Back to Journals » International Medical Case Reports Journal » Volume 13
Systemic Sclerosis Sine Scleroderma Manifested with Gastrointestinal Bleeding, Antiphospholipid Syndrome and Positive Anti-RNA Polymerase III Antibody: Case Report and Literature Review
Authors Alghamdi M ![]() , Derbes SJ
, Derbes SJ
Received 8 June 2020
Accepted for publication 18 July 2020
Published 4 August 2020 Volume 2020:13 Pages 323—326
DOI https://doi.org/10.2147/IMCRJ.S254859
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ronald Prineas
Mansour Alghamdi, Stephen J Derbes
Department of Medicine, Rheumatology Section, Louisiana State University School of Medicine, New Orleans, LA, USA
Correspondence: Mansour Alghamdi
Department of Medicine, Rheumatology Section, Louisiana State University School of Medicine, New Orleans, LA, USA
Email [email protected]
Abstract: Systemic sclerosis sine scleroderma (ssSSc) is a rare disease in which patients present with internal organ manifestations of systemic sclerosis in the absence of cutaneous findings. They tend to have serological markers characteristic of systemic sclerosis (SSc), including positive antinuclear antibodies (ANA) and anticentromere antibodies (ACA). The disease has been rarely reported in the literature, and the diagnosis can be easily missed due to a lack of relevant skin findings. Here we report a patient who presented with chronic gastrointestinal bleeding with angioectasia, antiphospholipid syndrome (APS), positive ANA, positive ACA, and positive anti-RNA polymerase III antibody. The constellation of all these findings has, to our knowledge, never been described in the literature. The purpose of presenting this case is to raise the clinician’s awareness of the occurrence of this disease when similar internal organs manifestations of scleroderma are encountered, and to monitor for the development of other internal manifestations and intervene promptly and accordingly.
Keywords: systemic sclerosis sine scleroderma, scleroderma, systemic sclerosis, angioectasia/angiodysplasia, anti-RNA polymerase III antibody
Introduction
Systemic sclerosis sine scleroderma (ssSSc) is characterized by internal organ involvement and serologic abnormalities in the absence of cutaneous manifestations of systemic sclerosis.1 Systemic sclerosis or scleroderma disorders comprise a heterogeneous group of conditions of unknown etiology and is characterized by the presence of thickened, sclerotic skin lesions.2 Here we report a patient with gastrointestinal manifestations and high specific serology of scleroderma without cutaneous manifestations.
The Case
A 59-year-old African American woman with a history of recurrent GI bleed, peripheral vascular disease (PVD), tobacco smoking, hepatitis C infection, and mitral valve stenosis presented to our rheumatology clinic in 2018 with a high positive ANA chromosomal centromere B of 1:640 by indirect immunofluorescence (IIF) and anticentromere antibody of > 8 AI. At our clinic, the patient was diagnosed with APS based on recurrent arterial thrombosis of both legs for which she underwent stenting in 2014, and the presence of very high triple-positive antiphospholipid antibodies on two occasions more than 12 weeks apart. See Table 1. By the time of APS diagnosis at our clinic in 2018, the patient had ongoing active upper GI bleeding with episodes of melena and dropping hemoglobin. The current bleeding was evaluated by endoscopy and showed gastric and duodenal angioectasia. The patient also reported symptoms of mild GERD and dysphagia but there were no features of CREST syndrome or symptoms of cutaneous scleroderma. She denied raynaud’s symptoms, skin thickening/tightness, skin rashes, or photosensitivity. Her obstetric history was remarkable only for one early pregnancy loss.
 |
Table 1 Laboratory Results |
She reported dyspnea on exertion and was found to have severe iron deficiency anemia secondary to GI bleeding which required therapy with iron infusion in the past. The initial presentation of PVD was four years ago before presenting to our clinic. She presented with complete occlusion of the Superficial Femoral Artery and the Ankle-Brachial Index (ABI) at that time was 0.38. She underwent stent placement and begun on dual antiplatelet therapy consisting of aspirin and clopidogrel. The first episode of GI bleeding occurred a few weeks later when angioectasia was seen in the duodenum for the first time. No definite underlying diagnoses for the gastrointestinal angioectasia or thrombotic episodes have been established till her immunologic markers were checked by vascular surgery and referred to us in 2018.
On physical exam, her blood pressure was normal, the patient has no signs of skin tightness/thickening, sclerodactyly, calcinosis, cutaneous telangiectasia, digital pitting or ulcers, and no signs of chronic liver diseases.
