Back to Journals » Drug Design, Development and Therapy » Volume 12
Synthesis of small peptide compounds, molecular docking, and inhibitory activity evaluation against phosphatases PTP1B and SHP2
Authors Kostrzewa T ![]() , Sahu KK, Gorska-Ponikowska M, Tuszynski JA
, Sahu KK, Gorska-Ponikowska M, Tuszynski JA ![]() , Kuban-Jankowska A
, Kuban-Jankowska A ![]()
Received 5 September 2018
Accepted for publication 24 October 2018
Published 5 December 2018 Volume 2018:12 Pages 4139—4147
DOI https://doi.org/10.2147/DDDT.S186614
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Qiongyu Guo
Tomasz Kostrzewa,1 Kamlesh K Sahu,2 Magdalena Gorska-Ponikowska,1 Jack A Tuszynski,3 Alicja Kuban-Jankowska1
1Department of Medical Chemistry, Medical University of Gdansk, Gdansk, Poland; 2Department of Medical Microbiology and Immunology, University of Alberta, Edmonton, AB, Canada; 3Department of Oncology, University of Alberta, Edmonton, AB, Canada
Background: The protein tyrosine phosphatases PTP1B and SHP2 are promising drug targets in treatment design for breast cancer. Searching for specific inhibitors of their activity has recently become the challenge of many studies. Previous work has indicated that the promising PTP inhibitors may be small compounds that are able to bind and interact with amino residues from the binding site.
Purpose: The main goal of our study was to synthesize and analyze the effect of selected small peptide inhibitors on oncogenic PTP1B and SHP2 enzymatic activity and viability of MCF7 breast cancer cells. We also performed computational analysis of peptides binding with allosteric sites of PTP1B and SHP2 phosphatases.
Methods: We measured the inhibitory activity of compounds utilizing recombinant enzymes and MCF7 cell line. Computational analysis involved docking studies of binding conformation and interactions of inhibitors with allosteric sites of phosphatases.
Results: The results showed that the tested compounds decrease the enzymatic activity of phosphatases PTP1B and SHP2 with IC50 values in micromolar ranges. We observed higher inhibitory activity of dipeptides than tripeptides. Phe-Asp was the most effective against SHP2 enzymatic activity, with IC50=5.2±0.4 µM. Micromolar concentrations of tested dipeptides also decreased the viability of MCF7 breast cancer cells, with higher inhibitory activity observed for the Phe-Asp peptide. Moreover, the peptides tested were able to bind and interact with allosteric sites of PTP1B and SHP2 phosphatases.
Conclusion: Our research showed that small peptide compounds can be considered for the design of specific inhibitors of oncogenic protein tyrosine phosphatases.
Keywords: breast cancer, protein tyrosine phosphatase PTP1B, SHP2, peptides, PTP inhibitors
Introduction
Breast cancer is one of the most common types of female tumors worldwide. Breast cancer therapy usually includes surgery, radiotherapy, and adjuvant chemotherapy. Disturbances in the course of tyrosine phosphorylation/dephosphorylation pathways is associated with numerous disorders, including breast cancer development.1
Protein tyrosine phosphatases (PTPs) form a large group of enzymes that remove phosphate groups from the tyrosine residues of proteins. Reversible tyrosine phosphorylation of proteins is regulated by a balance maintained by the antagonistic action of PTPs and tyrosine kinases.2 Phosphorylation/dephosphorylation of the tyrosine residues of proteins is an evolutionarily preserved mechanism of signal transduction in eukaryotic cells of fundamental importance in the regulation of cell physiology, such as proliferation, differentiation, migration, or tumorigenesis.
