Back to Journals » OncoTargets and Therapy » Volume 9
Synthetic treatment of intracranial peripheral primitive neuroectodermal tumor with multiple metastasis: a case report
Authors Zhang Y, Li H, Li Z, Liu M, Yang L, Fan L, Huang C, Li B ![]()
Received 10 January 2016
Accepted for publication 8 April 2016
Published 2 June 2016 Volume 2016:9 Pages 3327—3333
DOI https://doi.org/10.2147/OTT.S103988
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Min Li
Yang Zhang,1,2 Hongsheng Li,2 Zongjuan Li,3 Ming Liu,1,2 Linke Yang,2 Liyuan Fan,2 Chengsuo Huang,2 Baosheng Li2
1School of Medicine and Life Sciences, Jinan University-Shandong Academy of Medical Sciences, 2Department of Radiation Oncology, Shandong Cancer Hospital Affiliated to Shandong University, Shandong Academy of Medical Sciences, Jinan, Shandong, 3Department of Radiation Oncology, The Second Hospital of Dalian Medical University, Dalian, Liaoning, People’s Republic of China
Abstract: Ewing sarcomas (ES) and peripheral primitive neuroectodermal tumors (pPNET) are now thought to belong to the same tumor family. Ewing sarcoma family tumor (ESFT) members commonly originate in bones and soft tissues. However, a few published articles describe ESFT arising from cranial cavities. Pathologically, ES/pPNET are composed of small round cells. Unambiguous distinction between pPNET and other small round cell tumors, in particular central PNET, is of clinical significance. Definitive diagnoses of pPNET can be obtained through CD99 (MIC2 gene product) membrane positivities and molecular identifications of chromosomal rearrangements between EWS and ETS family genes. Multimodal approaches comprising surgical resections, radiotherapies, and chemotherapies are required for the treatment of ESFT. Decompressive medical measures are preferentially performed when epidural masses are compressing spinal cords. In cases of ES-induced brain herniations, emergent radiotherapies may serve as effective tools. We report a case of multiple disseminated intracranial ES/pPNET for which synthetic treatments were used.
Keywords: primitive neuroectodermal tumors, brain neoplasms, spinal tumors, neoplasm metastases
Introduction
Ewing sarcomas (ES) and peripheral primitive neuroectodermal tumors (pPNET) are currently classified as a tumor family because they share unifying histopathological, immunohistochemical, and molecular features.1,2 Typical sites of occurrence for ESFT are in bones and soft tissues. However, rare cases have reported pPNET originating from cranial cavities. Here, we report a case of intracranial pPNET with multiple metastases.
Case
Informed consent was obtained from the patient and her family. The study was approved by the Institutional Review Board of Radiation Oncology, Shandong Cancer Hospital Affiliated to Shandong University. A 28-year-old female presented with progressive back pains occurring over 2 months’ duration with pain radiating to the ipsilateral shoulder. She also had a 1-month history of upper-extremity weaknesses and a recent onset of slight headaches. No antecedent trauma had occurred in these areas. She denied having a history of nausea, vomiting, seizures, or losses of consciousness. Physical examination upon hospital admission revealed grade 4/5 power (Medical Research Council Scale) in the left upper limb and two palpable masses in the bilateral frontal regions (5 cm above the eyebrows). Further neurological examinations revealed no positive signs. Lactate dehydrogenase and alkaline phosphatase levels were both increased (353 and 478 U/L, respectively).
Magnetic resonance imaging (MRI; Figure 1) confirmed the presence of an extradural lesion at the spinal C7 level. The mass compressed the spinal cord, and a distinct heterogeneous enhancement was observed in the tumor. The MRI also revealed patch-like lesions with high signal intensity (in T2 images) on the C2–5, C7, T2–5 level vertebra. Further brain MRI evaluations (Figure 2) showed two extramedullary meningioma-like masses with bone involvements and frontal brain parenchymal compressions. A metastatic workup with a positron emission tomography and computed tomography scan was subsequently performed, and no other abnormalities were observed.
