Back to Journals » International Journal of Nanomedicine » Volume 14
Systemic exposure to a single dose of ferucarbotran aggravates neuroinflammation in a murine model of experimental autoimmune encephalomyelitis
Authors Hsiao YP, Huang CH, Lin YC, Jan TR ![]()
Received 1 October 2018
Accepted for publication 15 January 2019
Published 15 February 2019 Volume 2019:14 Pages 1229—1240
DOI https://doi.org/10.2147/IJN.S189327
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Mian Wang
Yai-Ping Hsiao,1 Chung-Hsiung Huang,2 Yu-Chin Lin,3 Tong-Rong Jan1
1Department and Graduate Institute of Veterinary Medicine, School of Veterinary Medicine, National Taiwan University, Taipei, Taiwan; 2Department of Food Science, National Taiwan Ocean University, Keelung, Taiwan; 3Department of Medicinal Botanicals and Health Applications, College of Biotechnology & Bioresources, Da-Yeh University, Changhua, Taiwan
Background: Medicinal preparations of iron oxide nanoparticles (IONPs) have been used as MRI contrast agents for the diagnosis of hepatic tumors and the assessment of neuroinflammation and blood–brain barrier integrity. However, it remains mostly unclear whether exposure to IONPs affects neuroinflammation under disease conditions. The present study aims to investigate the impact of IONPs on autoimmune-mediated neuroinflammation using a murine model of experimental autoimmune encephalomyelitis (EAE) that mimics human multiple sclerosis.
Methods: Mice were either left untreated or immunized with myelin oligodendrocyte glycoprotein on day 0 followed by two injections of pertussis toxin for EAE induction. The EAE mice were intravenously administered with a single dose of the carboxydextran-coated IONPs, ferucarbotran (20 mg Fe/kg) and/or saline (as vehicle) on day 18. Symptoms of EAE were daily monitored until the mice were killed on day 30. Tissue sections of the brain and spinal cord were prepared for histopathological examinations. Iron deposition, neuron demyelination and inflammatory cell infiltration were examined using histochemical staining. The infiltration of microglial and T cells, and cytokine expression were examined by immunohistochemical staining and/or reverse transcription polymerase chain reaction (RT-PCR).
Results: Iron deposition was detected in both the brain and spinal cord of EAE mice 3 days post-ferucarbotran treatment. The clinical and pathological scores of EAE, percentage of myelin loss and infiltration of inflammatory cells into the spinal cord were significantly deteriorated in EAE mice treated with ferucarbotran. Furthermore, ferucarbotran treatment increased the number of CD3+, Iba-1+, IL-6+, Iba-1+TNF-α+ and CD3+IFN-γ+ cells in the spinal cord of EAE mice.
Conclusion: A single exposure to ferucarbotran exacerbated neuroinflammation and disease severity of EAE, which might be attributed to the enhanced activation of microglia and T cells. These results demonstrated that the pro-inflammatory effect of ferucarbotran on the central nervous system is closely associated with the deterioration of autoimmunity.
Keywords: experimental autoimmune encephalomyelitis, ferucarbotran, iron oxide nanoparticles, microglia, neuroinflammation, T cell, multiple sclerosis
Introduction
The biomedical potential of iron oxide nanoparticles (IONPs) is substantiated by a wide range of applications of this nanomaterial in both basic research and clinical fields, such as MRI contrast enhancement, cell labeling, cancer therapy and drug delivery.1 Contrast-enhanced MRI has been used for the diagnosis of hepatic tumors, and the assessment of neuroinflammation and blood–brain barrier (BBB) integrity.2,3 For most clinical applications, IONPs are mainly administered via intravenous routes. Upon reaching the systemic circulation, IONPs are rapidly engulfed by the reticuloendothelial system, resulting in exposure of phagocytic cells to the nanoparticles.4,5 In addition, recent studies have shown that IONPs are capable of crossing the BBB following intranasal exposure.6,7 Hence, the impact of IONP exposure on the neuroimmune homeostasis of the central nervous system (CNS) may be of concern.
Microglia are macrophage-like immune cells ubiquitously distributed in the CNS, which play a critical role in maintaining the neuroimmune homeostasis. Under normal physiological conditions, they serve as resident phagocytes possessing immune surveillance functions. However, the pro-inflammatory activity of microglia may contribute to the pathophysiology of many CNS diseases, such as Alzheimer’s disease, multiple sclerosis (MS) and Parkinson’s disease.8 Previous studies reported that intranasal exposure of mice to IONPs resulted in the recruitment, activation and proliferation of microglia in the olfactory bulb, hippocampus and striatum.9 Repeated exposure of mice to IONPs via intraperitoneal administration induced ROS generation and cell apoptosis in the brain, and neurobehavioral toxicity.10 Direct exposure of cultured microglial cells to IONPs caused nitric oxide production and cytotoxicity, and stimulated the production of pro-inflammatory cytokines.11,12 In addition, IONPs affected lipopolysaccharide-induced IL-1β production by interfering with the secretory lysosomal pathway of cytokine processing in microglial cells.13 These results suggest that exposure to IONPs may cause neurotoxicity and influence the functionality of microglia. To date, the majority of studies addressing the effects of IONPs on microglia have been conducted primarily in cell culture models or healthy animals. Evidence pertaining to the impact of IONPs on microglial functions and neuroinflammation under disease conditions is scarce.
MS is the most common inflammatory demyelination disease in the CNS. Murine models of experimental autoimmune encephalomyelitis (EAE) induced by myelin oligodendrocyte glycoprotein (MOG), which mimic many characteristic inflammatory features of MS, have been widely employed for MS research.14 Microglia and CD4+ T-helper (Th) cells are critically involved in the immunopathology of EAE.15 During the development of EAE, activated microglia may promote the differentiation of autoreactive Th1 cells that participate in subsequent immune reactions causing focal inflammation and tissue destruction.16 It is noticed that MRI may be employed to non-invasively diagnose and monitor neuroinflammatory diseases. In particular, IONP-assisted MRI has been used to track macrophages and monitor neuroinflammation in rodent models of EAE.17,18 Moreover, medicinal preparations of IONPs are promising contrast agents for MRI imaging of the CNS in patients with MS.19,20 In this context, MS patients may be subjected to IONP exposure, yet the impact of IONPs on neuroinflammation under disease conditions remains mostly unclear.
The objective of the present study was to explore the potential impact of IONPs on neuroinflammation in EAE mice. We report here that systemic exposure of MOG-induced EAE mice to a single dose of the carboxydextran-coated IONPs ferucarbotran aggravates the disease severity, inflammatory reactions and the activation of microglia and Th1 cells.
Materials and methods
Reagents
All chemicals and reagents were purchased from Sigma Chemical Co. (St Louis, MO, USA) unless otherwise stated. Antibodies, enzymes and reagents used for immunohistochemistry (IHC) were obtained from Abcam (Cambridge, MA, USA) or BioLegend (San Diego, CA, USA). The commercial product of carboxydextran-coated INOPs ferucarbotran (Resovist®; Schering, Berlin-Wedding, Germany) was used, whose size property was previously confirmed to be a monodisperse population of particles with an average diameter of 58.7 nm.21
Animals
Female C57BL/6 mice, 11–13 weeks old, were obtained from the National Laboratory Animal Center (Taipei, Taiwan). On arrival, mice were housed in plastic cages (five mice per cage) with free access to standard diet and water. Mice were kept in a humidity- (60%±20%), light- (12-h light/dark cycle) and temperature (25°C±2°C)-controlled environment. All procedures involving animals were carried out in accordance with the guidelines for the care and use of laboratory animals and approved by the Institutional Animal Care and Use Committee of the National Taiwan University.