The participation of PTPs in the development of glioma, colorectal, lung, or breast cancer and multiple myeloma has been already proven. Phosphatases PTP1B and SHP2 are particularly important targets in the treatment of breast cancer.3 PTP1B dephosphorylates tyrosine kinases, which are essential for the induction of breast cancer, ie, HER1, Src, JAK, and STAT. PTP1B phosphatase is overexpressed in breast cancer cells and triggers tumor growth.4
PTP1B phosphatase inhibitors are promising compounds for treatment of metabolic diseases, eg, type 2 diabetes, obesity, and metabolic syndromes. SHP2 is found to be overexpressed in breast cancer cell lines and is usually involved with oncogenic signaling functions to promote growth factors and cytokines. Additionally, mutations of SHP2 have been observed in breast cancer cells. Due to oncogenic implications of SHP2, inhibition of these phosphatases can produce a favorable effect in anticancer therapy.5,6 Due to the key role of PTPs in cancer biology, they might be targeted for the development of new anticancer diagnostic and promising therapeutic strategies.7
PTPs have been challenging targets for inhibitor design, and there are already successful studies with utilization of peptidyl inhibitors against TPs.8 Utilizing medical chemistry in combination with molecular simulations reveals the key role of small molecules in designing new phosphatase inhibitors.9–11 There have recently been studies showing that a small molecule inhibitor of SHP2 can act as an allosteric modulator that stabilizes the inhibited conformation of SHP2.12 However, docking analyses performed by other groups of researchers revealed that the compounds tested by them exhibited IC50 values higher than expected and that the tested compounds were able to bind to other peripheral sites with lower free energy than when bound to the active or allosteric sites.13 For our studies, we selected simple dipeptides and tripeptides characterized by small compound size.
In the present work, we choose to study the effect of selected peptide compounds as potential PTP1B and SHP2 inhibitors, as there have been many recent studies showing therapeutic peptides as a promising approach to cancer treatment.14,15 Peptide compounds can be easily modified and rapidly synthesized, are atoxic, and are less immunogenic than, eg, recombinant antibodies.16,17 There are also many peptide-based drug conjugates utilized in cancer treatment.18 The Tat peptide conjugated to doxorubicin is highly effective against MCF7 and MCF7/ADR breast cancer cells, inducing cell death.19
Despite some disadvantages, such as poor solubility and membrane permeability, there are still important advantages, eg, high potency of action and target selectivity, as well as low accumulation in tissue.20,21 In the present work, we decided to synthesize and analyze the effect of selected small peptide inhibitors (Figure 1) on oncogenic PTP1B and SHP2 enzymatic activity and viability of MCF7 breast cancer cells. We also performed computational analysis of peptides binding with allosteric sites of PTP1B and SHP2 phosphatases.
 | Figure 1 Structures of synthesized and tested peptides. |
Methods
Synthesis of peptides
All peptides were synthesized on solid support using solid-phase peptide synthesis. Synthesis was carried out on 2′-chlorotrotyl resin (1.6 mmol/g) according to 9-fluorenylmethoxycarbonyl/tert-butyl (Fmoc/OtBu) chemistry with the side chain-protected amino acid derivatives Fmoc-Phe-OH and Fmoc-Asp(OtBu)-OH. Amino acid derivatives were attached to the growing peptide on the resin during deprotection and attachment stages, beginning the synthesis with the C-terminus amino acid. Before attachment of the C-terminal amino acid, the resin was activated by shaking for 30 minutes in a solvent mixture of dimethylformamide (DMF) and dichloromethane (DCM; 1:1, v:v) in a peptide synthesis vessel. Subsequently, 1 M Fmoc-AA and a fourfold excess of N,N-diisopropylethylamine (DIPEA) as the base, which were dissolved in a mixture of DMF:DCM (10:1, v:v), were introduced into the previously prepared resin. The reaction was carried out twice for 1 hour each time. When the reaction had completed, the solution was filtered under reduced pressure and then washed with DCM and DMF. To deactivate the remaining free linkers on the resin, solution containing DCM:DIPEA:MeOH (17:2:1, v:v:v) was added, and shaking was carried out in three cycles for 5, 15, and 30 minutes.