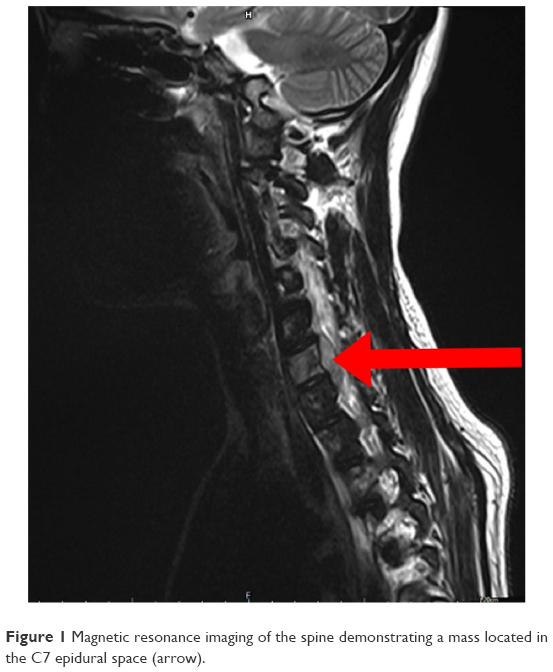 | Figure 1 Magnetic resonance imaging of the spine demonstrating a mass located in the C7 epidural space (arrow). |
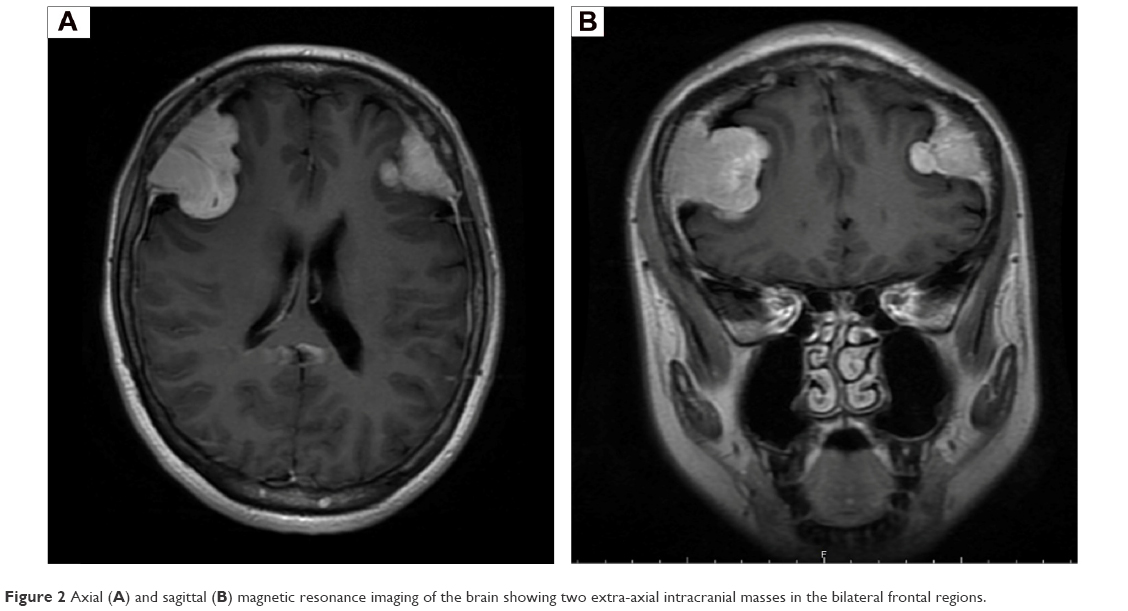 | Figure 2 Axial (A) and sagittal (B) magnetic resonance imaging of the brain showing two extra-axial intracranial masses in the bilateral frontal regions. |
In view of the severe spinal cord compression of the C7 level extradural lesion, the patient was subjected to laminectomy and mass removal. At laminectomy, a dark red mass was found in the epidural space. The mass had adhered slightly to the dura mater, compressing the adjacent dural sac and spinal cord. The tumor was easily separated from the adjoining dura mater, allowing a gross total resection to be performed. Postoperatively, symptoms of back pain and upper-extremity weakness were dramatically resolved. After the surgery, therapeutic agents were administered, including dehydrants, steroids, lansoprazole, and neuro nutrition. Gross visualizations by histopathological analyses showed that the epidural tumor was composed of dark-red soft tissue, which measured ~1.2 cm at its largest diameter. Upon microscopic analyses of hemotoxylin and eosin stained sections, the neoplasm displayed monotonous, closely packed small round blue cells with high nuclear-to-cytoplasmic ratios (Figure 3A). Typical Homer Wright rosettes were not observed. Using immunohistochemical analyses, the cells strongly expressed CD99 (Figure 3B), vimentin, and Bcl-2. Stains for neuron specific enolase (NSE), synaptophysin (Syn), and epithelial membrane antigen (EMA) were negative. For this case, further chromosomal translocation studies were not performed. Based on the collective histopathological and immunochemical findings, a diagnosis of ES/pPNET was made.
