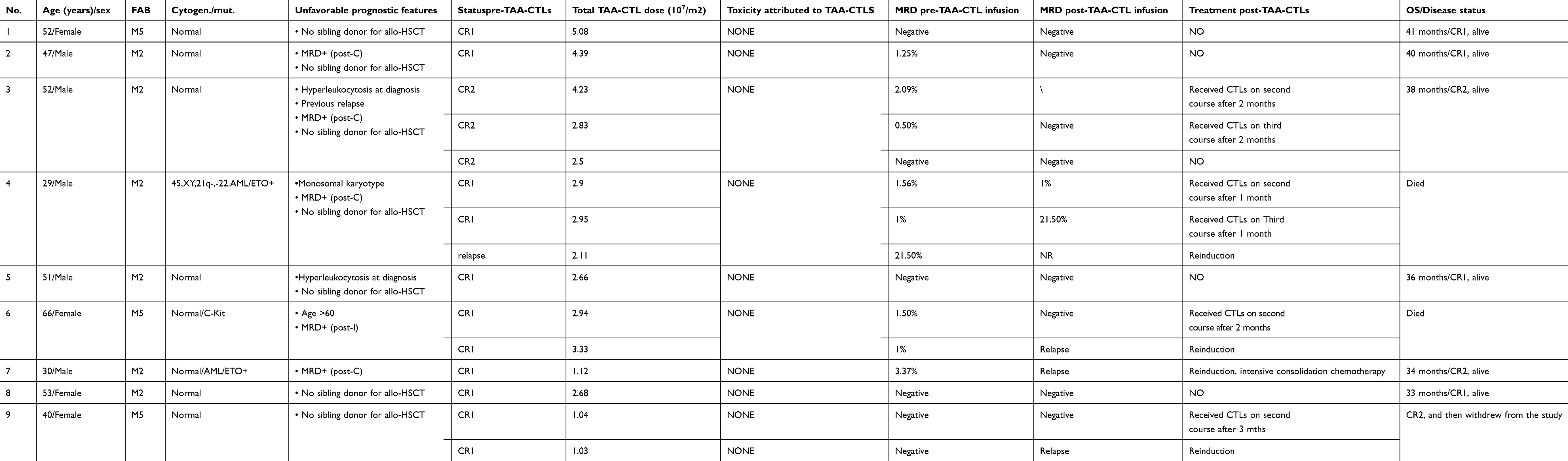 |
Table 1 Clinical characterization of 9 patients with TAA-CTL therapy |
Before the first TAA-CTL infusion, 5 patients were multiparameter flow cytometry (MFC) MRD positive, whereas 4 were MFC MRD negative. Although two different sensitive methods, real-time quantitative PCR (RQ-PCR) and MFC, can be used to monitor MRD in AML patients. In this study, only patient No. 4 and 7 had a molecular MRD marker (AML1-ETO) other than MRD assessment by MFC. Overall, there was a corresponding evolution between the AML1-ETO transcript levels and MRD assessment by MFC. If at least 4 weeks after the infusion there is no change or a reduction in the number of leukemia cells measured in the bone marrow, MRD-positive patients may receive up to two additional doses of the T cells at least 4 weeks apart. Total doses of TAA-CTLs received are shown in Table 1.
Patients received TAA-CTL infusion for 1–3 times, 1–5×107 cells/m2 for each time, and TAA-CTLs were well-tolerated. None of them had obvious adverse reactions during or post the infusion. Two patients had swelling at the injection site of IL-2 and fever; however, the symptoms disappeared after injection paused.
Patient No. 1, 5, 8, and 9 received TAA-CTLs as an adjuvant therapy, and their MRD were negative before TAA-CTL infusion. The MRD of these 4 patients remained negative without any other treatment. However, patient No. 9 recurred 4 months later and reached CR after one course of decitabine + cytarabine, aclarubicin, granulocyte colony stimulating factor (CAG) chemotherapy scheme.