EAE induction and IONP treatment
A commercial EAE induction kit (Hooke Laboratories, Lawrence, MA, USA) was used following the supplier’s instructions. In brief, mice were either left untreated (naïve; NA; n=8) or subcutaneously immunized with myelin oligodendrocyte glycoprotein (MOG)35–55 (0.2 mg/0.2 mL/mouse) emulsified in complete Freund’s adjuvant containing 4 mg/mL heat-inactivated Mycobacterium tuberculosis (H37RA) on the hind flank, followed by two intraperitoneal injections of pertussis toxin (PTX; 200 ng) at 3 and 27 hours post-MOG immunization (Figure 1). Clinical symptoms of EAE were scored and recorded daily from day 1 to day 30 in a blinded fashion, as follows: 0, no disease sign; 1, loss of tail tone; 2, hindlimb weakness; 3, hindlimb paralysis; 4, forelimb weakness or paralysis and hindlimb paralysis; 5, moribund or dead. The EAE mice were intravenously administered with a single dose of ferucarbotran (IONP; 20 mg/kg; 0.25 mL/mouse; n=11) and/or vehicle (VH; saline, 0.25 mL/mouse; n=10) on day 18. Three ferucarbotran-treated mice were killed on day 21 for the detection of iron deposition in the brain and spinal cord. The other mice were killed on day 30 and their spinal cords were isolated for further experimentation. Tissue blocks of the brain and spinal cord were fixed in 4% paraformaldehyde, embedded in paraffin and sectioned at a thickness of 5 μm.
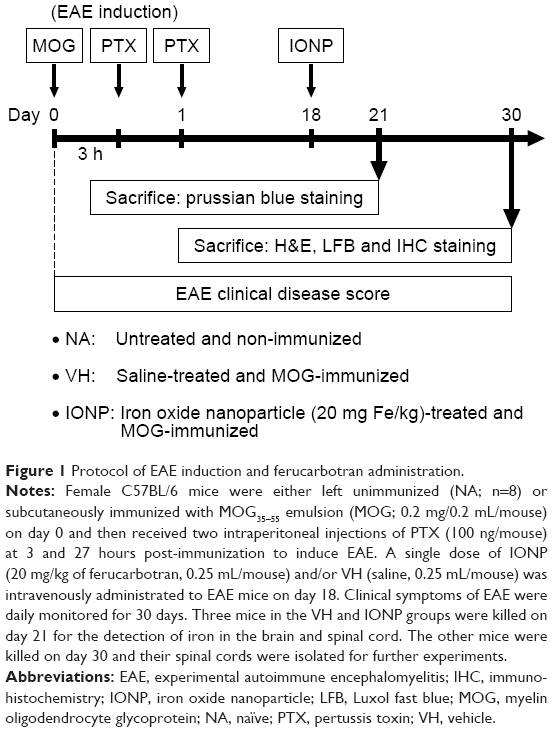 | Figure 1 Protocol of EAE induction and ferucarbotran administration. |
Prussian blue staining
Standard Prussian blue staining was employed to detect iron in the brain and spinal cord. In brief, tissue sections were incubated with 10% potassium ferrocyanide in 20% hydrochloric acid for 30 minutes and counterstained with nuclear fast red. After staining, the slides were visualized under an inverted microscope (Olympus IX83, Tokyo, Japan).
Histological examinations and neuronal demyelination
Tissue sections of the spinal cord were stained with H&E and Luxol fast blue (LFB) following standard protocols for histopathological examination and detection of myelin sheath, respectively.22,23 The degree of inflammatory cell infiltration was rated in H&E-stained sections in a blinded fashion using the following scoring scales from 0 to 3: 0, no inflammation; 1, small number of infiltrating cells; 2, numerous infiltrating cells; 3, widespread infiltration. The density of LFB-positive signals was measured using ImageJ image processing and analysis program (Bethesda, MD, USA). The percentage of myelination was expressed as the ratio between the myelinated plaque area and the total white matter area, and calculated as follows:
|
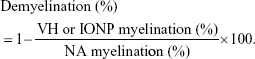
Immunohistochemistry
Tissue sections of the spinal cord were deparaffinized and then rehydrated following a standard procedure. The rehydrated slides were immersed in Trilogy™ (Cell Marque, Rocklin, CA, USA) at 121°C for 15 minutes for antigen retrieval. The endogenous peroxidase activity was quenched with 3% H2O2 in methanol and blocked with normal horse serum. Anti-mouse Iba-1 or CD3 antibody was applied on to each slide overnight at 4°C, followed by incubation with poly-horseradish peroxidase (HRP) at room temperature (RT) for 1 hour in the dark. For visualization, slides were treated with the HRP substrate 3,3′-diaminobenzidine for 3–7 minutes followed by hematoxylin counterstaining at RT for 5 minutes in the dark. Positive signals with a dark brown color on the entire spinal section of each mouse were counted manually in a blinded fashion.
For double staining, tissue sections were stained for Iba-1 or CD3 as described above without counterstaining. For the second staining, the Iba-1 and CD3-stained slides were incubated with anti-tumor necrosis factor-α (anti-TNF-α) and anti-interferon-γ (anti-IFN-γ) antibodies, respectively, overnight at 4°C, followed by incubation with a secondary antibody conjugated with alkaline phosphatase (AP) for 1 hour. For visualization, the slides were incubated with the AP substrate 3-amino-9-ethylcarbazole solution. Double positive signals with brown and dark red colors on the entire spinal section of each mouse were enumerated manually.
All slides were washed three times with PBS between each step after dewaxing. The slides were visualized using an inverted microscope (Olympus IX83, Tokyo, Japan).
Reverse transcription polymerase chain reaction (RT-PCR)
Total RNA from the spinal cord tissues was extracted using TRI reagent (Sigma) following the supplier’s instructions. The mRNA expression of IFN-γ, IL-6, TNF-α and β-actin was measured by RT-PCR. All isolated RNA samples were confirmed to be free of DNA contamination, as determined by the absence of products after PCR amplification in the absence of reverse transcriptase. For reverse transcription, 2 μg of total RNA was reverse-transcribed into cDNA using the SansiFASTTM.cDNA synthesis Kit (Bioline, London, UK) following the supplier’s instructions. The reverse transcription proceeded at 25°C for 10 minutes, 42°C for 15 minutes and then 85°C for 5 minutes. MyTaq™ Red Mix (Bioline, London, UK) and 20 μM of forward and reverse primers specific for the gene of interest were added to each cDNA sample for PCR. The PCR primers used were IFN-γ, 5′-CATGAAAATCCTGCAGAGCC-3′ and 5′-GGACAATCTCTTCCCCAGCC-3′; IL-6, 5′-TTGCCTTCTTGGGACTGATGCT-3′ and 5′-GTATCTCTCTGAAGGACTCTGG-3′; TNF-α, 5′-TACTGAACTTCGGGGTGATTGGTCC-3′ and 5′-CAGCCTTGTCCCTTGAAGAGAACC-3′; β-actin, 5′-ACTCATCGTACTCCTGCTTGCTGA-3′ and 5′-AGGGAAATCGTGCGTGACATCAAA-3′. PCR products were electrophoresed in 1.5% agarose gels and visualized by HealthView Nucleic Acid Stain (Genomics BioSci & Tech, Taiwan). Quantification was performed by assessing the optical density for the DNA bands (cytokine mRNA of interest) using Gel Doc EZ System (Bio-Rad Laboratories, Hercules, CA, USA) and ImageJ image processing and analysis program (Bethesda, MD, USA). The results are expressed as the density ratio between the gene of interest and the reference standard (β-actin).
