")
Back to Journals » International Journal of Nanomedicine » Volume 11
Synthetic high-density lipoproteins for delivery of 10-hydroxycamptothecin
Authors Yuan Y, Wen J, Tang J , Kan Q, Ackermann R, Olsen K, Schwendeman A
Received 15 May 2016
Accepted for publication 10 September 2016
Published 22 November 2016 Volume 2016:11 Pages 6229—6238
DOI https://doi.org/10.2147/IJN.S112835
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Carlos Rinaldi
Yue Yuan,1,2 Jian Wen,3 Jie Tang,2 Qiming Kan,1 Rose Ackermann,2 Karl Olsen,2 Anna Schwendeman2
1Department of Pharmaceutics, School of Pharmacy, Shenyang Pharmaceutical University, Shenyang, People’s Republic of China; 2Department of Pharmaceutical Sciences, Biointerfaces Institute, College of Pharmacy, University of Michigan, 3Department of Internal Medicine, Division of Molecular Medicine and Genetics, University of Michigan Medical School, Ann Arbor, MI, USA
Abstract: The purpose of this study was to develop a novel synthetic high-density lipoprotein (sHDL) nanoparticle delivery system for 10-hydroxycamptothecin (HCPT) for treatment of colon carcinoma. HDL is recognized by scavenger receptor B-I (SR-BI) over-expressed in colon carcinomas 5- to 35-fold relative to the human fibroblasts. The sHDL nanoparticles were composed of apolipoprotein A-I mimic peptide (5A) and contained 0.5%–1.5% (w/w) of HCPT. An optimized HCPT-sHDL formulation exhibited 0.7% HCPT loading with 70% efficiency with an average size of 10–12 nm. Partitioning of HCPT in the sHDL lipid membrane enhanced drug stability in its active lactone form, increased solubilization, and enabled slow release. Cytotoxicity studies in HT29 colon carcinoma cells revealed that the IC50 of HCPT-sHDL was approximately 3-fold lower than that of free HCPT. Pharmacokinetics in rats following intravenous administration showed that the area under the serum concentration-time curve (AUC0–t) and Cmax of HCPT-HDL were 2.7- and 6.5-fold higher relative to the values for the free HCPT, respectively. These results suggest that sHDL-based formulations of hydrophobic drugs are useful for future evaluation in treatment of SR-BI-positive tumors.
Keywords: apolipoprotein A-I mimic peptide, phospholipids, SR-BI receptor, colon carcinoma, camptothecin, nanoparticle, nanodisc
Introduction
Cancer cells commonly over express the scavenger receptor B-I (SR-BI), primarily found in the liver, in order to obtain extra cholesterol necessary for tumor growth.1 SR-BI recognizes high-density lipoproteins (HDLs), the natural nanoparticles (8–10 nm) that remove excess cholesterol from peripheral tissues and shuttle it to the liver for elimination in a process called reverse cholesterol transport (RCT).2 Several synthetic HDL (sHDL) infusion products have reached the clinic as potential therapies to treat acute coronary syndrome patients by promoting RCT.3–5 Clinically tested sHDLs are composed of protein, such as apolipoprotein A-I (ApoA-I), ApoA-I mutant, or ApoA-I mimetic peptides, which is wrapped around with a lipid bilayer to form a nanodisk.3–5 In the clinic, sHDL products have been dosed at 8–10 g lipid–protein mixture per administration (eg, dosed weekly intravenously over 5–6 weeks) and are known to have long circulating half-lives (8–24 h).3–5
Many hydrophobic drugs bind to HDL following administration, including paclitaxel and camptothecins (CPTs).6,7 Several groups have utilized the ability of HDL to retain hydrophobic anticancer drugs and selectively deliver chemotherapeutics to tumors.1,7–11 A long circulation half-life and a very small particle size allow for sHDL to effectively accumulate in a tumor region and to penetrate extracellular matrix.12 Yet the clinical translation of this concept is lacking due to high cost and technical difficulty in manufacturing of pure ApoA-I protein either recombinantly or by plasma-purification and to form homogeneous sHDL nanoparticles.1,7–9,13 Even when synthetic ApoA-I mimic peptide was used to assemble HDL drug delivery carriers, chromatographic purification of nanoparticles was required postassembly.10,14 In this study, we used a different amphipathic helix peptide, 5A, which is designed to bind lipids with high affinity and form stable sHDL particles.15 This peptide also has minimal membrane lytic ability and, thus, is nontoxic following intravenous administration.15
10-Hydroxycamptothecin (HCPT) is a hydrophobic anticancer drug and a topoisomerase I inhibitor.16 In addition to poor solubility, it exhibits a pH-dependent conversion from the active lactone form to its inactive carboxylate. HCPT exists fully as lactone at a pH below 5, but at physiological pH it rapidly converts to the carboxylate form (t1/2 ~20 min).17,18 The lactone form of HCPT binds to lipid membranes in plasma, including HDL, low-density lipoprotein (LDL), and red blood cells membrane. This binding increases the overall lactone-to-carboxylate ratio. Several formulation strategies have utilized preferential partitioning of the lactone into the hydrophobic lipid phase to improve lactone stability and CPT efficacy including liposomes, emulsions, nanoparticles, and microspheres.13,19–22 It is also reported that HDL reconstituted with ApoA-I cysteine mutants was used as delivery vehicles for HCPT.7
The goal of this study was to develop a novel 5A-peptide-based sHDL delivery system for treatment of colon carcinoma. The optimum formulation was selected based on drug loading (DL) and encapsulation efficiency (EE). The stability of HCPT in sHDL, in vitro drug release kinetics, cytotoxicity in colon carcinoma, and pharmacokinetics in normal rats were examined for selected HCPT-HDL nanoparticles and compared with free HCPT.
Materials and methods
Materials
HCPT (>99%, purity) was purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Sphingomyelin (SM), 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), and 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) were donated by Nippon Oil & Fat Corporation (Osaka, Japan). An ApoA-I mimetic peptide, 5A (acetate salt), was produced by Bachem (Torrance, CA, USA).
Preparation of HCPT-loaded HDL
HCPT-loaded HDL was prepared by the thin-film dispersion method. Briefly, 15 mg lipid was dissolved in 2 mL chloroform and mixed with a 2.5 mg/mL HCPT stock dimethyl sulfoxide (DMSO) solution. The organic solvent was evaporated and 0.7 mL buffer (50 mM acetate buffer, pH 5.0) was added to the flask to hydrate the film by probe sonication in 30 s intervals using a VibraCell ultrasonic processor (Sonics, Newtown, CT, USA). 5A peptide of 7.5–15 mg was dissolved in 0.3 mL buffer and mixed with the lipid suspension. The mixture was incubated in 50°C water bath for 5 min and cooled at room temperature for 5 min. The temperature was cycled three times to form sHDL.