Patient No. 2, 3, 4, 6, and 7 were MRD positive before TAA-CTL infusion. There was a demonstrable antileukemic effect of the TAA-CTL infusion alone without any concomitant chemotherapy in 2 of 5 patients, as demonstrated by the negative of MRD in bone marrow (No. 2 and 3). Until now, no other treatment has been used and patients remain in MRD remission.
Patient No. 6, a 66-year old female who was MRD+, received decitabine (15 mg/m2, day 6~day 2) followed by infusion of TAA-CTLs; 4 weeks later, MRD changed to negative. However, flow cytometry of bone marrow cells detected positive MRD 2 months after TAA-CTL cell therapy. This patient received decitabine (15 mg/m2/d, day 6~day 2) followed by reinfusion of TAA-CTLs, but the MRD was found to have progressed 4 weeks later. Although she then received induction chemotherapy, the primary disease remained unresolved and died due to infection 2 months later. The remaining 2 MRD+ patients (No. 4 and 7) received decitabine (15 mg/m2/d) chemotherapy on day −7 to −2 before TAA-CTL infusion; however they developed disease progression (months 2 and 1 post-TAA-CTLs, respectively). Patient No. 4 then received induction chemotherapy; however, the primary disease remained unresolved, and the patient died due to infection after 3 months. Patient No. 7 reached CR after one course of decitabine + CAG chemotherapy scheme. Afterward, the patient was given intensive consolidation chemotherapy treatment and is still in complete remission until now.
Characteristics of TAA –CTLs
At the time of cryopreservation, TAA-CTLs were examined through flowcytometry. As shown in the Figure 1, 93.5% of the ex-vivo expanded T cells were identified to be CD3+ (range, 56.5–99%) and varying distribution of CD4+ (mean 37.4%, range 3.2–57.1%) or CD8+ (mean 43.2%, range 5.5–64%) was found. While, few natural killer cells (CD3−CD56+, mean 4.9%, range 0.3%-9.8%) and rare residual B cells (CD19+, mean 0.3%, range 0–0.6%) were found. Moreover, the majority of both CD4+ (mean 46.7%, range 5.6–63.3%) and CD8+ (mean 50.2%, range 3.9–78%) T cells were composed of CD45RO+CD62L−CCR7− T cells in accordance with an effector-memory phenotype. Few naive CD45RA+CD62L+CCR7+cells (CD4+, mean 3.9%, range 0–8.9%; CD8+, mean 2.1%, range 0.5–3.9%) and central-memory CD45RO+CD62L+CCR7+cells (CD4+: mean 5.7%, range 0–12.3%; CD8+: mean 13.5%, range 0.8–21%) presented after two restimulations.