 | Figure 3 Hematoxylin and eosin staining (A) showing small round blue cells with a high nuclear-to-cytoplasmic ratios, and immunostaining (B) demonstrating positivity for CD99. Magnification ×100. |
On day 10 after the surgery, the patient’s consciousness level deteriorated gradually and headache intensity increased. Upon examination, her right pupil was dilated to 5 mm, and bilateral papilledema was detected. A subsequent head computed tomography scan indicated cerebral hernia with ventricle compression. The patient was immediately transferred to our treatment center.
When arriving at our unit, the patient was in a state of stupor. Given the special symptoms she presented and the radiosensitivity of ES, the oncology team performed emergent radiotherapy. One hour later, the first four Gy were given to the whole brain. Concomitant infusion treatment strategies were also given, and included dehydrants, hormones, and parenteral and neuro nutritions. After the delivery of eight Gy/two fractions to the whole brain, her consciousness level gradually improved. Therefore, subsequent boosts of 21 Gy/seven fractions were carried out on the cerebral epidural masses. After implementation of radiation therapies, the patient underwent eight total cycles of multiagent chemotherapy treatments (ie, vincristine, cyclophosphamide, and doxorubicin) every 21 days. The patient tolerated these treatments well with the exception for myelosuppression. A complete response was observed after four cycles of chemotherapy. Subsequently, the patient has been on regular follow-up and, 13 months after diagnosis, has remained disease-free.
Discussion
ES and pPNET are now commonly considered to be one tumor family because they share histopathological features and molecular properties and have been renamed Ewing sarcoma family tumor (ESFT).1,2 ESFT preferentially afflicts children and adolescents, exhibiting a slight predilection for females. Typically, ESFT occur in bones or soft tissues; origination from cranial cavities is rare. To our knowledge, only 29 cases have been reported in the body of English ESFT literature (Table 1). Despite the present case acts as multifocal disease, it was still reasonable to regard the intracranial lesions as the primary sites, because 1) size discrepancy surely exists between the intracranial masses and the spinal epidural lesion, 2) most spinal PNETs are caused by “drop” metastasis from an intracranial tumor through cerebrospinal fluid circulation,3 3) 2 years prior to symptom of spinal tumor development, frontal masses had been palpated, and 4) at laminectomy, the spinal epidural mass was only slightly adhered to the surrounding tissue and could be easily separated. Conversely, the cerebral lesions were significantly invasive, as indicated by MRI.