Determination of drug-loading capacity and EE
The un-encapsulated HCPT was removed by a Zeba desalting column (Thermo Fisher Scientific, Waltham, MA, USA). A 2.5 mL aliquot of HCPT-loaded HDL was added to a 5 mL desalting column, centrifuged at 1,000 rpm, and the eluent was collected. The HCPT-HDL solutions before and after desalting were analyzed by high performance liquid chromatograph (HPLC) for drug content. EE and DL were calculated according to the following formulas:
|
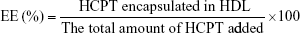
DL(%) = Theoretical loading × EE% |
Analyses of HCPT concentration and lactone-to-carboxylate ratio
The HPLC method to detect HCPT concentration and lactone-to-carboxylate ratio was adapted from Liu et al.23 The HPLC instrument consisted of a Waters 2695 system equipped with an Ascentis C18 separation column (15 cm ×4.6 mm, 5 μm). The mobile phase consisted of 50 mM phosphate buffer (pH 6.0), acetonitrile, and tetrahydrofuran (80:18:2, v/v). The carboxylate and lactone forms of HCPT were separated by isocratic elution at a flow rate of 0.75 mL/min at 25°C column temperature and detected by UV at 380 nm.
The ultra performance liquid chromatography (UPLC) method was developed based on an HPLC protocol reported by Warner and Burke.24 The lactone and carboxylate forms of HCPT were separated on a Waters Acquity UPLC system equipped with a fluorescence detector. The mobile phase consisted of 18:82 (v/v) acetonitrile and 1% (v/v) triethylamine acetate (pH 5.5). Waters Acquity UPLC BEH C18 column (1.7 μm, 2.1×100 mm2) was eluted in isocratic mode at 0.5 mL/min. An excitation wavelength of 380 nm and emission wavelength of 570 nm were used for fluorescence detection.
Dynamic light scattering
Mean diameter and size distribution of sHDL samples were analyzed by dynamic light scattering (DLS) with a Zetasizer Nano ZS (Malvern Instruments, Malvern, UK). sHDL samples were diluted to a concentration of 0.5 mg/mL of ApoA-I peptide in phosphate-buffered saline (PBS) buffer prior to analysis. Volume-weighted size distributions were reported.
Electron microscopy
A 3 μL aliquot of sHDL preparation was adsorbed on glow-discharged Quantifoil R2/2 200 mesh grids negatively stained with uranyl formate solution. Samples were imaged with a Tecnai F20 transmission electron microscope (TEM) equipped with a field emission gun operated at 120 kV. Images were recorded at a magnification of ×60,000-150,000 on a Gatan US4000 charge-coupled device (CCD) camera and defocus value of approximately −1 μm.
Drug release in vitro
One milliliter aliquot of HCPT-loaded sHDL or free HCPT (0.06 mg/mL) was added to a Float-A-Lyzer dialysis insert (molecular weight (MW) cutoff 12 kDa, Spectrum Labs) and subsequently placed in a flask containing 75 mL of PBS (pH 7.4). The medium was stirred at 100 rpm at 37°C in an incubator. A 0.4 mL aliquot of release media was removed at each time point and replaced with the same volume of fresh PBS buffer. The amount of HCPT released was measured by fluorescence spectroscopy analysis (excitation: 380 nm, emission: 570 nm). The data was expressed as the percentage of released HCPT relative to the total HCPT.
Degradation kinetics of free HCPT and HCPT-loaded sHDL
The degradation kinetics of free HCPT and HCPT-sHDL was examined by incubation in PBS (pH 7.4) at 37°C. The PBS was warmed to 37°C prior to addition of drug samples to attain a final concentration of 5 μg/mL. A total of 20 μL aliquots were taken out after preselected times (5, 10, 20, 40, 60, 75, and 120 min), added 180 μL of dry ice-chilled methanol (approximately −50°C), vortexed for 30 s and then centrifugation at 9,000 rpm for 1 min. The supernatant was diluted 10-fold in the mobile phase and either directly injected in UPLC or kept frozen on dry ice until the analysis. It has been previously established that no further lactone to carboxylate conversion occurred during drug extraction and analysis.18
In vitro cytotoxicity assay
HT29 cells were purchased from American Type Culture Collection (Manassas, VA, USA). The cells were cultured in McCoy’s 5A medium (1×) supplemented with 4 mM L-glutamine, 0.1 mM nonessential amino acids, 1.0 mM sodium pyruvate, and 10% fetal bovine serum (Thermo Fisher Scientific) and maintained at 37°C in a 5% CO2 incubator. For the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazonium bromide (MTT) assay, cells were plated at 5,000/well in 96-well plates. Cells were incubated overnight to allow for attachment and treated with free HCPT, blank sHDL, physical mixture of sHDL and HCPT or HCPT-loaded sHDL at 0.0025, 0.025, 0.25, 2.5, and 25 μg/mL HCPT concentration for 48 h. For blank HDL and HCPT-sHDL, these drug concentrations corresponded to 0.175, 1.75, 17.5, 175, and 1,750 μg/mL of 5A peptide or sHDL. Following the treatment, 20 μL of the MTT solution (0.5 mg/mL) was added to each well and incubated for 4 h. Thereafter, medium with the MTT solution was removed and 200 μL DMSO was added to each well to solubilize formazan. The formazan levels were measured by the UV absorbance at 560 nm. Cytotoxicity was expressed as the percentage of absorbance of treated cells relative to untreated cells.
Pharmacokinetics of HCPT-sHDL in rats
Sprague Dawley rats, weighing ~370 g, were fed a standard laboratory rodent diet with an access to water ad libitum and housed in a temperature-controlled room with a 12:12 h light/dark cycle. The rats were fasted for 12 h prior to experiments and randomly divided into three groups (n=3/group). Two groups received tail vein intravenous injections of either free HCPT and HCPT-loaded HDL at a dose of 0.7 mg/kg. The third group was administered with HCPT-loaded HDL by intraperitoneal (IP) injection at the same dose.
Blood samples (0.4 mL) were drawn from the jugular vein at 5, 15, 30 min, 1, 2, 4, and 8 h for intravenous dosing and 15, 30 min, 1, 2, 4, 8, and 12 h following IP administration of test articles. The blood samples were acidified by addition of 25 μL of 10% phosphoric acid to 0.4 mL of blood to adjust the pH to 5 and convert the HCPT to its lactone form. The samples were transferred into ice-cold heparinized collection tubes and plasma was separated by centrifugation. The plasma samples were flash frozen and stored at −20°C until analysis.