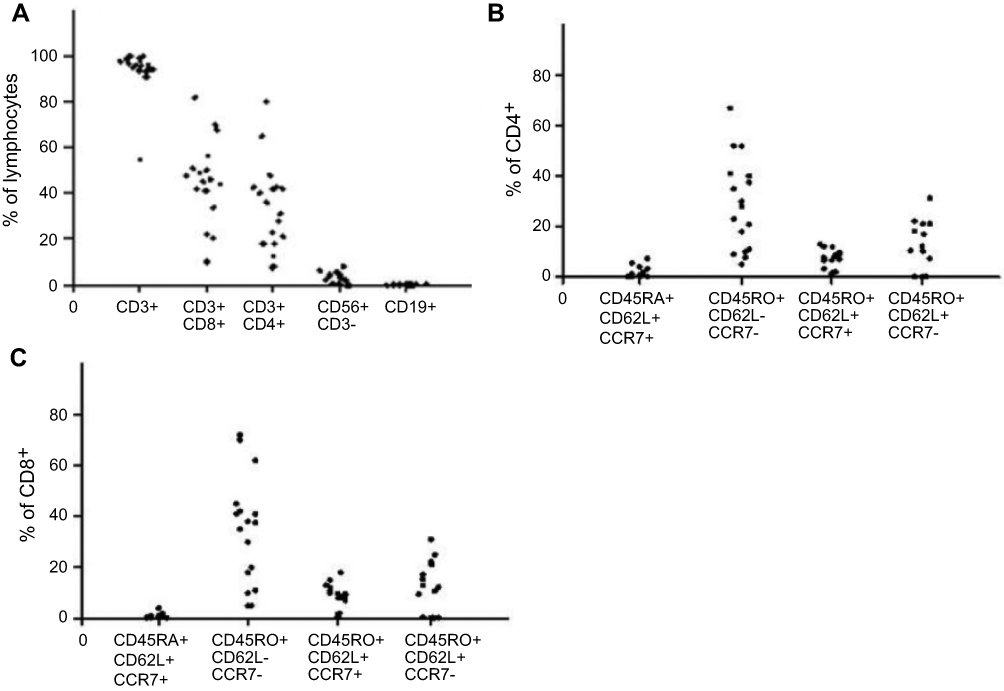 |
Figure 1 Phenotype analysis of TAA-CTLs generated from patients with AML. (A) Phenotyping of the ex vivo expanded TAA-CTLs after the second stimulation, gated on lymphocytes. (B) T-cell subsets within the CD3+/CD4+ subpopulation. (C) T-cell subsets within the CD3+/CD8+ subpopulation.Abbreviation: TAA-CTLs, Tumor associated antigen specific cytotoxic T lyphocytes. |
Immunomonitoring
Immunomonitoring was performed on PBMCs obtained before and after TAA-CTL infusion. Followed by stimulation of antigen peptide mixtures (NY-ESO-1, MAGE-A3, WT1, Survivin, and PRAME), we measured changes in the frequency of multiple TAA-CTLs in the PBMC by using IFN-γ ELISpot assays. As indicated in Figure 2, the SFC levels of most patients gradually increased and the peak appeared about 2–3 weeks after the infusion. Neither responders nor nonresponders had a concomitant rise in TAA-CTLs. Six of the patients showed a transient high value of IFN-γ up to 180–250 SFCs per 2×105 cells. Of note, in all relapsing patients (No. 4, 6, 7, and 9), recurrence of AML was preceded and accompanied by decreased frequency of circulating TAA-CTLs.
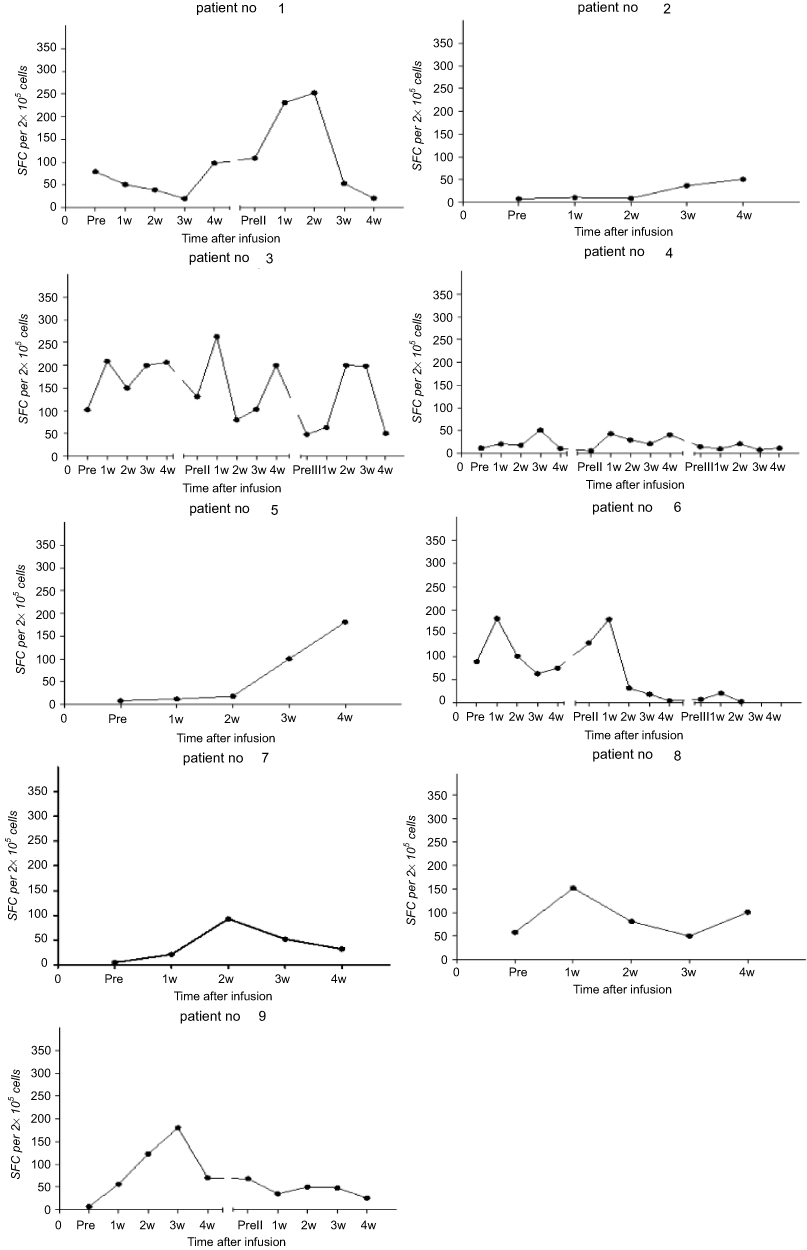 |
Figure 2 Detection of SFCs in patients re-infusion with TAA-CTLs. Followed by stimulation of antigen peptide mixtures (NY-ESO-1, MAGE-A3, WT1, Survivin, and PRAME), the frequency of multiple TAA-CTLs was measured in PBMCs in patients recieving TAA-CTL infusion by using ELISpot assays. Points represent the number of IFN-γ spot-forming cells (SFCs) per 2×105 PBMCs. The SFC levels of most patients gradually increased and the peak appeared about 2–3 weeks after the infusion. Lines represent individual CTL variation tendency. |
Discussion
With an ever-increasing number of cancer immunotherapeutics entering the clinic, immunotherapy is now on the forefront of oncologic clinical research and patient care.11 TAAs were identified as valuable agents and have been applied for the development of novel immunotherapeutic approach. Clinical studies have revealed that TAA-CTLs were capable of treating hematological malignancies, especially the patients suffered from relapsed refractory or high risk of recurrence.8,9,12