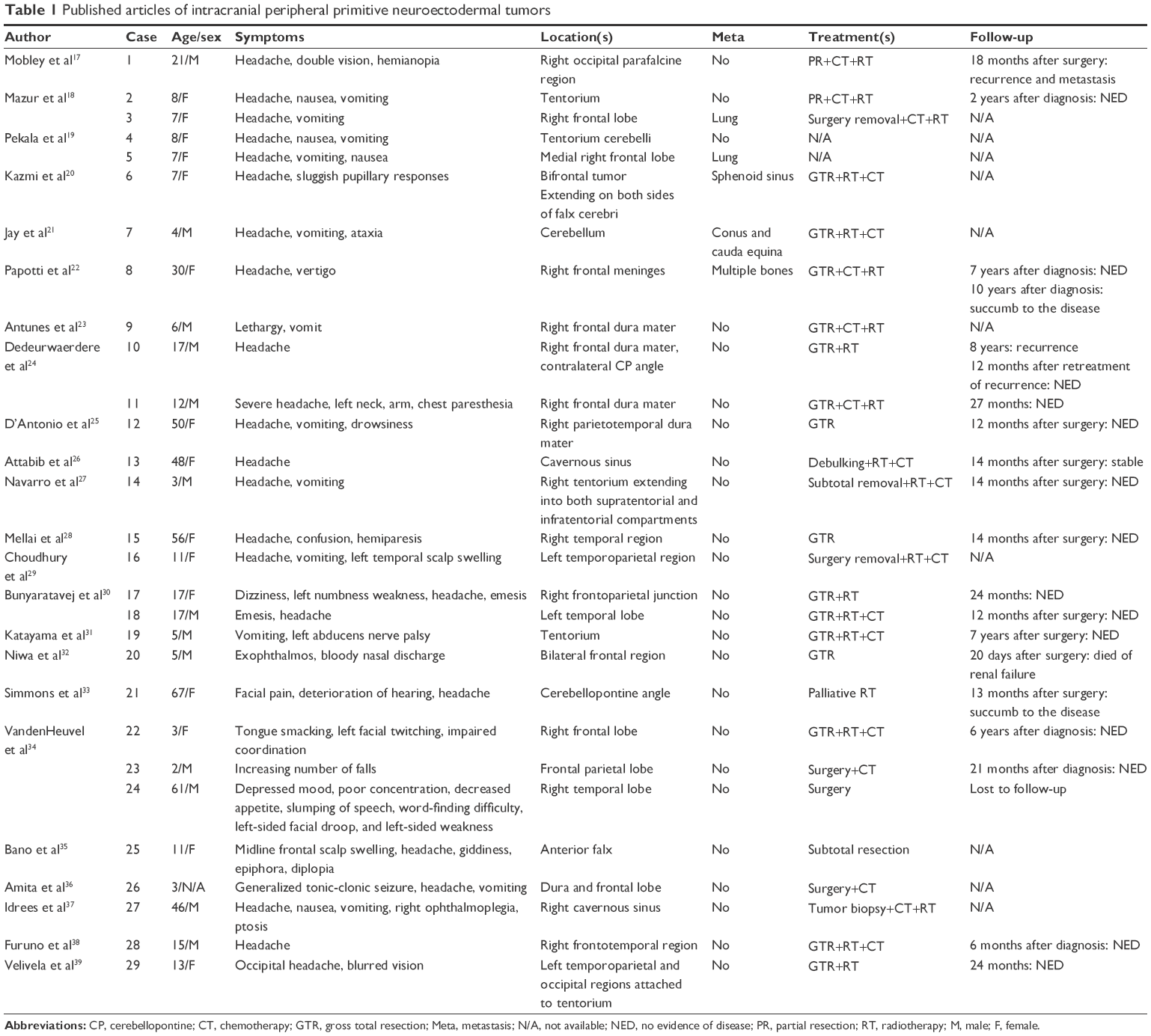 | Table 1 Published articles of intracranial peripheral primitive neuroectodermal tumors |
Unambiguous distinction between pPNET and other small round cell tumors, in particularly central PNET, is of clinical significance. CD99-positive membrane staining is detected in nearly all cases of pPNET, which is a highly reliable biomarker, but on no account pathognomonic; CD99 also identifies other small round cell tumors, albeit with staining patterns that diverge from those observed in pPNET.4 Advanced molecular detection of EWS–ETS gene fusions are confirmatory for final pPNET diagnoses.5
Multimodal strategies, including surgery, radiation therapy, and chemotherapy, are required for the treatment of intracranial ESFT with epidural space metastases.6 Priority must be given to laminectomies and tumor removals when epidural masses present with spinal cord compressions. These techniques aim to obtain specimens for pathologic diagnoses and avoid permanent neurological dysfunctions.7,8 In these cases, close attention should be made to intracranial tumor masses to prevent disease progression due to cure delays and surgical stressors. In the present case, symptoms and signs of intracranial neoplasms became suddenly severe with subsequent development of herniation. For such cases, typical care includes immediate surgical measures to relieve cerebral hernias. Under certain circumstances, feasibility and safety of such surgeries may be dramatically limited (eg, tumors are in high-risk locations or patient rejections of further traumatic treatments). Thus, in some cases, nonsurgical methods may be useful treatment modalities. In this case, radiotherapy was chosen, with some uncertainty, as the primary treatment modality given the highly radiosensitivity of ES. In addition to radiation, other therapies, in particular dehydrants and hormone treatments, were also clinically crucial. First, they directly reduced intracranial pressures. Moreover, cellular edema occurring in initial stages of radiotherapy was alleviated by these adjuvant therapies. Overall, this case provided a novel demonstration of emergent radiation as a feasible treatment for intracranial radiosensitive tumor-induced hernias. Given that this study presents a single case, there are limitations that warrant further investigations.