Statistical analyses
Data are expressed as mean ± standard error (SE) for each experimental group. Data of clinical and pathological scores, the percentage of demyelinated areas and RT-PCR were analyzed using the Mann–Whitney test to compare the difference between the IONP group and the VH control. All others were analyzed using one-way ANOVA and Student’s t-test. A P-value <0.05 was defined as statistical significance.
Results
Iron deposition was detected in the CNS of ferucarbotran-treated EAE mice
It has been shown that IONPs administered via intranasal and intraperitoneal routes can enter the CNS and cause detrimental effects.7,10 In the present study, we first examined whether IONPs could reach the CNS of EAE mice after a single intravenous injection. The dose of 20 mg Fe/kg was chosen according to previous reports showing its effectiveness in affecting antigen-specific antibody production and inducing hepatic toxicity in mice.24,25 EAE mice were treated with ferucarbotran (IONP) and/or the vehicle (VH; saline) on day 18. Positive signals of Prussian blue staining were observed in both brain and spinal cord tissue sections of the IONP group obtained on day 21, whereas no positive staining were found in the VH group (Figure 2A and B). We also stained spinal sections obtained on day 30 and the results were negative (data not shown). These results demonstrated that IONPs could transfer from the bloodstream into the CNS in EAE mice, and the distributed iron was later cleared from the spinal cord.
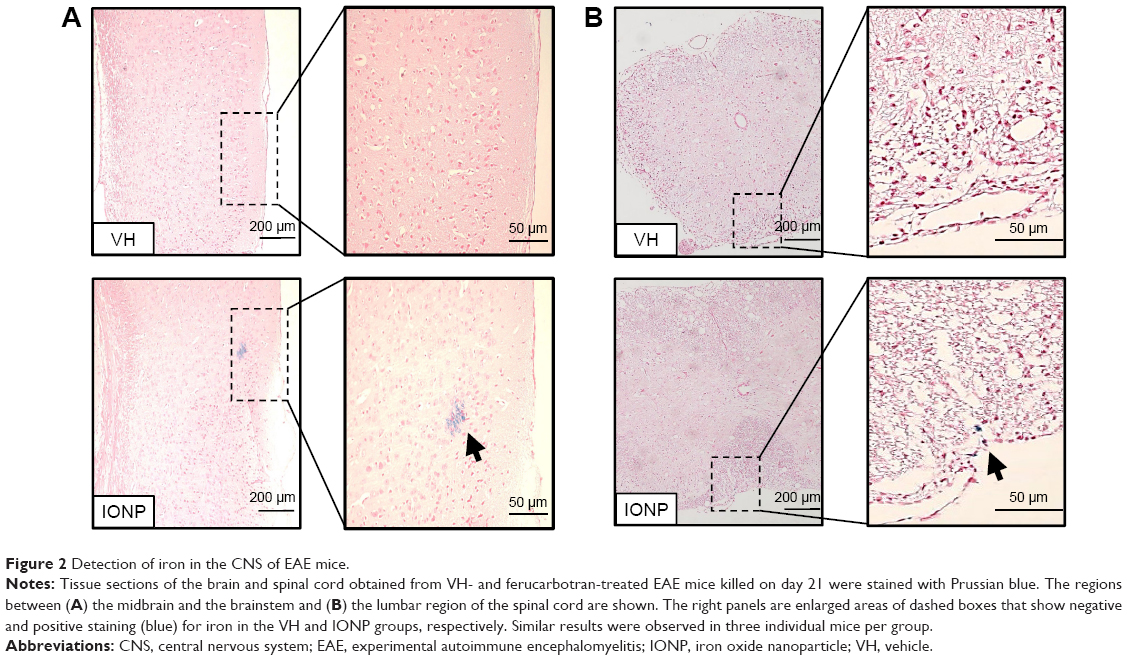 | Figure 2 Detection of iron in the CNS of EAE mice. |
Exposure to ferucarbotran aggravated the disease severity of EAE
We next addressed the impact of IONPs on the disease severity of EAE. Clinical symptoms of EAE were daily monitored throughout the entire experimental period. The onset of EAE symptoms was observed on day 12 in MOG-immunized mice, whereas mice in the NA group remained free of clinical symptoms, demonstrating a successful induction of EAE. The clinical score of EAE mice progressively increased and reached the peak phase approximately on day 18, on which a single dose of ferucarbotran (20 mg Fe/kg) and/or VH (saline) was administered via the tail vein. Notably, the daily, mean and cumulative clinical scores of the IONP group were significantly elevated from day 23 to day 30, compared to those of the VH group (Figure 3A–C).
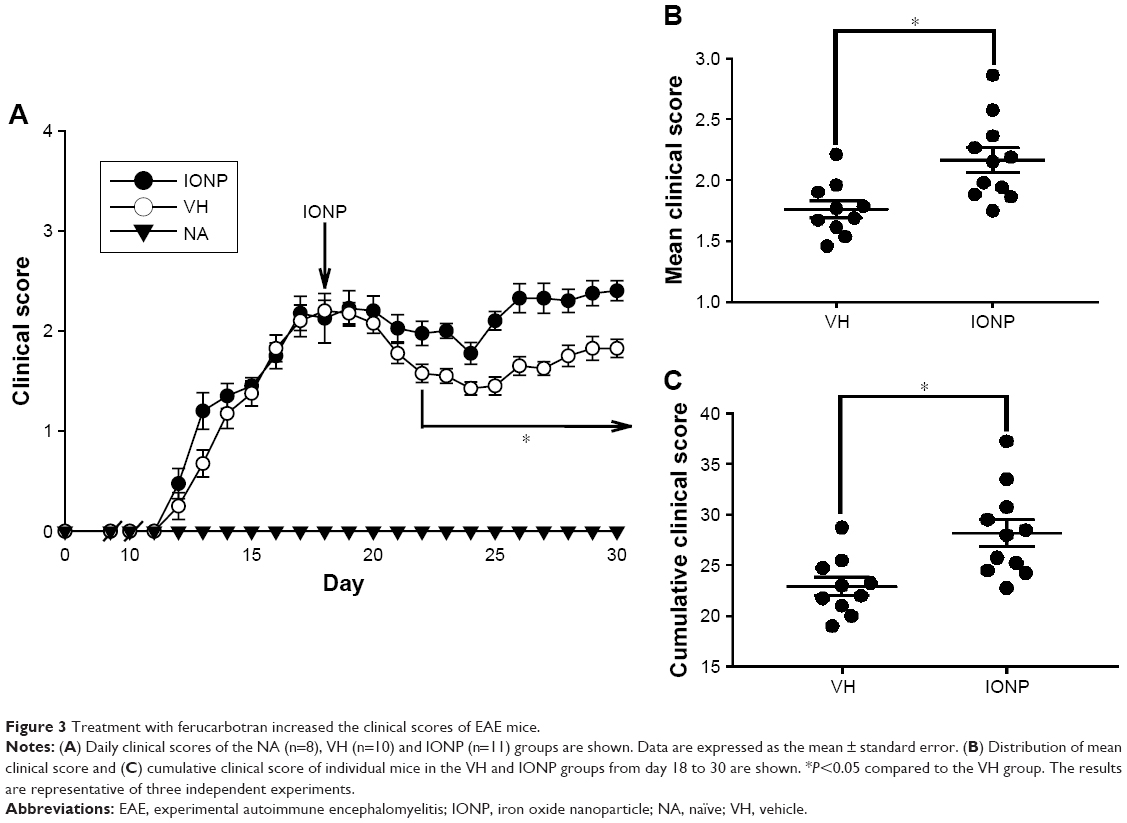 | Figure 3 Treatment with ferucarbotran increased the clinical scores of EAE mice. |
In addition to clinical symptoms on motor functions, demyelination in the CNS is another hallmark of EAE, which is commonly detected in patients with long-standing MS.26 The severity of demyelination in the spinal cords of EAE mice was examined by LFB staining. Images of stained sections showed several regions with manifest demyelination in EAE mice of both IONP and VH groups (Figure 4A). Notably, the percentage of demyelinated areas in the IONP group was significantly increased, compared to that in the VH group (Figure 4B).