Endoscopy revealed 2 mm non-bleeding angioectasias in the second portion of the duodenum and two non-bleeding angioectasias in the stomach, treated with argon beam coagulation, Figure 1. Capsule endoscopy reported two large angioectasias in the ileum, a single lesion in the jejunum, and several small bowel angioectasias, Figure 2. Colonoscopy was entirely normal. Transthoracic echocardiogram estimated the systolic pulmonary artery pressure value of 40–45 mmHg. This was interpreted by the cardiologist as secondary to mitral stenosis. Pulmonary function studies were significant for severe diffusion impairment with DLCO of 29 mL/min/mmHg with no evidence of obstruction or restriction. HRCT showed evidence of emphysematous changes and no features of interstitial lung diseases.
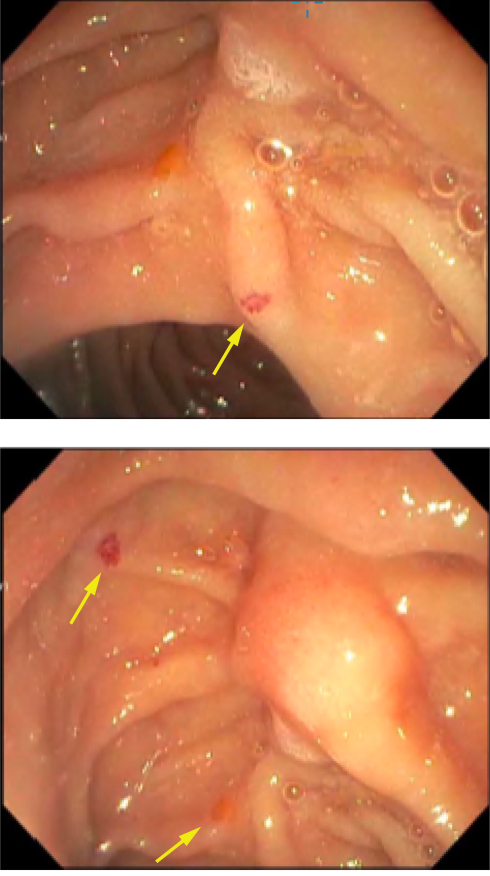 |
Figure 1 Duodenal angioectasia (arrows). |
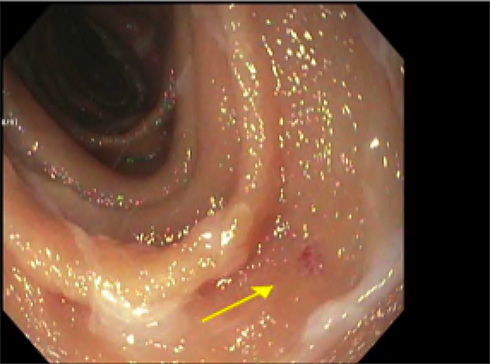 |
Figure 2 Jejunal angioectasia (arrow). |
The patient was started on hydroxychloroquine 400 mg daily. Antiplatelet therapy was discontinued because of active GI bleeding. Hematology recommended only antiplatelet for APS due to the high risk of bleeding and clopidogrel was resumed later when her GI bleed treated and resolved. She failed to develop any skin findings concerning for scleroderma for two years of follow up in our clinic, and no further thrombosis occurred. Her GI bleed was eventually controlled after endoscopic and Octreotide infusion by gastroenterology and no further episodes were encountered.
Discussion
The first report of ssSSc in the literature was in 1954 when Abrams, et al, described a 42-year-old woman with ssSSc and gastrointestinal involvement.3 Later, Rodnan, et al, in 1962 published a case series of four patients who presented with unexplained symptoms of malabsorption and dysphagia. Internal organ sclerosis found on postmortem examinations in all cases. There was neither clinical nor definite pathological evidence of scleroderma except for one patient with minimal changes of localized morphea on the chest. The gastrointestinal tract was the main organ involved in these patients, and in some, the only organ involved. The authors suggested that might explain cases with otherwise undiagnosed clinical problems of intestinal hypomotility or malabsorption and esophageal dysfunction.4 Whether this entity is part of limited cutaneous scleroderma (lcSSc) spectrum is still unclear. In one study, a series of 48 patients with ssSSc were compared with 507 patients with lcSSc, and those who subsequently developed scleroderma were excluded. The pulmonary hypertension was more frequent complications of ssSSc, there were no major differences between these two groups of patients regarding internal organ involvement and the most frequent antibodies reported in both groups were anticentromere. The authors suggested that ssSSc is considered to represent a portion of the spectrum of lcSSc where the scleroderma features are not yet developed.5
In a recent cohort, 45 patients with ssSSc and 186 patients with lcSSc were compared and showed a higher prevalence of pulmonary arterial hypertension (PAH) in ssSSc. The authors recommended that this should be considered a different subset within the scleroderma spectrum to avoid misdiagnosis and to be taken into account that in some syndromes, the PAH and ILD may be related to SSc with no skin changes.6 Based on a recent multicenter study, serological profiles of patients with ssSSc were similar to those with lcSSc but different from diffuse cutaneous systemic sclerosis (dcSSc) with higher rates of anticentromere antibodies (50% ssSSc versus 12% in dcSSc) and lower rate of anti-topoisomerase I antibodies (16% ssSSc versus 22% dcSSc).7