The crude peptide was cleaved from the resin using a mixed solution of 88% trifluoroacetic acid (TFA), 5% phenol, 5% deionized water, and 2% triisopropylsilane for 2 hours (10 mL per 1 g resin at room temperature for 2 hours). The obtained filtrate was then evaporated to approximately 2 mL on a rotary evaporator. Next, a cold diethyl ether was added dropwise to the prepared solution. The precipitate, suspended in ether, was transferred to centrifuge tubes and centrifuged (centrifugation program: relative centrifugal force=3,600 × g, 4°C, 15 minutes). The process of peptide centrifugation was repeated three times. The peptide obtained after centrifugation was transferred to a vacuum desiccator and dried for 24 hours over NaOH. The dried compound was then dissolved in water, frozen, and lyophilized.
Peptide purification
The crude peptide was dissolved in water and then purified by reversed-phase HPLC (RP-HPLC) on a Cosmosil C18 column with a grain diameter of 5 μm, a length of 25 cm and a cross-section of 10 mm. The mobile phase was water containing 0.1% TFA and acetonitrile (ACN) containing 0.1% TFA, flow rate: 4.0 mL/min. Gradient was determined depending on the peptides, which are presented in Table 1. Purification was monitored by ultraviolet absorption at a wavelength of 214 nm.
 | Table 1 Analysis of purity of compounds |
Purity analysis of compounds
We performed purity analysis of the synthesized compounds. Purified peptide was first dissolved in water and then purified by RP-HPLC on a Cosmosil C18 column with a grain diameter of 5 μm, length of 25 cm, and cross-section of 10 mm. The mobile phase was water containing 0.1% TFA and ACN containing 0.1% TFA, with a flow rate of 1 mL/min. The gradient was determined based on the peptides, which are presented in Table 1. Analysis was monitored by ultraviolet absorption at a wavelength of 214 nm. The purity of all peptides was suitable for carrying out biological tests (Table 1). Analyses of the purified peptides were performed by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. The matrix used in the analysis was 2,5-dihydroxybenzoic acid. All scheduled peptide sequences were obtained. Values for the pseudomolecular ions are presented in Table 1.
Molecular modeling
Peptides were docked on selected sites of enzymes to predict the binding mode and supramolecular interactions. The initial structure of PTP1B was taken from the Research Collaboratory for Structural Bioinformatics protein data bank (www.pdb.org) with code 5K9V.pdb and SHP2 with code 5EHR.pdb. This structure was loaded into Molecular Operating Environment software (Chemical Computing Group, Montreal, Canada), removing water molecules. Polar hydrogen atoms were added. The structure was protonated at a temperature of 300 K, pH 7, and salt concentration of 0.1. Ligands were removed, and structures were optimized using the Amber10:EHT force field of the software. Peptide molecules were docked into the structures where the binding site was assumed to be the allosteric site. The side chains were kept free to move during force field refinement. Alpha PMI was the placement method used with default settings. The top 30 docking conformations were retained for each peptide, and these poses were ranked by London dG scoring function to estimate the free energy of binding of peptide conformers. The pose with the lowest score (most stable pose) was chosen from the top conformation, and its binding orientation was used to calculate binding interactions.
Inhibitory activity analysis
The inhibitory activity of compounds was measured utilizing recombinant PTP1B and SHP2 phosphatases with final concentration in tested samples (1.5 μg/mL; 3.3 nM) in a solution of 10 mM HEPES buffer (pH 7.4). Phosphatase samples of final volume 200 μL were untreated (control) or treated with solutions of peptides. Inhibitory analysis was performed in 96-well microplates at 37°C. The activity of enzymes was read at 405 nm using a Jupiter microplate reader (Biogenet, Jozefow, Poland) and DigiRead Communication Software (Asys Hitech, Eugendorf, Austria). The activity of phosphatases was able to be observed due to chromogenic substrate para-nitrophenyl phosphate (2 mM).