The HCPT extraction from plasma was performed according to Zhao et al.25 The plasma (0.1 mL) was mixed with 10 μL of 0.5 μg/mL CPT internal standard solution and 10 μL of phosphoric acid. HCPT and CPT were extracted from plasma by addition of 0.5 mL of ethyl acetate, followed by 1 min vortexing and 5 min shaking. The mixture was centrifuged at 15,000 rpm for 5 min at 4°C, the upper organic layer was collected and evaporated to dryness with N2 at 35°C. The residue was reconstituted in 50 μL of UPLC mobile phase, vortexed for 2 min and centrifuged before analysis. The supernatant samples (10 μL) were analyzed by UPLC as described earlier. The pharmacokinetic data were fitted by noncompartmental analysis using a Topfit 2.0 software package (Thomas GmbH, Langenselbold, Germany). The Cmax (μg/mL), tmax (h), t1/2 (h), and AUC0–t (μg/mL·h) were calculated for three groups. Animal protocol was reviewed and approved by University of Michigan Institutional Animal Care and Use Committee to assure compliance with the federal policies and guidelines specified by the the United States Department of Agriculture; as well as guidelines specified by the Association for Assessment and Accreditation of Laboratory Animal Care, International.
Statistical analysis
All the results are expressed as mean ± standard deviation. Statistical analysis was performed using Graphpad Prism 5. For IC50 analysis, the data were analyzed using nonlinear regression analysis. Unpaired t-test was used for comparison between two groups. For multiple comparisons, results were analyzed using one-way analysis of variance followed by Tukey’s posttest analysis. P<0.05 was considered statistically significant.
Results
Selection of phospholipid composition of sHDL
The phospholipid composition of native HDL particles is a rather complex mixture of SM, phosphotidyl choline (PC), triglyceride, and cholesterol.26 For the simplicity of pharmaceutical preparations, a single phospholipid, such as soybean PC, egg PC, POPC, or SM, is used in most sHDL4. Due to a relatively small space available for incorporation of drug molecule inside sHDL nanoparticle lipid membrane, molecular interactions between the drug and phospholipid are rather important. The phospholipid type, fatty acid chain length, and degree of saturation affect fluidity of sHDL membrane and spacing available for drug insertion. Due to some structural similarity between HCPT and cholesterol, we investigated SMs and several PCs known to have different affinities for cholesterol.27,28 For the initial lipid screening, we have fixed the theoretical DL at 1% (w/w) and fixed the weight ratio of 5A peptide to lipids as 1–1.5. A schematic representation of the preparation of HCPT-loaded sHDL is shown in Figure 1. This weight ratio of 5A to phospholipid is known to form homogeneous sHDL nanoparticles of 8–12 nm diameter.29 The particle size and size homogeneity of sHDL made with different phospholipids were similar (Table 1). The highest HCPT EE (71%) was achieved for SM-based sHDL, followed by DPPC-based (49%), POPC (16%), and DMPC (12%). The affinity of phospholipids to cholesterol follows similar trend as HCPT EE% of SM having greater affinity relative to PC and saturated longer fatty acid chains PC having greater affinity relative to unsaturated and short chain (SM > DPPC > DMPC/POPC).27,28 Another possible reason for large differences in EE between various sHDL lipid compositions is the difference in lipid transition or melting temperature. The transition temperatures for SM (Tm=38°C) and DPPC (Tm=41°C) is higher than room temperature, which provides for a better trapping of HCPT in the lipid bilayer, while POPC (Tm=−3°C) and DMPC (Tm=23°C) membranes are fluid and allow for drug leakage and dissociation from membrane.30,31
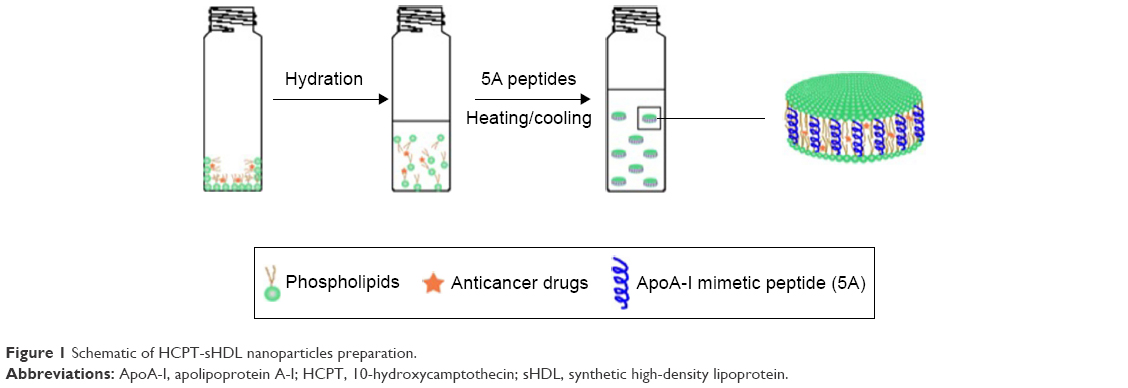 | Figure 1 Schematic of HCPT-sHDL nanoparticles preparation. |
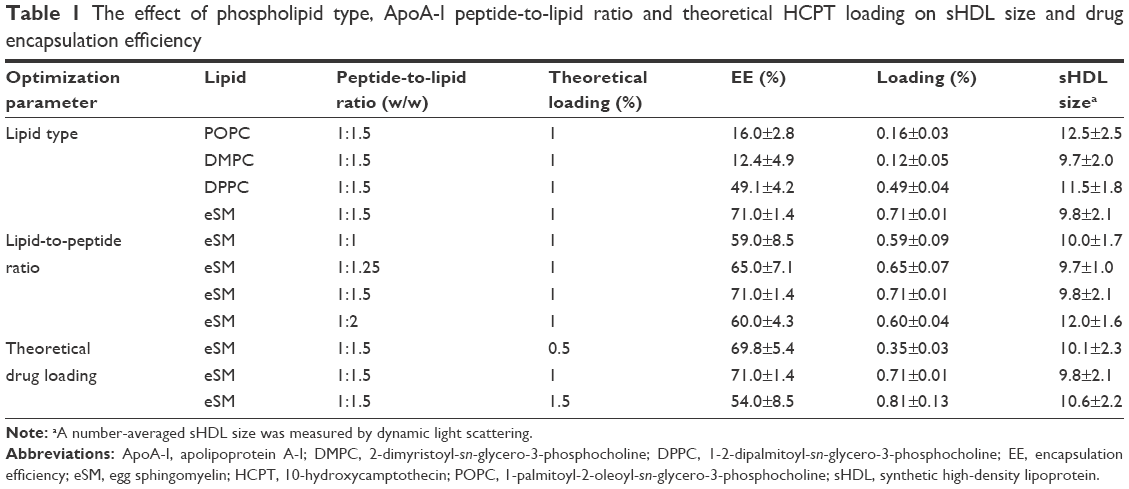 | Table 1 The effect of phospholipid type, ApoA-I peptide-to-lipid ratio and theoretical HCPT loading on sHDL size and drug encapsulation efficiency |
Optimization of DL and EE
While the structure of peptide-based sHDL nanoparticles has not been fully characterized yet, it is assumed that amphipathic helices of ApoA-I peptide surround the perimeter of phospholipid bilayer, as represented in Figure 1. By altering the ratio of 5A peptide to SM, we could slightly increase the diameter of the nanodisks and potentially improve the DL. The sHDL were prepared at 1:1–1:3 (w/w) ratio of 5A to SM at a theoretical HCPT loading of 1% (w/w). The average diameter of sHDL increased slightly with the increase of peptide-to-lipid ratio (Table 1). At the 1:3 (w/w) ratio of peptide to SM, a cloudy solution was observed, indicating insufficient amount of ApoA-I mimetic to solubilize lipid vehicles into sHDL. The drug EE increased only slightly with an increase of lipid to peptide ratio from 59% to 71%.