Prognostication of localized pPNET has been markedly improved by multidisciplinary collaboration in the development of therapeutic proposals. However, patients with primary disseminated multifocal ES still harbor very low survival rates. Previous reports revealed that sites of metastasis were overt, independent prognostic factors. Conversely, primary disseminated ES with single pulmonary metastases often have had much better outcomes compared with metastases to other sites.9–11 Additionally, negative prognoses for primary disseminated multifocal ES have correlated with relatively older patient ages (>14 years old,10 >15 years old9), larger primary masses volumes (>200 mL), bone marrow involvements, the presence and number of bone lesions, additional lung metastases, and fevers at diagnosis.9,10 ES are routinely characterized by gene fusions between EWS and ETS family genes. Retrospective studies have demonstrated different types of chromosomal rearrangements predicting divergent outcomes.12,13 One recent prospective cohort study found no prognostic value for characterization of any gene fusions.14 van Doorninck et al15 attributed these discrepancies regarding the value of type 1 fusions in ES prognostication to current intensive treatment proposals. Additionally, new biomarkers and molecularly detectable minimal disseminated diseases are completely novel areas for prognostication.16 Although, these new directions have potential promise, their clinical utilities require further study.
Conclusion
Intracranial pPNET are rare, but serious, diseases. Classifications of pPNET and central PNET should be completely differentiated as they display unique treatment proposals and prognostications. Emergent medical measures ought to be performed when metastatic neoplasms present as spinal cord compressions. Radiotherapy may be an effective choice to alleviate brain herniations induced by radiosensitive intracranial pPNET.
Acknowledgments
This work was supported by the Taishan Scholars Program of Shandong Province, People’s Republic of China (Grant No Ts20120505) and Shandong Province Natural Science Foundation (ZR2012HL33, ZR2013HL049).
Disclosure
The authors report no conflicts of interest in this work.