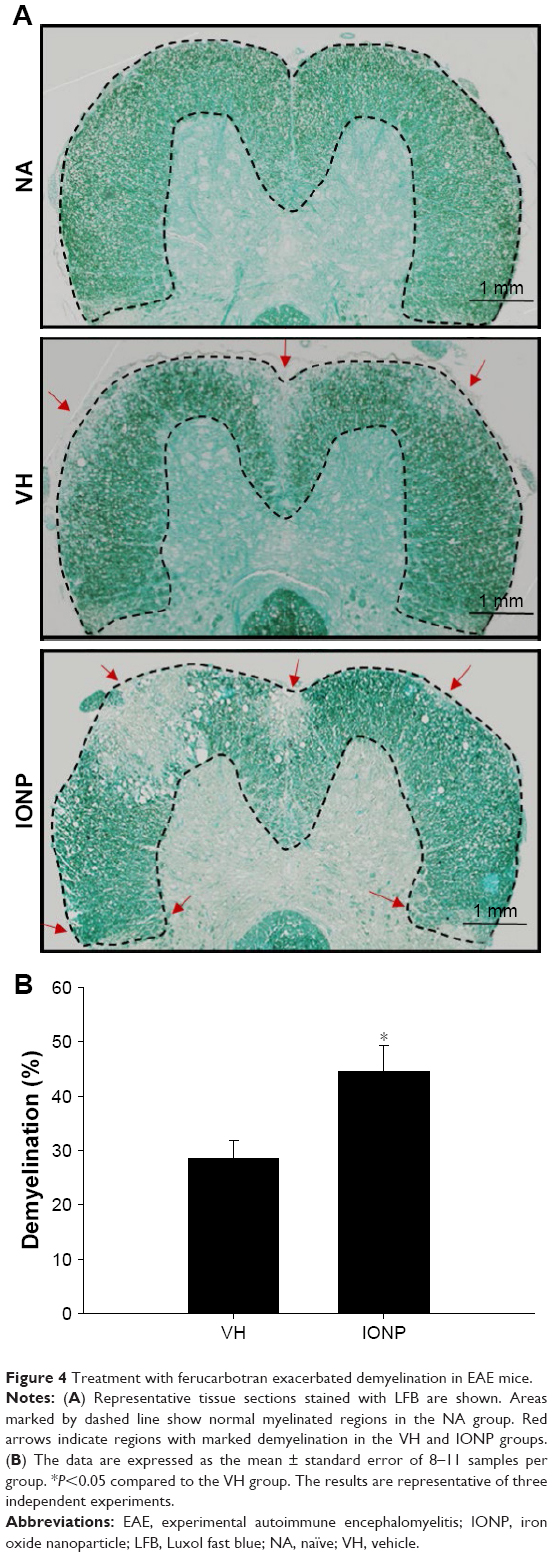 | Figure 4 Treatment with ferucarbotran exacerbated demyelination in EAE mice. |
Exposure to ferucarbotran enhanced inflammation associated with EAE
To explore the impact of IONPs on neuroinflammation in the EAE mice, histological analysis using H&E staining was performed. The results showed a marked infiltration of inflammatory cells in the myelin region of the spinal cords (VH vs NA) (Figure 5A), and a more extensive infiltration in the IONP group (IONP vs VH) (Figure 5A). The pathological score in the IONP group was significantly greater than that in the VH group (Figure 5B).
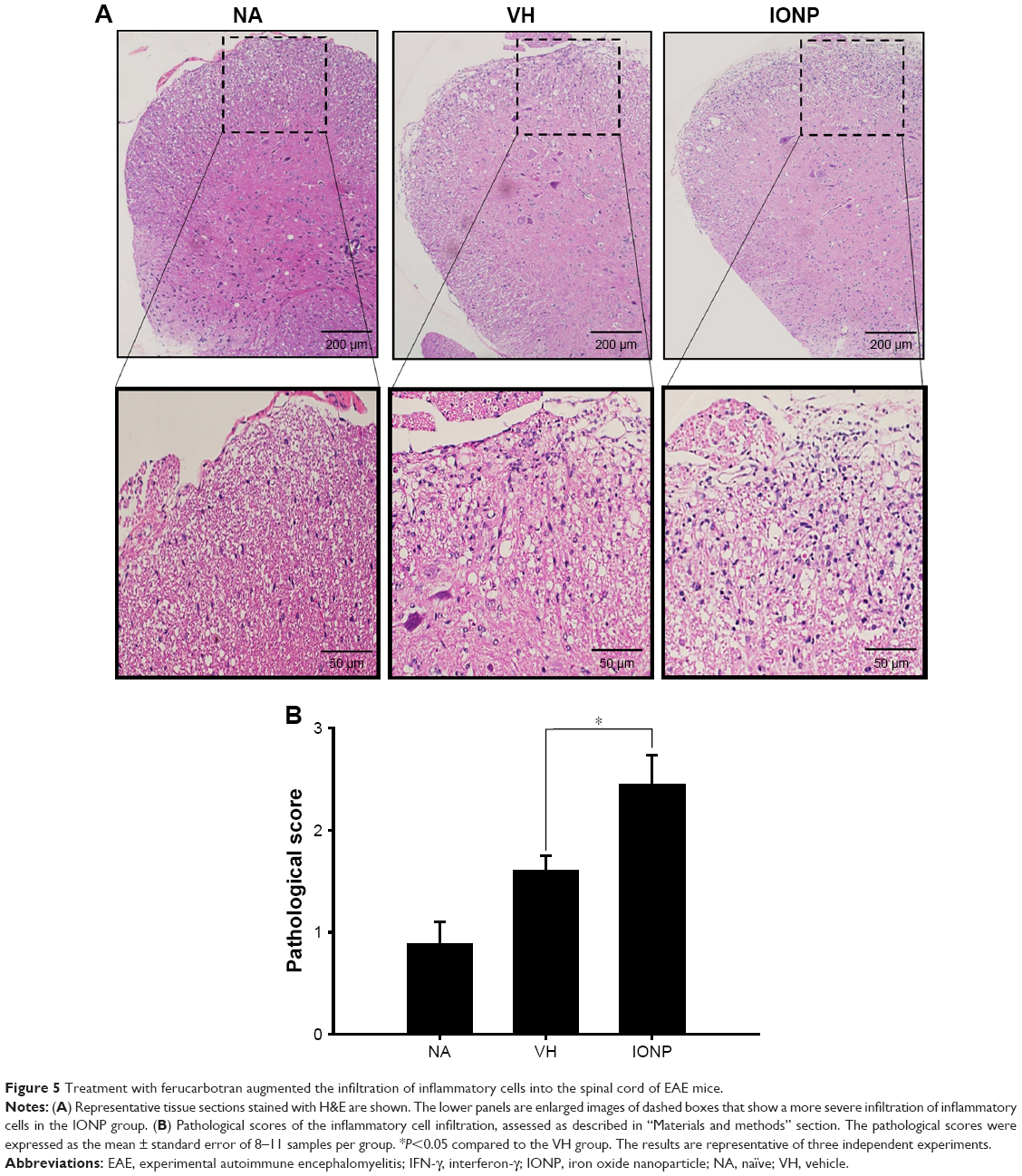 | Figure 5 Treatment with ferucarbotran augmented the infiltration of inflammatory cells into the spinal cord of EAE mice. |
Activated microglia are involved in the neuroinflammation of EAE by secreting pro-inflammatory cytokines.27 Hence, we examined the activation of microglia in the spinal cords using IHC staining. Treatment with ferucarbotran increased the expression of IL-6 and TNF-α, and the expression of Iba-1, the surface marker of activated microglia (Table 1). We next investigated whether the cytokines were expressed by activated microglia using IHC double staining. Positive signals of double staining were observed in the VH and IONP groups, whereas the NA group was negative (Figure 6A). The number of infiltrated Iba-1+TNF-α+ cells was significantly increased in the IONP group, compared to that in the VH group (Figure 6B). We further examined the mRNA expression of IL-6 and TNF-α in the spinal cord using RT-PCR, and the results confirmed the enhanced expression of these two pro-inflammatory cytokines in the IONP group (Figure 7A–C).