In order to increase DL, we have prepared sHDL at 1:1.5 of 5A-to-SM ratio at the theoretical loading of 0.5%, 1%, and 1.5% (Table 1). With the increase in theoretical loading, the actual loading also increased from 0.35% to 0.81%. However, a greater loss of the drug during the sHDL preparation (54% EE) was observed for 1.5% loading relative to 0.5% and 1% (69.8% and 71.0% EE). In the current design of HCPT-sHDL nanoparticles, we rely on the physical binding of HCPT to the HDL lipid membrane for incorporation. Thus, only relatively small amounts of HCPT could be loaded into sHDL particles and 1% theoretical loading for 1:1.5 (w/w) peptide-SM sHDL was selected as an optimal formulation for further evaluation.
Analytical characterization of sHDL-HCPT
The sizes of all drug-loaded sHDL determined by DLS are summarized in Table 1. All sHDL formulations had number-averaged particle size between 9.5 and 12.5 nm. Examples of DLS profiles for blank and HCPT-loaded sHDL are shown in Figure 2 for the selected formulation of 1:1.5 5A:SM containing 0.7% (w/w) of HCPT. The resulting sHDL had narrow size distribution without purification post-sHDL formation. The homogeneity of the size was due to selection of ApoA-I mimetic with favorable lipid binding properties. The optimization of peptide-to-lipid ratio also allowed us to avoid the presence of both free peptide and liposomal lipid dispersions. The morphologies of the same two sHDL preparations were examined by electron microscopy (Figure 2). Both preparations displayed characteristic discoidal shapes of cholesterol-poor prebeta HDL and appeared to be very homogeneous with an approximate diameter of 10 nm.32
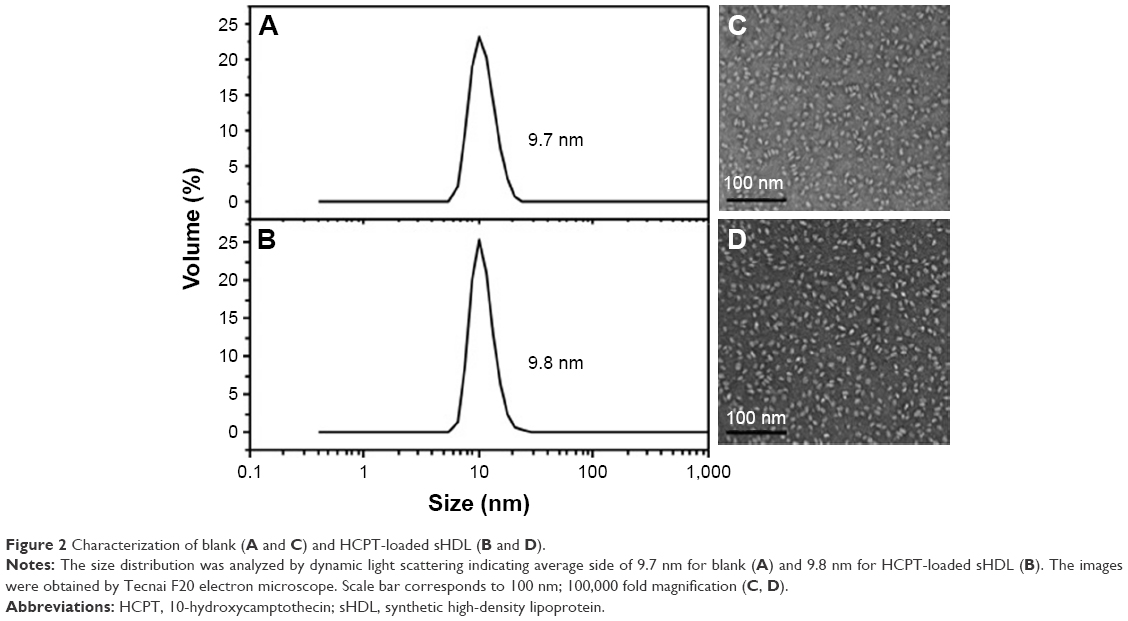 | Figure 2 Characterization of blank (A and C) and HCPT-loaded sHDL (B and D). |
The effect of sHDL incorporation on HCPT lactone stability
CPTs undergo a reversible and pH-dependent conversion from pharmacologically active lactone form to an inactive carboxylate form.18 At a pH of less than 5, HCPT exists fully in its lactone form and at pH over 8, it is found in carboxylate form. Because of this conversion, CPT injections are formulated at acidic pH. To examine if incorporation of HCPT in sHDL improved it stability, solutions of free HCPT and sHDL-HCPT were diluted in PBS pH 7.4 and the kinetics of lactone to carboxylate conversion was examined by HPLC (Figure 3A). Free HCPT rapidly converted to ~85% carboxylate form, while the conversion was slower for sHDL-HCPT and the equilibrium carboxylate level was ~60%. The hydrophobic lactone form is known to partition in lipid phase of lipoproteins and red blood cells membrane and this partitioning shifts equilibrium toward lactone.18 Still, conversion of the HCPT lactone to its carboxylate in the sHDL formulation strongly suggests that the drug dissociates from sHDL membrane and undergoes conversion to carboxylate in the buffer.