Cell viability analysis
MCF7 breast cancer cells were obtained from the European Collection of Cell Culture (Salisbury, UK). Cells were cultured in DMEM supplemented with 10% FBS, 100 μg/mL penicillin–streptomycin, and 2 mM L-glutamine form Sigma-Aldrich (St Louis, MO, USA). The culture was maintained at 37°C in an atmosphere containing 5% CO2. Viability was measured using MTT for cell-metabolism activity. Cells (106 cells/mL) were untreated (control) or treated with dipeptide solutions in 96-well plates. After 24 hours of incubation, a solution of 5 mg/mL MTT in DMEM without phenol red was added. Samples were then incubated for 3–4 hours at 37°C. When the purple precipitate was clearly visible under microscopy, samples were dissolved in 100 μL of dimethyl sulfoxide, and the plate with cover was left in the dark for 15 minutes. Viability was measured as absorbance at 570 nm in a microplate reader.
Statistical analysis
All experiments were performed three times or more. Data obtained were then incorporated and analyzed using GraphPad Prism version 4 (GraphPad Software, Inc., La Jolla, CA, USA). Statistical analyses were performed utilizing ANOVA in combination with Tukey’s test or Student’s t-test in combination with Wilcoxon test. Data are showed as mean±SD. Differences between means were considered significant at P<0.05.
Results
Small peptides decreased activity of PTP1B and SHP2
We performed inhibitory activity analysis of the tested compounds against PTP1B and SHP2 phosphatases. We calculated IC50 values of dipeptides and tripeptides tested, which are presented in Table 2. We found that selected peptides were capable of decreasing the enzymatic activity of phosphatases PTP1B and SHP2. Inhibitory activity was estimated with IC50 values in micromolar ranges. We observed higher inhibitory activity for dipeptides than tripeptides. Especially, Phe-Asp was the most effective against SHP2 enzymatic activity, with an IC50 value of around 5 μM.
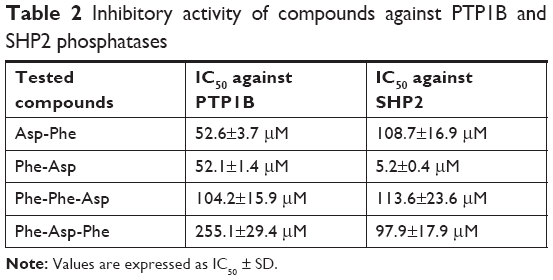 | Table 2 Inhibitory activity of compounds against PTP1B and SHP2 phosphatases |
We performed inhibition activity assays as screening for many concentrations of peptides and incubation times. Here, we present (Figure 2) the enzymatic activity of PTP1B and SHP2 pretreated for 15 minutes with 100 μM peptides and incubated for 30 minutes with a substrate.
 | Figure 2 Enzymatic activity of PTP1B and SHP2 after treatment with peptides. |
Inhibitory properties of dipeptides against breast cancer cell viability
Since we observed higher inhibitory activity for dipeptides than tripeptides, we performed breast cancer cell viability analysis after treatment with dipeptides. We discovered that micromolar concentrations of the dipeptides used were able to decrease the viability of MCF7 breast cancer cells after 24 hours of incubation (Figure 3). We also found that the Phe-Asp peptide had higher inhibitory properties than Asp-Phe. While 50 μM Asp-Phe revealed no significant effect on MCF7 viability, even 5 μM Phe-Asp still significantly decreased the viability of cells.
 | Figure 3 Viability of MCF7 breast cancer cells after treatment with Phe-Asp and Asp-Phe peptides. |
Molecular docking
We then performed molecular docking studies using dipeptide molecules. Peptide molecules were docked on the 3-D structures of PTP1B and SHP2 phosphatases to investigate the possibility of binding and conformation. Figure 4 shows the docking for Asp-Phe and Phe-Asp on PDB 5K9V (PTP1B [1-301], open state). Figure 5 shows the docking for Asp-Phe and Phe-Asp on PDB 5EHR (non-receptor SHP2 in complex with allosteric inhibitor SHP099). The docking was done on sites of allosteric inhibitors, as already shown in previous studies for the small molecule inhibitor SHP099.22 We obtained the top 30 conformations from runs of flexible docking. In all 30 conformations, dipeptides were shown to be able to be bound to allosteric sites of PTP1B and SHP2, as shown in Figures 4 and 5. The docking studies showed that selected peptides were not precluded from binding to allosteric sites of PTP1B and SHP2.