AML cells express a multitude of TAAs, including WT-1, PRAME, NY-ESO-1, MAGEA3, and Survivin.13–15 Peptide-based vaccine approaches have also been utilized to generate WT1-specific T cells in AML patients.16 Vaccination of AML patients with WT1 mRNA-electroporated DCs can effectively prevent or delay relapse after standard chemotherapy by the induction of WT1-specific CD8+ T-cell response.6 Similarly, T cells recognizing LAA (WT1, PRAME, Survivin, NY-ESO, and p53) can be detected in healthy subjects.17
In our previous study, we had demonstrated that it is possible to expand multi-TAA-CTLs ex vivo by peptide stimulation from healthy donors, and subsequent experiments confirmed its specific killing function in vitro.10 In this trial, TAA-CTL production from peripheral blood was successful for all patients. We observed preliminary indications of activity and safety after administration of autologous TAA-CTLs in patients with AML. MDR was detected by flow cytometry in 2 patients (No. 2 and 3) before TAA-CTL administration. Strikingly, we found that, even without adjunct chemotherapy, TAA-CTL infusion alone eradiated MDR in the bone marrow in these patients. These data further supported the antileukemic effect of TAA-CTLs.
We found that the obtained CTLs were CD3+ T cells (average 93.5%) in majority, including CD8+ T cells 43.2% and CD4+ T cells 37.4%. Moreover, the majority of both CD4+ and CD8+ T cells were composed of CD45RO+CD62L−CCR7− T cells in accordance with an effector-memory phenotype. Memory T cells can be divided into Tcm (central memory T cells) and Tem (effect memory T cells) according to two criteria.18 Tem exhibited a rapid effector function and mediated protective memory activity. The TAA-CTLs cultured in the study was mainly Tem, indicating that it can quickly proliferate after infusion into body and recognize tumor antigens to exert immune response and provide persistent immune memory protection. Previous studies have described that cytokines have a fundamental role in the differentiation of memory T cells.19 In particular, IL-15 and IL-7 can significantly enhance the function of human CD4+ and CD8+ memory T cells and contribute to their long survival,20–22 and IL-7 and IL-12 showed synergistic effects on human T-cell activation.23 Hence, we added IL-7, IL-15, and IL-12 to culture medium to promote CTL production.