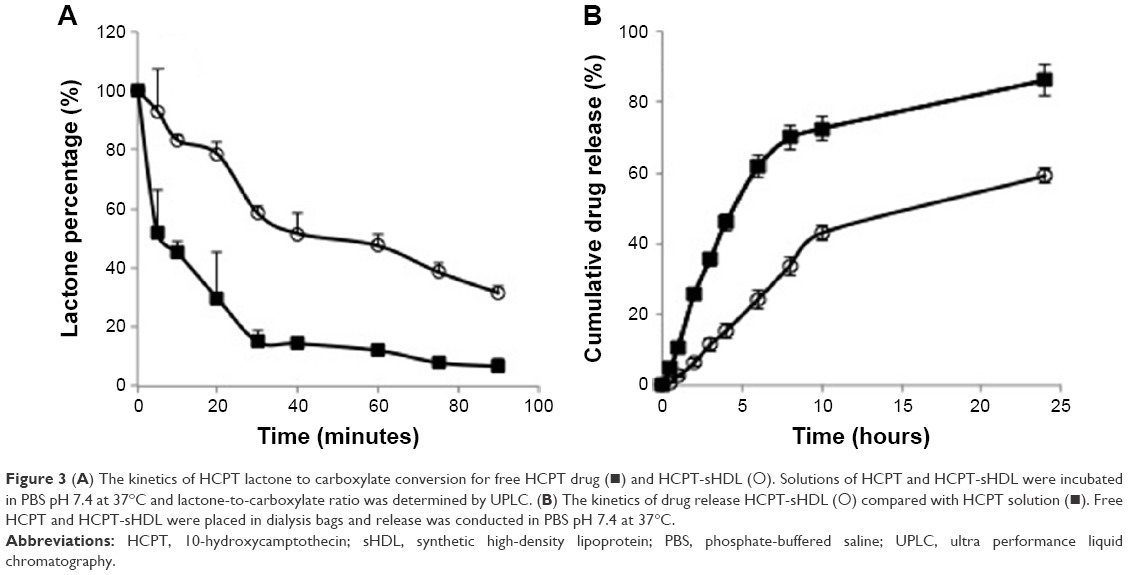 | Figure 3 (A) The kinetics of HCPT lactone to carboxylate conversion for free HCPT drug (◼) and HCPT-sHDL (◯). Solutions of HCPT and HCPT-sHDL were incubated in PBS pH 7.4 at 37°C and lactone-to-carboxylate ratio was determined by UPLC. (B) The kinetics of drug release HCPT-sHDL (◯) compared with HCPT solution (◼). Free HCPT and HCPT-sHDL were placed in dialysis bags and release was conducted in PBS pH 7.4 at 37°C. |
Characterization of drug release from sHDL
The in vitro release profiles of free HCPT and HCPT-sHDL nanoparticles in PBS (pH 7.4) are shown in Figure 3B. Due to the small size of the sHDL preparation, 10 kDa MW cutoff dialysis tubes were used in the study. The release rate is limited by dialysis membrane diffusion with free HCPT exhibiting more than 70% release from the dialysis bag to the medium by 10 h. The release of HCPT from sHDL nanoparticles was slower with ~40% of the drug released over the same time frame. The steadier and slower release from HCPT-sHDL formulation may be attributed to effective encapsulation of drug in nanoparticle and preference of lactone form of HCPT to lipid membrane of sHDL.
Cytotoxicity of HCPT-sHDL
The cytotoxicity of free HDL, blank sHDL, a physical mixture of HCPT and sHDL and HCPT-sHDL were examined in colon carcinoma cells HT29 using an MTT assay as described previously.16,21,33 HT29 was selected based on the reported high expression level of SR-BI in this cell line (10-fold higher expression levels over fibroblast cells). The IC50 of free HCPT and HCPT-sHDL were 0.56±0.01 and 0.17±0.01 μg/mL, respectively (Figure 4). The HCPT-sHDL was approximately 3-fold more potent than free HCPT (P<0.05). Interestingly, the blank sHDL also showed some antitumor activity but at a significantly higher concentration of 5A (IC50=0.21 mg/mL of 5A) than that of HCPT-sHDL (IC50=0.012 mg/mL of 5A and 0.17 μg/mL of HCPT). This activity could be possibly due to cholesterol depletion of colon carcinoma cells by sHDL-mediated cholesterol efflux. In contrast, the physical mixture of HCPT and sHDL exhibited cytotoxicity with IC50 value of 0.42±0.03 μg/mL, which was not statistically significant from free HCPT.
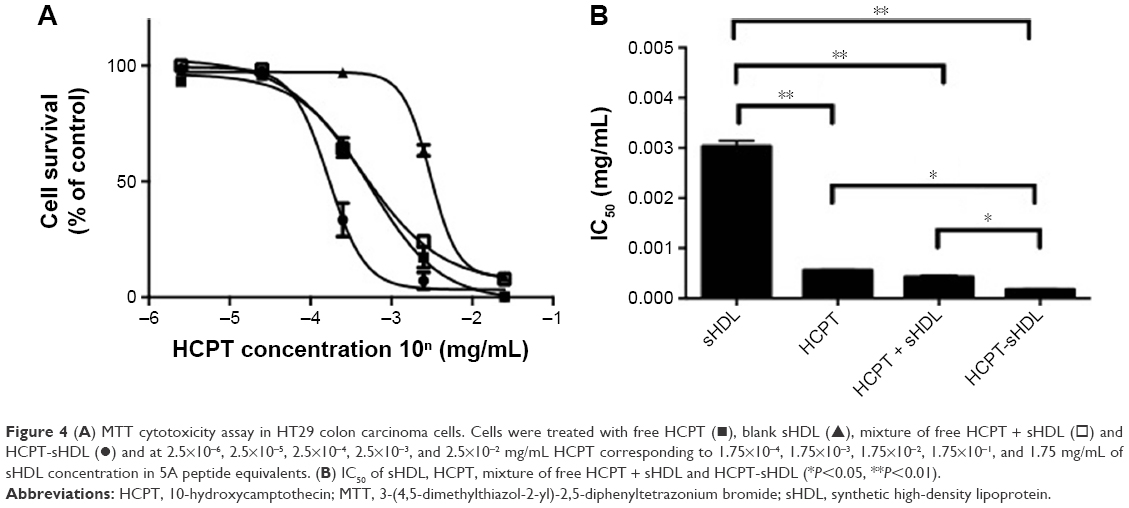 | Figure 4 (A) MTT cytotoxicity assay in HT29 colon carcinoma cells. Cells were treated with free HCPT (◼), blank sHDL (▲), mixture of free HCPT + sHDL (◻) and HCPT-sHDL (●) and at 2.5×10−6, 2.5×10−5, 2.5×10−4, 2.5×10−3, and 2.5×10−2 mg/mL HCPT corresponding to 1.75×10−4, 1.75×10−3, 1.75×10−2, 1.75×10−1, and 1.75 mg/mL of sHDL concentration in 5A peptide equivalents. (B) IC50 of sHDL, HCPT, mixture of free HCPT + sHDL and HCPT-sHDL (*P<0.05, **P<0.01). |
Pharmacokinetics in rats
The mean plasma drug concentration–time profiles of free HCPT in HCPT-sHDL nanoparticles after intravenous administration to rats at a single dose of 0.7 mg/kg are shown in Figure 5 and their pharmacokinetic parameters are summarized in Table 2. The plasma maximum concentration (4.62±1.49 μg/mL) and AUC0–t (1.96±0.42 μg/mL·h) of HCPT following administration of HCPT-HDL were significantly greater than those following administration of free HCPT (Cmax of 0.71±0.70 μg/mL and AUC0–t of 0.72±0.36 μg/mL·h) (P<0.01). This increased availability may have been due to the fact that the HDL lipid membrane facilitates the incorporation of hydrophobic compounds like HCPT into the carrier and have tight affinity. This affinity results in extended drug circulation, characteristic of the HDL carrier avoidance of the reticulo endothelial system, and a finite HDL desorption rate of the drug into the blood, which effectively delays availability of the drug in the blood compartment.