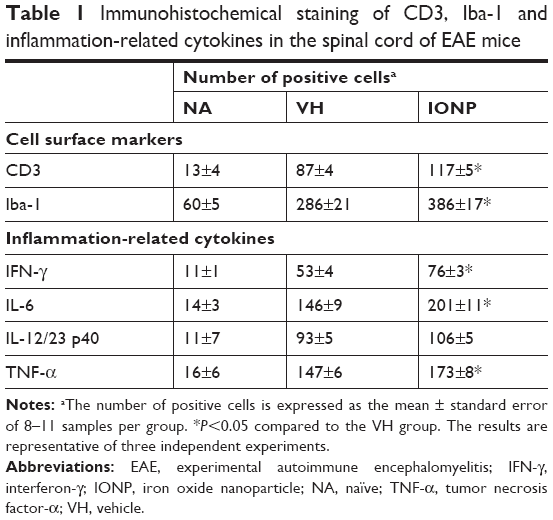 | Table 1 Immunohistochemical staining of CD3, Iba-1 and inflammation-related cytokines in the spinal cord of EAE mice |
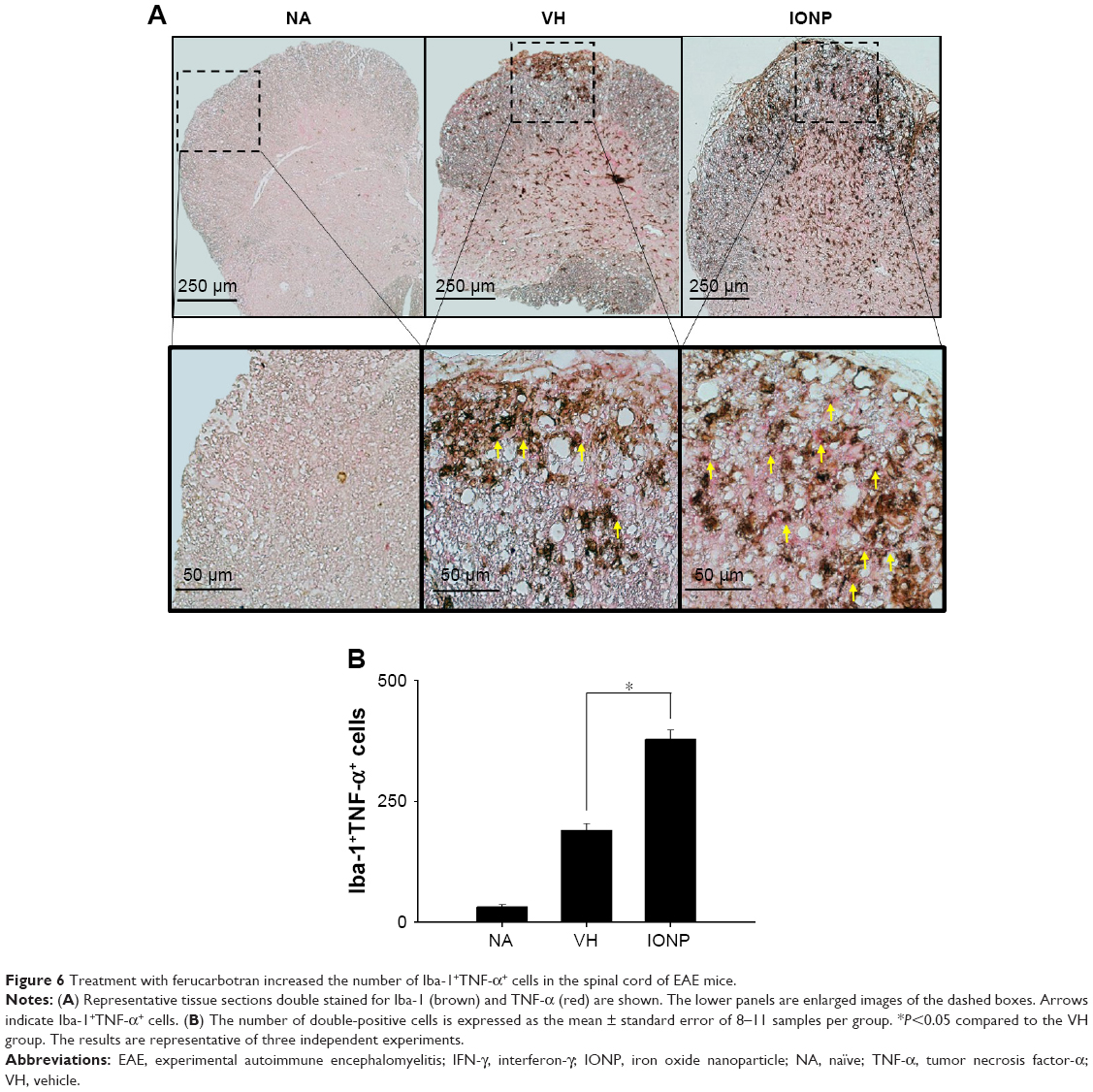 | Figure 6 Treatment with ferucarbotran increased the number of Iba-1+TNF-α+ cells in the spinal cord of EAE mice. |
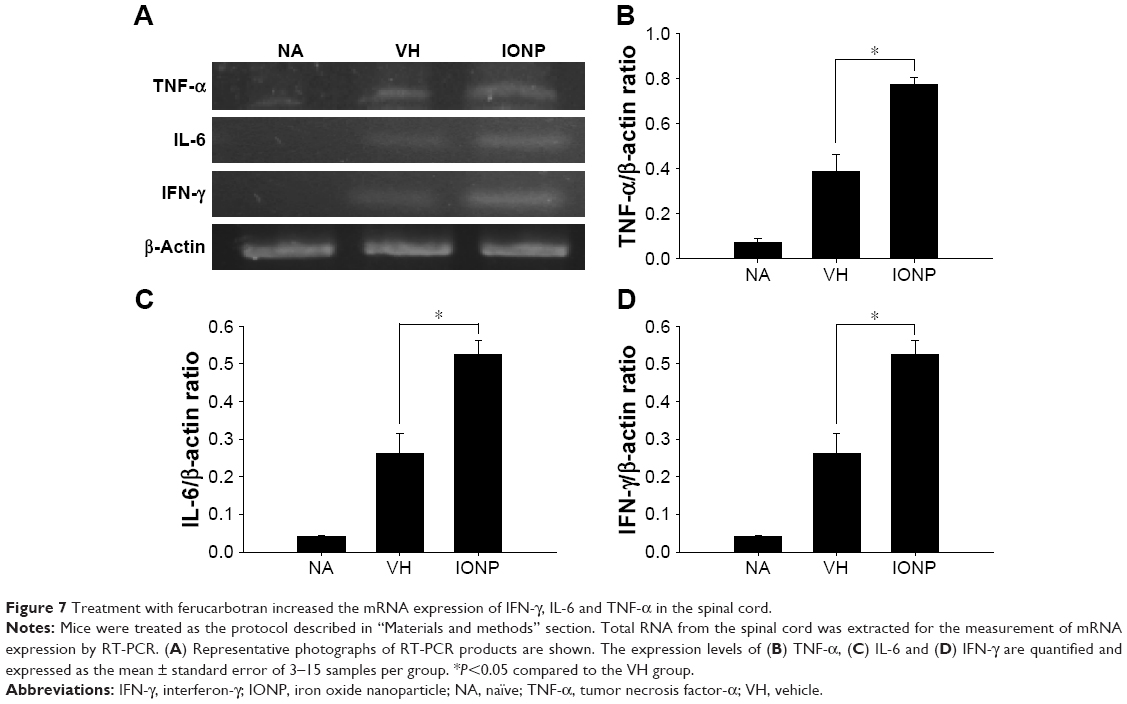 | Figure 7 Treatment with ferucarbotran increased the mRNA expression of IFN-γ, IL-6 and TNF-α in the spinal cord. |
Exposure to ferucarbotran increased the infiltration of CD3+IFN-γ+ T cells
In addition to microglia, Th1 and Th17 cells play a critical role in the pathophysiology of EAE.28 Hence, we further investigated the effect of ferucarbotran on the expression of the T-cell marker CD3 and the associated cytokines, including IL-12, IL-17 and IFN-γ. IL-12/23 p40 has been reported to play a crucial role in the pathogenesis of MS by driving the polarization of naïve CD4+ T cells toward Th1 and Th17 cells.29,30 Inhibition of the expression of IL-12/23 p40 dampened Th1 and Th17 immune responses and alleviated the severity of EAE.31 Our results showed that treatment with ferucarbotran did not affect the expression of IL-12/23 subunit p40, but significantly increased the number of CD3+ cells (Table 1). The mRNA expression of the signature Th1 cytokine IFN-γ in the spinal cord was augmented in the IONP group, compared to that in the VH group (Figure 7A and D). Moreover, results from IHC double staining revealed a heavy infiltration of CD3+ cells capable of expressing IFN-γ in the IONP group, as evidenced by a non-negligible number of CD3+IFN-γ+ cells (Figure 8A and B). However, treatment with ferucarbotran did not influence the expression of IL-17, the major Th17 cell-derived cytokine (data not shown).