During this study, peripheral blood was collected per week from patients who received TAA-CTL therapy, and the mononuclear cells were extracted for ELISpot assay. The cellular immune responses induced by mixed antigen was evaluated through analyzing IFN-γ level secreted by T cells. Dynamic changes of peripheral blood SFC in TAA-CTL-treated patients were measured, and the results suggested that the SFC curve gradually increased over time and the average peak occurred 2–3 weeks after infusion. The other patient’s SFC level gradually increased and maintained at a high level within 4 weeks after one course of treatment. It also showed that SFC number was increased in continuous remission patients, up to 253 per 2×105 cells with 2–3 times of cell therapy. Of note, in all relapsing patients (No. 4, 6, 7, and 9), recurrence of AML was preceded and accompanied by decreased circulating TAA-CTLs. Therefore, it indicated that the higher the level of SFC, the stronger antitumor activity of CTL remained in patients.
Nine patients treated with TAA-CTLs in this study showed distinct outcomes. The immune escape may take place due to down-regulation of MHC class I molecules on cancer cell surfure, the failure of type II molecules related CLIP binding to HLA-II antigen pockets, and the inability of antigen presentation.24 Thus tumor cells with low expression of tumor antigens might not be effectively detected and killed by tumor-specific T cells, which likely resulted in the escape of leukemia cells from TAA-CTLs monitoring.25 To decrease the possibility of cancer immune evasion, we included a combination of 5 tumor antigens to boost CTL-mediated antitumor immunity in the current study. Although this strategy had clinical benefits in most patients recruited in our study, it remains to be determined which antigen is the main inducer for IFN-γ production by T cells in the future study. A detailed assessment of the correlation of clinical outcomes with the inductive effect of individual antigens would provide valuable information to understand the underlying mechanism and to improve the design for clinical trails.
In addition, decitabine can upregulate mRNA and protein expression levels of CTA gene family (NY-ESO-1 and MAGEA3/A6) in tumor cells and reduce the degree of methylation of NY-ESO-1 gene promoter in myeloid cells, thereby enhancing the killing activities of TAA-CTLs against leukemia cells.26,27 In decitabine-treated MDS patients with increased expression of NY-ESO-1 in malignant myeloid cells, Griffiths EA et al demonstrated that vaccinating against NY-ESO-1 can provoke an NY-ESO-1-specific MDS-directed cytotoxic T-cell immune response.28 Therefore, given the potential importance of eliciting a T-cell response to TAAs, decitabine was added in patients with high-risk and/or recurrent AML before TAA-CTL infusion. Because the numbers were small, we are not certain whether the combination of decitabine with TAA-CTL treatment can improve outcome for patients with AML, which requires further investigation in future.
Conclusion
We conducted a clinical trial using TAA-CTLs in 9 patients with AML. TAA-CTLs were well tolerated in all patients and eliminate residual leukemic cells, decrease the likelihood of AML relapse, and improve survival. The ultimate question of clinical efficacy, however, will need to be addressed in a larger trial with larger homogeneous patient population.
Ethics approval and consent to participate
This study was approved by the Medical Ethics Committee of the First Affiliated Hospital of USTC (Anhui Provincial Hospital), Hefei, China (Approval ID #2014-43). The patient and donor gave their written informed consent in accordance with the Declaration of Helsinki.