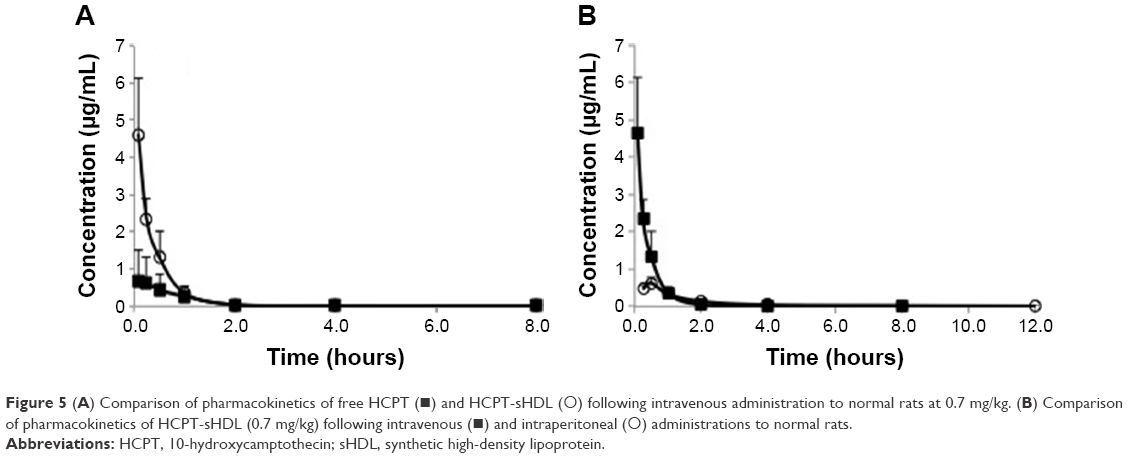 | Figure 5 (A) Comparison of pharmacokinetics of free HCPT (◼) and HCPT-sHDL (◯) following intravenous administration to normal rats at 0.7 mg/kg. (B) Comparison of pharmacokinetics of HCPT-sHDL (0.7 mg/kg) following intravenous (◼) and intraperitoneal (◯) administrations to normal rats. |
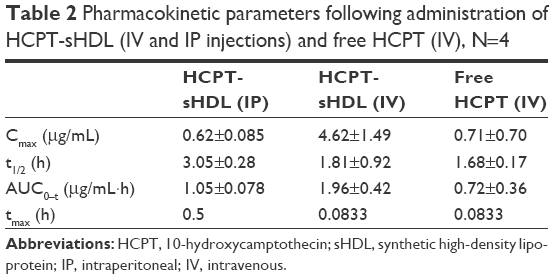 | Table 2 Pharmacokinetic parameters following administration of HCPT-sHDL (IV and IP injections) and free HCPT (IV), N=4 |
The mean plasma concentration–time profiles of HCPT after intravenous administration of HCPT-sHDL nanoparticles were also compared with that of IP, as seen in Figure 5 and Table 2. The elimination half-life and tmax after IP injection were extended longer than those after intravenous (IV) administration of HCPT-sHDL nanoparticles. By contrast, the AUC0–t and Cmax after IP injection were lower than those after IV administration.
Discussion
The stability, release, and pharmacokinetics of HCPT were altered by incorporation into synthetic HDL nanoparticles. The hydrophobicity and lipid binding properties of HCPT’s active lactone isoform were exploited for incorporation into sHDL’s lipid bilayer. Without using covalent binding of drug to nanocarrier, ~0.7% (w/w) of HCPT was loaded in 5A-SM sHDL. Because of the unusually high maximally tolerated doses of sHDL recently used in human clinical trials,3,4 the loading level of these formulations is reasonable. The preferential partitioning of HCPT lactone in sHDL lipid bilayers was found to improve drug stability, nanoparticle retention, and cytotoxicity relative to solutions of unencapsulated HCPT. The incorporation of HCPT into sHDL resulted in a 3-fold higher AUC relative to the free drug following IV administration in rats.
The in vitro experiments revealed the ability of black sHDL to cause cytotoxicity in HT29 cells with IC50 of 0.012 mg/mL. Several recent studies indicate that the drug-free sHDL exhibits certain intrinsic anticancer activities. This anticancer activity is likely due to sHDL’s ability to efflux cholesterol from cancer cell by passive efflux from cellular membrane or through SR-BI and facilitating cell death through cholesterol depletion. The SR-BI receptor is bidirectional and could serve to either uptake the cargo of sHDL or 10-HCPT or efflux cholesterol to cholesterol-free sHDL nanoparticles.34 The 5A-SM has been shown to exhibit strong SR-BI-mediated efflux deemed beneficial for antiatherosclerotic properties of this sHDL.29 Normally the antitumor activity of sHDL is observed at relatively high dosages, such as 0.1–0.2 mg/mL of 22A-sHDL, is required to generate in vitro cytotoxicity in NCI-H295R and SW13 cells35 and 250 mg/kg of R4F-sHDL to inhibit the tumor growth in vivo.36 The improved activity of HCPT-sHDL is unlikely to be attributed to a synergetic effect between topoisomerase I inhibition by HCPT and cholesterol efflux by sHDL based on no statistically significant improvement in cytotoxicity of the physical mixture of sHDL and HCPT. However, the synergetic effect might be seen at other concentrations of sHDL and HCPT and it could not be ruled completely. The improved cytotoxicity of HCPT-sHDL relative to HCPT itself is likely due to improved delivery by SR-BI and potentially higher lactone stability of the nanoparticle drug in cell culture media.