References
Delattre O, Zucman J, Melot T, et al. The Ewing’s family of tumors: a subgroup of small-round-cell tumors defined by specific chimeric transcripts. N Engl J Med. 1994;331(5):294–299. | ||
Ijichi K, Tsuzuki T, Adachi M, Murakami S. A peripheral primitive neuroectodermal tumor in the larynx: a case report and literature review. Oncol Lett. 2016;11(2):1120–1124. | ||
Pezeshkpour GH, Henry JM, Armbrustmacher VW. Spinal metastases. A rare mode of presentation of brain tumors. Cancer. 1984;54(2):353–356. | ||
Hasegawa SL, Davison JM, Rutten A, Fletcher JA, Fletcher CD. Primary cutaneous Ewing’s sarcoma: immunophenotypic and molecular cytogenetic evaluation of five cases. Am J Surg Pathol. 1998;22(3):310–318. | ||
Navarro R, Laguna A, de Torres C, et al. Primary Ewing sarcoma of the tentorium presenting with intracranial hemorrhage in a child. J Neurosurg. 2007;107(5 Suppl):411–415. | ||
Musahl V, Rihn JA, Fumich FE, Kang JD. Sacral intraspinal extradural primitive neuroectodermal tumor. Spine J. 2008;8(6):1024–1029. | ||
Kobayashi S, Takahashi J, Sakashita K, Fukushima M, Kato H. Ewing sarcoma of the thoracic epidural space in a young child. Eur Spine J. 2013;22:S373–S379. | ||
Tong X, Deng X, Yang T, et al. Clinical presentation and long-term outcome of primary spinal peripheral primitive neuroectodermal tumors. J Neurooncol. 2015;124(3):455–463. | ||
Oberlin O, Rey A, Desfachelles AS, et al. Impact of high-dose busulfan plus melphalan as consolidation in metastatic Ewing tumors: a study by the Société Française des Cancers de l’Enfant. J Clin Oncol. 2006;24(24):3997–4002. | ||
Ladenstein R, Pötschger U, Le Deley MC, et al. Primary disseminated multifocal Ewing sarcoma: results of the Euro-EWING 99 trial. J Clin Oncol. 2010;28(20):3284–3291. | ||
Luksch R, Tienghi A, Hall KS, et al. Primary metastatic Ewing’s family tumors: results of the Italian Sarcoma Group and Scandinavian Sarcoma Group ISG/SSG IV study including myeloablative chemotherapy and total-lung irradiation. Ann Oncol. 2012;23(11):2970–2976. | ||
Zoubek A, Dockhorn-Dworniczak B, Delattre O, et al. Does expression of different EWS chimeric transcripts define clinically distinct risk groups of Ewing tumor patients? J Clin Oncol. 1996;14(4):1245–1251. | ||
de Alava E, Kawai A, Healey JH, et al. EWS-FLI1 fusion transcript structure is an independent determinant of prognosis in Ewing’s sarcoma. J Clin Oncol. 1998;16(4):1248–1255. | ||
Le Deley MC, Delattre O, Schaefer KL, et al. Impact of EWS-ETS fusion type on disease progression in Ewing’s sarcoma/peripheral primitive neuroectodermal tumor: prospective results from the cooperative Euro-E.W.I.N.G. 99 trial. J Clin Oncol. 2010;28(12):1982–1988. | ||
van Doorninck JA, Ji L, Schaub B, et al. Current treatment protocols have eliminated the prognostic advantage of type 1 fusions in Ewing sarcoma: a report from the Children’s Oncology Group. J Clin Oncol. 2010;28(12):1989–1994. | ||
Gaspar N, Hawkins DS, Dirksen U, et al. Ewing sarcoma: current management and future approaches through collaboration. J Clin Oncol. 2015;33(27):3036–3046. | ||
Mobley BC, Roulston D, Shah GV, et al. Peripheral primitive neuroectodermal tumor/Ewing’s sarcoma of the craniospinal vault: case reports and review. Hum Pathol. 2006;37(7):845–853. | ||
Mazur M, Gururangan S, Bridge J, et al. Intracranial Ewing’s sarcoma. Pediatr Blood Cancer. 2005;45(6):850–856. | ||
Pekala JS, Gururangan S, Provenzale JM, Mukundan S Jr. Central nervous system extraosseous Ewing sarcoma: radiologic manifestations of this newly defined pathologic entity. AJNR Am J Neuroradiol. 2006;27(3):580–583. | ||