Abbreviation list
AML, acute myeloid leukemia; TAA-CTLs, tumor-associated antigen-specific cytotoxic T lymphocytes; MRD, minimal residual disease; HSCT, hematopoietic stem cell transplantation; CTL, cytotoxic T lymphocytes; CML, chronic myeloid leukemia; DLI, donor lymphocyte infusion; DC, dendritic cell; LAA, leukemia-associated antigens; PBMC, peripheral blood mononuclear cell; GM-CSF, granulocyte macrophage colony-stimulating factor; TAA, tumor-associated antigen; SFCs, spot-forming cells.
Acknowledgments
The authors would like to thank all members of the study team, the patient, and their family. This work was supported by the Science and Technology Planning Project of Anhui Province, China (Grant No. 1604a0802071), the National Natural Science Foundation (Grant No. 81270573), and by the Major Special Science and Technology Project of Anhui Province, China (Grant No. 18030801126).
Author contributions
Drs. XW and LX had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. XW contributed to the study concept and design. YH, JW, and LX performed the experiments or provided the study patients. All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that they have no competing interests in this work.
References
1. Acheampong DO, Adokoh CK, Asante DB, et al. Immunotherapy for acute myeloid leukemia (AML): a potent alternative therapy. Biomed Pharmacother. 2018;97:225–232. doi:10.1016/j.biopha.2017.10.100
2. Ravandi F, Walter RB, Freeman SD. Evaluating measurable residual disease in acute myeloid leukemia. Blood Adv. 2018;2(11):1356–1366. doi:10.1182/bloodadvances.2018016378
3. Kolb HJ, Schattenberg A, Goldman JM, et al. Graft-versus-leukemia effect of donor lymphocyte transfusions in marrow grafted patients. Blood. 1995;86:2041–2050.
4. Toprak SK. Donor lymphocyte infusion in myeloid disorders. Transfus Apher Sci. 2018;57(2):178–186. doi:10.1016/j.transci.2018.04.018
5. Van Tendeloo VF, Van de Velde A, Van Driessche A, et al. Induction of complete and molecular remissions in acute myeloid leukemia by Wilms’ tumor 1 antigen-targeted dendritic cell vaccination. Proc Natl Acad Sci U S A. 2010;107(31):13824–13829. doi:10.1073/pnas.1008051107
6. Anguille S, Van de Velde AL, Smits EL, et al. Dendritic cell vaccination as postremission treatment to prevent or delay relapse in acute myeloid leukemia. Blood. 2017;130(15):1713–1721. doi:10.1182/blood-2017-04-780155
7. Weber G, Gerdemann U, Caruana I, et al. Generation of multi-leukemia antigen-specific T cells to enhance the graft-versus-leukemia effect after allogeneic stem cell transplant. Leukemia. 2013;27:1538–1547. doi:10.1038/leu.2013.66
8. Lulla P, Naik S, Tzannou I, et al. Safety and efficacy of adoptively transferred donor-derived tumor-directed T cells with relapsed AML post-allogeneic HSCT. Blood. 2017;130:164.
9. Leen A, Tzannou I, Bilgi M, et al. Immunotherapy for Lymphoma Using T Cells Targeting Multiple Tumor Associated Antigens. Blood. 2015;126:186.
10. Zhang Q, Liu X, Wang XB, et al. Amplification ex Vivo and cytocidal activity of leukemia tumor-associated antigen-specific cytotoxic T lymphocytes. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2015;23(3):814–820. doi:10.7534/j.issn.1009-2137.2015.03.041
11. Cruz CR, Bollard CM. Adoptive immunotherapy for leukemia with ex vivo expanded T cells. Curr Drug Targets. 2017;18(3):271–280. doi:10.2174/1389450117666160209143529
12. Bollard CM, Tripic T, Cruz CR, et al. Tumor-specific T-cells engineered to overcome tumor immune evasion induce clinical responses in patients with relapsed hodgkin lymphoma. J Clin Oncol. 2018;36(11):1128–1139. doi:10.1200/JCO.2017.74.3179
13. Martínez A, Olarte I, Mergold MA, et al. mRNA expression of MAGE-A3 gene in leukemia cells. Leuk Res. 2007;31(1):33–37. doi:10.1016/j.leukres.2006.05.009
14. Greiner J, Ringhoffer M, Simikopinko O, et al. Simultaneous expression of different immunogenic antigens in acute myeloid leukemia. Exp Hematol. 2000;28(12):1413–1422.