The incorporation of HCPT in sHDL was performed by Jia et al6 and Zhang and Chen 7 by using full-length ApoA-I or its multiple mutants and soybean PC phospholipid. In that study, the obtained particles were spherical at 22–25 nm in diameter (LDL-range) and contained 4% HCPT loading. Increase cytotoxicity of sHDL-formulation over the free drug was observed in cell culture. Following IV dosing at 10 mg/kg, the detectable drug levels were found in plasma for longer periods and drug levels were higher in kidney, liver, and spleen for sHDL formulations relative to free HCPT. Several groups including us have reported accumulation of fluorescent dye-loaded sHDL in subcutaneous tumor xenographs as well as liver, lungs, and spleen.10,11 The uniqueness of this study is that we have merged the ability to prepare very homogeneous pharmaceutical quality peptide-based synthetic HDL nanoparticles with loading of a hydrophobic drug. Several investigators have successfully loaded hydrophobic drugs, peptides and siRNA, and vaccine antigens in sHDL.1,7–9,14 A number of start-up companies had been formed on the basis of HDL-drug delivery idea including DLVR Therapeutics (Toronto, ON, Canada), Lypro Biosciences (Berkeley, CA, USA), Signablok Inc. (Shrewsbury, MA, USA), and possibly others. While all companies have secured funding through government grants and private investments, the clinical translation has been limited due to difficulties in manufacturing pharmaceutical grade sHDL particles. In the current study, HCPT-sHDL particles assemble by hydrophobic interaction of phospholipids, amphipathic peptide, and drug. The purity of the sHDL particles is controlled by adjustment of the sHDL ingredients ratios and selection of phospholipids, ApoA-I peptide, and drug with unique physicochemical properties (hydrophobic momentum and length for peptide, chain length and saturation for lipid, and hydrophobicity/size for the drug). Without covalent binding and using manufacturing methodologies exploited for other clinically tested sHDL, we were able to show the improvement in pharmacokinetic properties, HCPT stability, and cytotoxicity. The observation of the alteration of the pharmacokinetic profile following physical binding of drug to sHDL is broadly applicable because several Food and Drug Administration-approved pharmaceuticals tend to physically bind to endogenous patient HDL following oral or parenteral administrations.37 Hence, the drug doses might need to be adjusted on a patient-by-patient basis due to significant inter-patient differences in lipoprotein levels. In addition, the drug–HDL physical binding might be partially responsible for drug-induced dyslipidemia that occurs in lipoprotein binding drugs, such as amphotericin B, cyclosporine A, clozapine, and paclitaxel.
Limitations
The drawback of the current formulation is a low DL and the need of administering large doses of sHDL because of it. In the current design of HCPT-sHDL nanoparticles, we rely on physical binding of HCPT to the HDL lipid membrane for incorporation. A similar interaction of sHDL is observed with free cholesterol, as it is well known that infusion of sHDL causes rapid mobilization of free (unesterified) cholesterol.4 In the body, sHDL cholesterol undergoes esterification by lecithin–cholesterol acyltransferase. Cholesterol esters are significantly more hydrophobic and are able to partition into the HDL core, which causes a transformation of the initially discoidal HDL into its spherical form.3 One potential way to significantly increase DL into sHDL particles is to modify CPT with ester moieties to form spherical sHDL particles containing drug inside particle core rather than phospholipid membrane. Similar strategy was attempted by de Vrueh et al for incorporation of Hepatitis B replication inhibitor in the core of sHDL particles38 and Nikanjam et al for incorporation of paclitaxel oleate ester in LDL nanoparticles.39 However, this approach is outside of the scope for the current article, and may be investigated in the future.
Conclusion
In this study, the composition of sHDL was optimized to promote formation of pharmaceutical quality nanoparticles by selection of an appropriate ApoA-I mimic-to-phospholipid ratio. The optimal formulation was screened of 5A peptide and SM (1:1.5, w/w ratio, 0.7% HCPT loading), with the highest EE of 71%. In vitro cytotoxicity study indicated that the colon carcinoma cell lines HT29 were more sensitive to the HCPT-sHDL nanoparticles than to free HCPT (P<0.05). Stability of sHDL-incorporated HCPT was improved in vitro by retention of the active and hydrophobic lactone in the lipid bilayer. The affinity of HCPT to sHDL membrane allowed for extended drug circulation characteristic of sHDL carriers, slower desorption rate to the blood compartment, which all resulted in higher drug exposure in vivo. Further work is ongoing to increase DL and to assess therapeutic effect of HCPT-sHDL on tumor-bearing mice. The use of peptide-based sHDL with an optimized composition in order to obtain pharmaceutical grade nanoparticles is a valuable approach to overcome current barriers for clinical translation of this drug delivery platform.
Acknowledgments
The authors would like to acknowledge Dr Georgios Skiniotis and Ms Annie Dosey from Life Sciences Institute of University of Michigan for their help with TEM imaging.
Disclosure
This work was supported by the University of Michigan’s Michigan Translational Research and Commercialization (MTRAC) grant, NIH R21NS091555, and an Upjohn research award to AS. YY was supported by a grant 81202481 from National Natural Science for Youth Foundation of China, grant L2015527 of Education Department of Liaoning Province, and Scientific Research Foundation for the Returned Overseas Chinese Scholars by State Education Ministry and Shenyang Pharmaceutical University grant GGJJ2014102. The authors report no other conflicts of interest in this work.