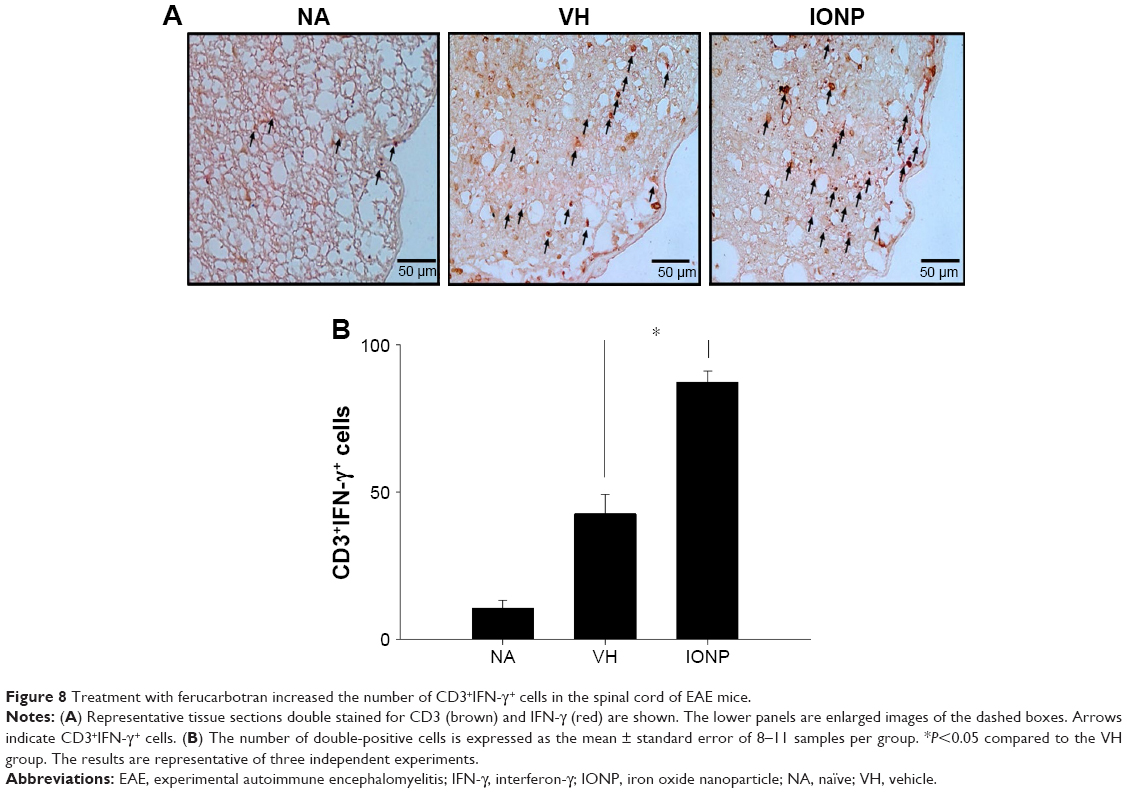 | Figure 8 Treatment with ferucarbotran increased the number of CD3+IFN-γ+ cells in the spinal cord of EAE mice. |
Discussion
Previous studies reported potential immunotoxic and neurotoxic properties of IONPs. For example, exposure of normal mice to a single intravenous dose of IONPs caused iron accumulation and detrimental effects in the liver, lungs and spleen, accompanied by increased levels of white blood cells, neutrophils and inflammation-related factors in the blood.32 In addition, intranasal and intraperitoneal administration with IONPs induced neurotoxicity in mice, such as neurobehavioral impairment and neurodendron degeneration.10,33 To date, little is known regarding the impact of IONPs on the pathophysiology of neuroinflammatory diseases, such as MS. In the present study, a murine model of EAE was employed to address whether IONPs affect the inflammatory responses under disease conditions. Our results showed that a single intravenous injection of ferucarbotran on the peak phase of EAE exacerbated the disease severity, which included the infiltration of inflammatory cells, the expression of pro-inflammatory cytokines and the demyelination of the spinal cord. In addition, we further demonstrated that the increased EAE severity caused by the IONPs might be attributed to the activation of both microglia and Th1 cells, which are crucial immune cells involved in the immunopathology of EAE.