Pulmonary artery hypertension was the presenting and the only isolated feature of ssSSc in a young woman who has a positive ANA, 1/2560 titer with nucleolar staining pattern on IIF, and positive anti-U3 RNP (fibrillarin) autoantibodies.8 Furthermore, Positive anti-RNA polymerase III in association with Scleroderma renal crisis was the initial presentation of a patient who initially diagnosed with ssSSc and eventually developed cutaneous manifestation.9
Our patient findings are similar to those patients reported in the literature with ssSSc, including high positive ANA and ACA, absence of cutaneous findings of scleroderma, and the presence of internal organ pathology. The patient reduced DLCO is multifactorial, per pulmonary, smoking, mitral valve stenosis, pulmonary hypertension and, emphysema/COPD. There was no evidence of scleroderma lung on imaging. An important feature in our patient is the positivity of anti-RNA polymerase III which has been very rarely reported.5,10 In a recent case report in 2016, the author reported the first coexistence of ssSSc and anti-RNA polymerase III positivity.10 The anti-RNA polymerase III has been linked to a higher risk of GI angioectasia in scleroderma.11 Although antiphospholipid syndrome coexistence with scleroderma has been very rarely reported; our patient has antiphospholipid syndrome which would be the second reported case in the literature associated with ssSSc.12
In conclusion, ssSSc should always be considered in patients presenting with scleroderma internal organ features in the absence of cutaneous manifestations and the specific immunologic markers should be checked to avoid misdiagnosis and to monitor for other organs involvement.
Ethics and Consent
Institutional approval was not required for a case report. Written informed consent has been provided by the patient to have the case details published.
Disclosure
The authors report no funding and no conflicts of interest for this work.
References
1. Kucharz EJ, Kopeć-Mędrek M. Systemic sclerosis sine scleroderma. Adv Clin Exp Med. 2017;26(5):875–880. doi:10.17219/acem/64334
2. Black CM. Scleroderma–clinical aspects. J Intern Med. 1993;234:115. doi:10.1111/j.1365-2796.1993.tb00719.x
3. Abrams HL, Carnes WH, Eaton J. Alimentary tract in disseminated scleroderma with emphasis on small bowel. Arch Intern Med. 1954;94(1):61–81. doi:10.1001/archinte.1954.00250010067006
4. Rodnan GP, Fennel RH. Progressive systemic sclerosis sine scleroderma. JAMA. 1962;180(8):665–670. doi:10.1001/jama.1962.03050210027006
5. Poormoghim H, Lucas M, Fertig N, Medsger TA
6. Simeón-Aznar CP, Tolosa-Vilella C, Gabarró-Juliá L, et al. Systemic sclerosis sine scleroderma and limited cutaneous systemic sclerosis: similarities and differences. Clin Exp Rheumatol. 2014;6 Suppl 86:
7. Diab S, Dostrovsky N, Hudson M, et al. Systemic sclerosis sine scleroderma: a multicenter study of 1417 subjects. J Rheumatol. 2014;41(11):2179–2185. doi:10.3899/jrheum.140236
8. Pauling JD, Gunawardena H, Coghlan JG, Easaw J, Suntharalingam J, McHugh NJ. Pulmonary artery hypertension as the presenting feature of systemic sclerosis sine scleroderma. Rheumatology (Oxford). 2008;47(9):1431–1432. doi:10.1093/rheumatology/ken241
9. Molina JF, Anaya JM, Cabrera GE, Hoffman E, Espinoza LR. Systemic sclerosis sine scleroderma: an unusual presentation in scleroderma renal crisis. J Rheumatol. 1995;22(3):557–560.
10. Lee CM, Girnita D, Sharma A, Khanna S, Elwing JM. A unique presentation of anti-RNA Polymerase III positive systemic sclerosis sine scleroderma. Case Rep Rheumatol. 2016;2016:8536341.
11. Graf SW, Hakendorf P, Lester S, et al. South Australian Scleroderma Register: autoantibodies as predictive biomarkers of phenotype and outcome. Int J Rheum Dis. 2012;15(1):102–109. doi:10.1111/j.1756-185X.2011.01688.x
12. Leite P, de Carvalho JF. Systemic sclerosis sine scleroderma associated with antiphospholipid syndrome. Rheumatol Int. 2012;32(10):3265–3268. doi:10.1007/s00296-010-1482-6
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.