References
Mooberry LK, Nair M, Paranjape S, McConathy WJ, Lacko AG. Receptor mediated uptake of paclitaxel from a synthetic high density lipoprotein nanocarrier. J Drug Target. 2010;18(1):53–58. | ||
Rosenson RS, Brewer HB Jr, Davidson WS, et al. Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation. 2012;125(15):1905–1919. | ||
Kingwell BA, Chapman MJ, Kontush A, Miller NE. HDL-targeted therapies: progress, failures and future. Nat Rev Drug Discov. 2014;13(6):445–464. | ||
Krause BR, Remaley AT. Reconstituted HDL for the acute treatment of acute coronary syndrome. Curr Opin Lipidol. 2013;24(6):480–486. | ||
Kuai R, Li D, Chen YE, Moon JJ, Schwendeman A. High-density lipoproteins: nature’s multifunctional nanoparticles. Acs Nano. 2016;10(3):3015–3041. | ||
Jia J, Xiao Y, Liu J, et al. Preparation, characterizations, and in vitro metabolic processes of paclitaxel-loaded discoidal recombinant high-density lipoproteins. J Pharm Sci. 2012;101(8):2900–2908. | ||
Zhang X, Chen B. Recombinant high density lipoprotein reconstituted with apolipoprotein AI cysteine mutants as delivery vehicles for 10-hydroxycamptothecin. Cancer Lett. 2010;298(1):26–33. | ||
Singh AT, Evens AM, Anderson RJ, et al. All trans retinoic acid nanodisks enhance retinoic acid receptor mediated apoptosis and cell cycle arrest in mantle cell lymphoma. Br J Haematol. 2010;150(2):158–169. | ||
Ghosh M, Singh AT, Xu W, Sulchek T, Gordon LI, Ryan RO. Curcumin nanodisks: formulation and characterization. Nanomedicine. 2011;7(2):162–167. | ||
Glickson JD, Lund-Katz S, Zhou R, et al. Lipoprotein nanoplatform for targeted delivery of diagnostic and therapeutic agents. Mol Imaging. 2008;7(2):101–110. | ||
Cui L, Lin Q, Jin CS, et al. A PEGylation-Free Biomimetic Porphyrin Nanoplatform for Personalized Cancer Theranostics. ACS Nano. 2015;9(4):4484–4495. | ||
Gullotti E, Yeo Y. Extracellularly activated nanocarriers: a new paradigm of tumor targeted drug delivery. Mol Pharm. 2009;6(4):1041–1051. | ||
Remaley AT, Amar M, Sviridov D. HDL-replacement therapy: mechanism of action, types of agents and potential clinical indications. Exp Rev Cardiovasc Ther. 2008;6(9):1203–1215. | ||
Yang M, Jin HL, Chen JA, et al. Efficient cytosolic delivery of siRNA using HDL-mimicking nanoparticles. Small. 2011;7(5):568–573. | ||
Amar MJ, D’Souza W, Turner S, et al. 5A apolipoprotein mimetic peptide promotes cholesterol efflux and reduces atherosclerosis in mice. J Pharmacol Exp Ther. 2010;334(2):634–641. | ||
Wall ME. Camptothecin and taxol: discovery to clinic. Med Res Rev. 1998;18(5):299–314. | ||
Wang A, Li S. Hydroxycamptothecin-loaded nanoparticles enhance target drug delivery and anticancer effect. BMC Biotechnol. 2008;8:46. | ||
Mi Z, Burke TG. Differential interactions of camptothecin lactone and carboxylate forms with human blood components. Biochemistry. 1994;33(34):10325–10336. | ||
Shi K, Tian Y, Jiang Y, Wang L, Cui F. Modified hydrolysis kinetics of the active lactone moiety of 10-hydroxycamptothecin by liposomal encapsulation. Pharm Dev Technol. 2010;15(6):644–652. | ||
Lundberg BB. Biologically active camptothecin derivatives for incorporation into liposome bilayers and lipid emulsions. Anticancer Drug Des. 1998;13(5):453–461. | ||
Yang L, Cui F, Cun D, Tao A, Shi K, Lin W. Preparation, characterization and biodistribution of the lactone form of 10-hydroxycamptothecin (HCPT)-loaded bovine serum albumin (BSA) nanoparticles. Int J Pharm. 2007;340(1–2):163–172. | ||
Tong W, Wang L, D’Souza MJ. Evaluation of PLGA microspheres as delivery system for antitumor agent-camptothecin. Drug Dev Ind Pharm. 2003;29(7):745–756. | ||
Liu J, Jiang ZZ, Zhang SM, Saltzman WM. Poly(omega-pentadecalactone-co-butylene-co-succinate) nanoparticles as biodegradable carriers for camptothecin delivery. Biomaterials. 2009;30(29):5707–5719. | ||
Warner DL, Burke TG. Simple and versatile high-performance liquid chromatographic method for the simultaneous quantitation of the lactone and carboxylate forms of camptothecin anticancer drugs. J Chromatogr B Biomed Sci Appl. 1997;691(1):161–171. | ||
Zhao YX, Liu DX, Liang WQ, Ye ZW. In-vivo pharmacokinetics, tissue distribution and anti-tumour effect of hydroxycamptothecin delivered in oil-in-water submicron emulsions. J Pharm Pharmacol. 2012;64(6):783–791. | ||
Kontush A, Lhomme M, Chapman MJ. Unraveling the complexities of the HDL lipidome. J Lipid Res. 2013;54(11):2950–2963. | ||
Ohvo-Rekila H, Ramstedt B, Leppimaki P, Slotte JP. Cholesterol interactions with phospholipids in membranes. Prog Lipid Res. 2002;41(1):66–97. | ||
Ramstedt B, Slotte JP. Interaction of cholesterol with sphingomyelins and acyl-chain-matched phosphatidylcholines: a comparative study of the effect of the chain length. Biophys J. 1999;76(2):908–915. | ||
Schwendeman A, Sviridov DO, Yuan W, et al. The effect of phospholipid composition of reconstituted HDL on its cholesterol efflux and anti-inflammatory properties. J Lipid Res. 2015;56(9):1727–1737. | ||
Silvius JR, McElhaney RN. Molecular properties of membrane lipids and activity of a membrane adenosine triphosphatase from Acholeplasma laidlawii B. Rev Infect Dis. 1982;4(Suppl):S50–S58. | ||
Barenholz Y, Thomas TE. Sphingomyelin: biophysical aspects. Chem Phy Lipids. 1999;102(1–2):29–34. | ||
van Antwerpen R, Chen GC, Pullinger CR, et al. Cryo-electron microscopy of low density lipoprotein and reconstituted discoidal high density lipoprotein: imaging of the apolipoprotein moiety. J Lipid Res. 1997;38(4):659–669. | ||
Tian Q, Zhang J, Tan TM, et al. Human multidrug resistance associated protein 4 confers resistance to camptothecins. Pharm Res. 2005;22(11):1837–1853. | ||
Yancey PG, de la Llera-Moya M, Swarnakar S, et al. High density lipoprotein phospholipid composition is a major determinant of the bi-directional flux and net movement of cellular free cholesterol mediated by scavenger receptor BI. J Biol Chem. 2000;275(47):36596–36604. | ||
Subramanian C, Kuai R, Zhu Q, et al. Synthetic high-density lipoprotein nanoparticles: A novel therapeutic strategy for adrenocortical carcinomas. Surgery. 2016;159(1):284–294. | ||
Zheng Y, Liu Y, Jin H, et al. Scavenger receptor B1 is a potential biomarker of human nasopharyngeal carcinoma and its growth is inhibited by HDL-mimetic nanoparticles. Theranostics. 2013;3(7):477–486. | ||
Wasan KM, Brocks DR, Lee SD, Sachs-Barrable K, Thornton SJ. Impact of lipoproteins on the biological activity and disposition of hydrophobic drugs: implications for drug discovery. Nat Rev Drug Discov. 2008;7(1):84–99. | ||
de Vrueh RL RE, Sliedregt LA, Biessen EA, van Berkel TJ, Bijsterbosch MK. Synthesis of a lipophilic prodrug of 9-(2-phosphonylmethoxyethyl)adenine (PMEA) and its incorporation into a hepatocyte-specific lipidic carrier. Pharm Res. 1999;16(8):1179–1185. | ||
Nikanjam M, Gibbs AR, Hunt CA, Budinger TF, Forte TM. Synthetic nano-LDL with paclitaxel oleate as a targeted drug delivery vehicle for glioblastoma multiforme. J Control Release. 2007;124(3):163–171. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.