Kazmi SA, Perry A, Pressey JG, et al. Primary Ewing sarcoma of the brain: a case report and literature review. Diagn Mol Pathol. 2007;16(2):108–111. | ||
Jay V, Zielenska M, Lorenzana A, Drake J. An unusual cerebellar primitive neuroectodermal tumor with t(11;22) translocation: pathological and molecular analysis. Pediatr Pathol Lab Med. 1999;16(1):119–128. | ||
Papotti M, Abbona G, Pagani A, et al. Primitive neuroectodermal tumor of the meninges: an histology, immunohistochemical, ultrastructural and cytogenetic study. Endocr Pathol. 1998;3(1): 275–280. | ||
Antunes NL, Lellouch-Tubiana A, Kalifa C, et al. Intracranial Ewing sarcoma/‘peripheral’ primitive neuroectodermal tumor of dural origin with molecular genetic confirmation. J Neurooncol. 2001;51(1):51–56. | ||
Dedeurwaerdere F, Giannini C, Sciot R, et al. Primary peripheral PNET/Ewing’s sarcoma of the dura: a clinicopathologic entity distinct from central PNET. Mod Pathol. 2002;15(6):673–678. | ||
D’Antonio A, Caleo A, Garcia JF, Marsilia GM, De Dominicis G, Boscaino A. Primary peripheral PNET/Ewing’s sarcoma of the dura with FISH analysis. Histopathology. 2004; 45(6):651–654. | ||
Attabib NA, West M, Rhodes RH. Peripheral primitive neuroectodermal tumor of the cavernous sinus: case report. Neurosurgery. 2006; 58(5):E992. | ||
Navarro R, Laguna A, de Torres C, et al. Primary Ewing sarcoma of the tentorium presenting with intracranial hemorrhage in a child. J Neurosurg. 2007;107(5):411–415. | ||
Mellai M, Caldera V, Comino A, Fortunato M, Bernucci C, Schiffer D. PNET/ESFT of the cranial vault: a case report. Clin Neuropathol. 2010;29(6):372–377. | ||
Choudhury KB, Sharma S, Kothari R, Majumder A. Primary extraosseous intracranial Ewing’s sarcoma: Case report and literature review. Indian J Med Paediatr Oncol. 2011;32(2):118–121. | ||
Bunyaratavej K, Khaoroptham S, Phonprasert C, Tanboon J, Shuangshoti S. Primary intracranial peripheral primitive neuroectodermal tumor/Ewing’s sarcoma presenting with acute intracerebral hemorrhage. Clin Neuropathol. 2005;24(4):184–190. | ||
Katayama Y, Kimura S, Watanabe T, Yoshino A, Koshinaga M. Peripheral-type primitive neuroectodermal tumor arising in the tentorium: case report. J Neurosurg. 1999;90(1):141–144. | ||
Niwa J, Shimoyama N, Takahashi Y. Primitive neuroectodermal tumor involving the frontal skull base in an infant. Childs Nerv Syst. 2001;17(9):570–574. | ||
Simmons MA, Luff DA, Banerjee SS, Ramsden RT. Peripheral primitive neuroectodermal tumour (pPNET) of the cerebellopontine angle presenting in adult life. J Laryngol Otol. 2001;115(10):848–852. | ||
VandenHeuvel KA, Al-Rohil RN, Stevenson ME, et al. Primary intracranial Ewing’s sarcoma with unusual features. Int J Clin Exp Pathol. 2015;8(1):260–274. | ||
Bano S, Yadav SN, Garga UC. Case Report: Intracranial peripheral primitive neuroectodermal tumor-Ewing’s sarcoma of dura with transcalvarial–subgaleal extension: An unusual radiological presentation. Indian J Radiol Imaging. 2009;19(4):305–307. | ||
Amita R, Sandhyamani S, Nair S, Kapilamoorthy TR. Intracranial ewings sarcoma/peripheral primitive neuroectodermal tumor. Neurol India. 2014;62(4):432–433. | ||
Idrees M, Gandhi C, Betchen S, Strauchen J, King W, Wolfe D. Intracranial peripheral primitive neuroectodermal tumors of the cavernous sinus: a diagnostic peculiarity. Arch Pathol Lab Med. 2005;129(1): e11–e15. | ||
Furuno Y, Nishimura S, Kamiyama H, et al. Intracranial peripheral-type primitive neuroectodermal tumor. Neurol Med Chir (Tokyo). 2008;48(2):72–76. | ||
Velivela K, Rajesh A, Uppin MS, Purohit AK. Primary intracranial peripheral PNET-a case report and review. Neurol India. 2014;62(6):669–673. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.