In light of the increasingly medical applications of IONPs for the diagnosis of CNS diseases, the potential immunological effect of IONPs on neuroinflammation is a relevant health issue. Medicinal preparations of IONPs as MRI contrast agents have been used to evaluate the status of neurological diseases, suggesting that IONPs can penetrate the BBB.34 In the present study, it was confirmed that iron deposition could be detected in both the brain and spinal cord of EAE mice after a single intravenous injection with ferucarbotran. Iron accumulation in the CNS has been reported to cause a number of adverse outcomes, such as disturbing the function of the BBB and brain damage.35,36 Furthermore, exposure of healthy mice to IONPs resulted in defective motor coordination and demyelination.6,10 In line with these results, the present study showed that exposure to ferucarbotran aggravated the neuroinflammation and demyelination associated with EAE. Collectively, these lines of evidence demonstrates that exposure to IONPs may have severe impacts on the CNS in both normal and diseased animals.
The present study was conducted using the murine EAE model, the results of which cannot be directly extrapolated to humans. Whether exposure of MS patients to IONPs will cause a similar pro-inflammatory effect is currently unclear, which is an intriguing issue to be further explored in clinical settings. However, as IONPs have been used for brain imaging in clinical settings,19,20,37,38 our findings showing the aggravated disease severity in ferucarbotran-treated EAE mice implicate a potential health concern for MS patients exposed to these IONPs.
Ferucarbotran comprises magnetic iron oxide particles coated with carboxydextran, with an average diameter of 58.7 nm.21 Other medicinal preparations of IONPs are also used clinically, such as ferumoxytol. The iron oxide core of ferumoxytol is coated with carboxymethyldextran, and the mean hydrodynamic diameter of the particles is 30 nm. These two IONPs are coated with similar carbohydrate molecules, but their particle sizes are slightly different. It will be interesting to determine whether IONPs with similar coatings, but different sizes, induce similar pro-inflammatory effects on EAE neuroinflammation. This issue warrants a more comprehensive investigation to address the impact of various medicinal IONPs on neuroinflammation.
Microglia, the resident macrophage-like cells in the CNS, possess various immunological and neurobiological functions. Activated microglia may exhibit neuroprotective or harmful functions in the CNS. Microglia produce beneficial effects by the clearance of cell debris and myelin fragments; however, chronic and overactivated microglia may produce a large array of cytotoxic factors that contribute to the propagation of immune responses and inflammatory demyelination during EAE.39 Notably, IONPs have been shown to possess pro-inflammatory properties. For instance, IONPs could be taken up by monocytes and cause monocyte-driven endothelial cell dysfunction, leading to atherosclerosis.40 Treatment of macrophages with IONPs increased the production of ROS and the expression of activation markers, including CD40, CD80 and CD86.41 With regard to microglia, exposure to IONPs evoked cell proliferation, phagocytosis, and the generation of ROS, nitric oxide and pro-inflammatory cytokines.9,11 In the present study, exposure of EAE mice to ferucarbotran increased the number of activated microglia (Iba-1+ cells) and the expression of pro-inflammatory cytokines (IL-6 and TNF-α) in the spinal cord. We further revealed that the activated Iba-1+ microglia were capable of expressing TNF-α, indicating the functional activation of microglia. On the basis of these findings, we speculated that overactivation of microglia might be one of the underlying immunological mechanisms, which contributes to ferucarbotran-mediated aggravation of EAE.
In addition to microglia, autoreactive Th1 and Th17 cells are competent T-cell subsets involved in the autoimmune reactions of EAE.15 The present study showed that treatment with ferucarbotran promoted the infiltration of Th1 cells, as evidenced by the increased number of CD3+ cells capable of expressing IFN-γ+ in the spinal cord of EAE mice. However, the expression of IL-12/23 p40 and IL-17 was unaffected. These results suggest that Th1 cells may play a more dominant role in the IONP-mediated effects on EAE. Augmentation by IONPs of Th1-cell responses has been reported in several murine models. For example, intravenous administration of IONPs enhanced the production of Th1 cytokines, including IL-2 and IFN-γ, but had no influence on Th2 cytokines in the peripheral blood of normal mice.42 Pulmonary accumulation of IONPs augmented Th1 cell-mediated immunity by promoting the function of antigen-presenting cells in the lungs.43 IONP-induced exosomes targeted antigen-presenting cells to initiate Th1-type immune activation.44 On the basis of these lines of evidence, exposure to IONPs exhibited immunomodulatory effects favoring Th1 polarization, implicating the potential of IONPs to aggravate Th1-dominant immune disorders.
Conclusion
The present study demonstrated that a single intravenous administration of ferucarbotran markedly exacerbated EAE severity and enhanced the activation of microglial and Th1 cells. The IONP-mediated aggravation of neuroinflammation associated with EAE implicates a potential risk of IONP exposure on neuroimmunity in patients with MS.
Acknowledgments
This work was supported by grants 104-2320-B-002-024-MY3 and 107-2320-B-002-020 from the Ministry of Science and Technology, Executive Yuan, Taiwan.
Disclosure
The authors report no conflicts of interest in this work.
References
Ittrich H, Peldschus K, Raabe N, Kaul M, Adam G. Superparamagnetic iron oxide nanoparticles in biomedicine: applications and developments in diagnostics and therapy. Fortschr Röntgenstr. 2013;185(12):1149–1166. | ||
Weinstein JS, Varallyay CG, Dosa E, et al. Superparamagnetic iron oxide nanoparticles: diagnostic magnetic resonance imaging and potential therapeutic applications in neurooncology and central nervous system inflammatory pathologies, a review. J Cereb Blood Flow Metab. 2010;30(1):15–35. | ||
Busquets MA, Espargaró A, Sabaté R, Estelrich J. Magnetic nanoparticles cross the blood-brain barrier: when physics rises to a challenge. Nanomaterials. 2015;5(4):2231–2248. | ||
Hamm B, Staks T, Taupitz M, et al. Contrast-enhanced MR imaging of liver and spleen: first experience in humans with a new superparamagnetic iron oxide. J Magn Reson Imaging. 1994;4(5):659–668. | ||
Wang J, Chen Y, Chen B, et al. Pharmacokinetic parameters and tissue distribution of magnetic Fe(3)O(4) nanoparticles in mice. Int J Nanomedicine. 2010;5:861–866. | ||
Wang B, Feng WY, Wang M, et al. Transport of intranasally instilled fine Fe2O3 particles into the brain: micro-distribution, chemical states, and histopathological observation. Biol Trace Elem Res. 2007;118(3):233–243. | ||