15. Greiner J, Bullinger L, Guinn BA, Döhner H, Schmitt M. Leukemia-associated antigens are critical for the proliferation of acute myeloid leukemia cells. Clin Cancer Res. 2008;14(22):7161–7166. doi:10.1158/1078-0432.CCR-08-1102
16. Maslak PG, Dao T, Krug LM, et al. Vaccination with synthetic analog peptides derived from WT1 oncoprotein induces T-cell responses in patients with complete remission from acute myeloid leukemia. Blood. 2010;116:171–179. doi:10.1182/blood-2009-10-250993
17. Matko S, Manderla J, Bonsack M, et al. PRAME peptide-specific CD8+ T cells represent the predominant response against leukemia-associated antigens in healthy individuals. Eur J Immunol. 2018;48(8):1400–1411. doi:10.1002/eji.201747399
18. Mueller SN, Gebhardt T, Carbone FR, Heath WR. Memory T cell subsets, migration patterns, and tissue residence. Annu Rev Immunol. 2013;31:137–161. doi:10.1146/annurev-immunol-032712-095954
19. Schluns KS, Lefrançois L. Cytokine control of memory T-cell development and survival. Nat Rev Immunol. 2003;3(4):269–279. doi:10.1038/nri1052
20. Shmarov VA, Malashchenko VV, Meniailo ME, et al. Direct effects of interleukin-7 on the function of human T cells in vitro. Eur Cytokine Netw. 2016;27(4):102–107. doi:10.1684/ecn.2016.0385
21. Mueller YM, Makar V, Bojczuk PM, Witek J, Katsikis PD. IL-15 enhances the function and inhibits CD95/Fas-induced apoptosis of human CD4+ and CD8+ effector-memory T cells. Int Immunol. 2003;15(1):49–58. doi:10.1093/intimm/dxg013
22. Sato N, Patel HJ, Waldmann TA, Tagaya Y. The IL-15/IL-15Ralpha on cell surfaces enables sustained IL-15 activity and contributes to the long survival of CD8 memory T cells. Proc Natl Acad Sci U S A. 2007;104(2):588–593. doi:10.1073/pnas.0610115104
23. Mehrotra PT, Grant AJ, Siegel JP. Synergistic effects of IL-7 and IL-12 on human T cell activation. J Immunol. 1995;154(10):5093–5102.
24. van Luijn MM, Chamuleau ME, Thompson JA, et al. Class II-associated invariant chain peptide down-modulation enhances the immunogenicity of myeloid leukemic blasts resulting in increased CD4+ T-cell responses. Haematologica. 2010;95(3):485–493. doi:10.3324/haematol.2009.010595
25. Atanackovic D, Luetkens T, Kloth B, et al. Cancer-testis antigen expression and its epigenetic modulation in acute myeloid leukemia. Am J Hematol. 2011;86(11):918–922. doi:10.1002/ajh.22141
26. Almstedt M, Blagitko-Dorfs N, Duque-Afonso J, et al. The DNA demethylating agent 5-aza-2ʹ-deoxycytidine induces expression of NY-ESO-1 and other cancer/testis antigens in myeloid leukemia cells. Leuk Res. 2010;34(7):899–905. doi:10.1016/j.leukres.2010.02.004
27. Srivastava P, Paluch BE, Matsuzaki J, et al. Induction of cancer testis antigen expression in circulating acute myeloid leukemia blasts following hypomethylating agent monotherapy. Oncotarget. 2016;7(11):12840–12856. doi:10.18632/oncotarget.7326
28. Griffiths EA, Srivastava P, Matsuzaki J, et al. NY-ESO-1 vaccination in combination with decitabine induces antigen-specific T-lymphocyte responses in patients with myelodysplastic syndrome. Clin Cancer Res. 2018;24(5):1019–1029. doi:10.1158/1078-0432.CCR-17-1792
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.