Wu J, Ding T, Sun J. Neurotoxic potential of iron oxide nanoparticles in the rat brain striatum and hippocampus. Neurotoxicology. 2013;34:243–253. | ||
Gupta N, Shyamasundar S, Patnala R, et al. Recent progress in therapeutic strategies for microglia-mediated neuroinflammation in neuropathologies. Expert Opin Ther Targets. 2018;22(9):765–781. | ||
Wang Y, Wang B, Zhu MT, et al. Microglial activation, recruitment and phagocytosis as linked phenomena in ferric oxide nanoparticle exposure. Toxicol Lett. 2011;205(1):26–37. | ||
Dhakshinamoorthy V, Manickam V, Perumal E. Neurobehavioural toxicity of iron oxide nanoparticles in mice. Neurotox Res. 2017;32(2):187–203. | ||
Xue Y, Wu J, Sun J. Four types of inorganic nanoparticles stimulate the inflammatory reaction in brain microglia and damage neurons in vitro. Toxicol Lett. 2012;214(2):91–98. | ||
Petters C, Thiel K, Dringen R. Lysosomal iron liberation is responsible for the vulnerability of brain microglial cells to iron oxide nanoparticles: comparison with neurons and astrocytes. Nanotoxicology. 2016;10(3):332–342. | ||
Wu HY, Chung MC, Wang CC, Huang CH, Liang HJ, Jan TR. Iron oxide nanoparticles suppress the production of IL-1beta via the secretory lysosomal pathway in murine microglial cells. Part Fibre Toxicol. 2013;10:46. | ||
Constantinescu CS, Farooqi N, O’Brien K, Gran B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br J Pharmacol. 2011;164(4):1079–1106. | ||
Murphy AC, Lalor SJ, Lynch MA, Mills KHG. Infiltration of Th1 and Th17 cells and activation of microglia in the CNS during the course of experimental autoimmune encephalomyelitis. Brain Behav Immun. 2010;24(4):641–651. | ||
Legroux L, Arbour N. Multiple sclerosis and T lymphocytes: an entangled story. J Neuroimmune Pharmacol. 2015;10(4):528–546. | ||
Millward JM, Schnorr J, Taupitz M, Wagner S, Wuerfel JT, Infante-Duarte C. Iron oxide magnetic nanoparticles highlight early involvement of the choroid plexus in central nervous system inflammation. ASN Neuro. 2013;5(2):e00110:AN20120081. | ||
Kirschbaum K, Sonner JK, Zeller MW, et al. In vivo nanoparticle imaging of innate immune cells can serve as a marker of disease severity in a model of multiple sclerosis. Proc Natl Acad Sci U S A. 2016;113(46):13227–13232. | ||
Dousset V, Brochet B, Deloire MS, et al. MR imaging of relapsing multiple sclerosis patients using ultra-small-particle iron oxide and compared with gadolinium. AJNR Am J Neuroradiol. 2006;27(5):1000–1005. | ||
Toth GB, Varallyay CG, Horvath A, et al. Current and potential imaging applications of ferumoxytol for magnetic resonance imaging. Kidney Int. 2017;92(1):47–66. | ||
Shen CC, Liang HJ, Wang CC, Liao MH, Jan TR. A role of cellular glutathione in the differential effects of iron oxide nanoparticles on antigen-specific T cell cytokine expression. Int J Nanomedicine. 2011;6:2791–2798. | ||
Wang J, Chen F, Zheng P, et al. Huperzine A ameliorates experimental autoimmune encephalomyelitis via the suppression of T cell-mediated neuronal inflammation in mice. Exp Neurol. 2012;236(1):79–87. | ||
Scuteri A, Donzelli E, Rigolio R, et al. Therapeutic administration of mesenchymal stem cells abrogates the relapse phase in chronic relapsing-remitting EAE. J Stem Cell Res Ther. 2015;5(2):262. | ||
Shen CC, Wang CC, Liao MH, Jan TR. A single exposure to iron oxide nanoparticles attenuates antigen-specific antibody production and T-cell reactivity in ovalbumin-sensitized BALB/c mice. Int J Nanomedicine. 2011;6:1229–1235. | ||
Sadeghi L, Babadi VY, Espanani HR. Toxic effects of the Fe2O3 nanoparticles on the liver and lung tissue. Bratislava Medical Journal. 2015;116(06):373–378. | ||
Lassmann H. Multiple sclerosis pathology. Cold Spring Harb Perspect Med. 2018;8(3):a028936. | ||
Luo C, Jian C, Liao Y, et al. The role of microglia in multiple sclerosis. Neuropsychiatr Dis Treat. 2017;13:1661–1667. | ||
Jäger A, Dardalhon V, Sobel RA, Bettelli E, Kuchroo VK. Th1, Th17, and Th9 effector cells induce experimental autoimmune encephalomyelitis with different pathological phenotypes. J Immunol. 2009;183(11):7169–7177. | ||
Brahmachari S, Pahan K. Role of cytokine p40 family in multiple sclerosis. Minerva Med. 2008;99(2):105–118. | ||
Lim HX, Hong HJ, Jung MY, Cho D, Kim TS. Principal role of IL-12p40 in the decreased Th1 and Th17 responses driven by dendritic cells of mice lacking IL-12 and IL-18. Cytokine. 2013;63(2):179–186. | ||
Guo W, Wang C, Wang X, et al. A novel human truncated IL12rβ1-Fc fusion protein ameliorates experimental autoimmune encephalomyelitis via specific binding of p40 to inhibit Th1 and Th17 cell differentiation. Oncotarget. 2015;6(30):28539–28555. | ||
Park EJ, Oh SY, Kim Y, et al. Distribution and immunotoxicity by intravenous injection of iron nanoparticles in a murine model. J Appl Toxicol. 2016;36(3):414–423. | ||
Wang B, Feng W, Zhu M, et al. Neurotoxicity of low-dose repeatedly intranasal instillation of nano- and submicron-sized ferric oxide particles in mice. Journal of Nanoparticle Research. 2009;11(1):41–53. | ||
Yarjanli Z, Ghaedi K, Esmaeili A, Rahgozar S, Zarrabi A. Iron oxide nanoparticles may damage to the neural tissue through iron accumulation, oxidative stress, and protein aggregation. BMC Neurosci. 2017;18(1):51. | ||
Sripetchwandee J, Pipatpiboon N, Chattipakorn N, Chattipakorn S. Combined therapy of iron chelator and antioxidant completely restores brain dysfunction induced by iron toxicity. PLoS One. 2014;9(1):e85115. | ||
Schneider SA. Neurodegeneration with brain iron accumulation. Curr Neurol Neurosci Rep. 2016;16(1):9. | ||
Wáng YX, Idée JM. A comprehensive literatures update of clinical researches of superparamagnetic resonance iron oxide nanoparticles for magnetic resonance imaging. Quant Imaging Med Surg. 2017;7(1):88–122. | ||
Maarouf A, Ferré JC, Zaaraoui W, et al. Ultra-small superparamagnetic iron oxide enhancement is associated with higher loss of brain tissue structure in clinically isolated syndrome. Mult Scler. 2016;22(8):1032–1039. | ||
Rodgers JM, Miller SD. Cytokine control of inflammation and repair in the pathology of multiple sclerosis. Yale J Biol Med. 2012;85(4):447–468. | ||
Zhu MT, Wang B, Wang Y, et al. Endothelial dysfunction and inflammation induced by iron oxide nanoparticle exposure: risk factors for early atherosclerosis. Toxicol Lett. 2011;203(2):162–171. | ||
Mulens-Arias V, Rojas JM, Pérez-Yagüe S, Morales MP, Barber DF. Polyethylenimine-coated SPIONs trigger macrophage activation through TLR-4 signaling and ROS production and modulate podosome dynamics. Biomaterials. 2015;52:494–506. | ||
Chen BA, Jin N, Wang J, et al. The effect of magnetic nanoparticles of Fe3O4 on immune function in normal ICR mice. Int J Nanomedicine. 2010;5:593–599. | ||
Park EJ, Oh SY, Lee SJ, et al. Chronic pulmonary accumulation of iron oxide nanoparticles induced Th1-type immune response stimulating the function of antigen-presenting cells. Environ Res. 2015;143(Pt A):138–147. | ||
Zhu M, Tian X, Song X, et al. Nanoparticle-induced exosomes target antigen-presenting cells to initiate Th1-type immune activation. Small. 2012;8(18):2841–2848. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.