 | Figure 4 Top 30 binding conformations of peptides with PTP1B allosteric site. |
 | Figure 5 Top 30 binding conformations of peptides with SHP2 allosteric site. |
We also found that dipeptides were able to interact with some essential residues in allosteric sites of PTP1B and SHP2. Figure 6 shows possible interactions between dipeptides and Asp181 residue from the WPD loop and essential Arg221 in the PTP1B allosteric site. Possible interactions in the allosteric SHP2 site are shown in Figure 7.
 | Figure 6 Binding interactions of peptides with PTP1B allosteric site. |
 | Figure 7 Binding interactions of peptides with SHP2 allosteric site. |
Discussion
Apart from being one of the most common female tumor types, breast cancer usually occurs with complicated etiology and multiple-organ metastasis. The number of breast cancer cases has been growing in recent years in spite of numerous advances in detection and therapy.23
PTP1B and SHP2 phosphatases play critical roles in regulation of many cellular processes by controlling signaling pathways using PTP catalytic activity. Dysfunctions of PTP1B and SHP2 phosphatases result in pathogenic mutations and aberrant expression that lead to the dysregulation of numerous signaling pathways, thus contributing to different pathologies. PTP1B and SHP2 are also identified as oncogenic TPs, as their crucial role in the development of cancer has been demonstrated.24 Therefore, the pharmacological inhibition of SHP2 is an important therapeutic approach for the treatment of cancers.25 There have already been studies performed that searched for allosteric inhibitors of SHP2 with therapeutic potential for cancer treatment.26
The significant role of PTP1B phosphatase in breast cancer development has already been demonstrated. Elevated levels of PTP1B phosphatase relative to normal control breast cells were found in several human breast cancer cell lines with increased cSrc activity and confirmed the ability of PTP1B to dephosphorylate and activate cSrc kinase. The studies indicated that PTP1B was the primary phosphatase dephosphorylates cSrc in several human breast cancer cell lines and suggest a regulatory role of PTP1B in the control of cSrc-kinase activity.27
PTP1B was identified to be overexpressed in more than half of breast cancer tissue. It was also shown that tumor size and lymph node metastasis were significantly higher in patients with a higher level of PTP1B. The proliferation and migration of MCF7 cells were found to be inhibited after knocking down the gene of PTP1B.28
PTP1B is positively associated with lymph node metastasis and estrogen receptor status. In vitro, disturbing PTP1B expression attenuates cell migration and invasion. PTP1B overexpression increases migration and invasion of breast cancer cells. All these data indicate that PTP1B may play a crucial role in the development of breast cancer.29
Because PTP1B and SHP2 are associated with multiple cancer-related diseases, as well as SHP2 is a potential immunomodulator, which alters autoimmunity and related immunopathology, controlling PTP1B and SHP2 activities is of significant therapeutic interest.30,31 Studies have indicated that some natural diterpenes reveal significant inhibitory effects on the PTP1B enzyme and are considered an anti-breast cancer agents.32
All peptide compounds synthesized by us contained carboxyl groups, which are able to interact with amino acids in the binding sites of TPs, as was also shown in previous studies, where an extensive hydrogen bond network with a carboxyl group and van der Waals interactions stabilized the protein–ligand complexes of PTP1B and triterpenic acids.33 There have been many studies showing the inhibitory properties of compounds with carboxyl groups attached against the enzymatic activity of SHP2 and PTP1B.34–36
It was found that allosteric inhibition blocked closure of the WPD loop. The WPD loop has been shown to play a critical role at two stages of the catalytic cycle. First, Asp181 of the WPD loop serves as the proton donor during cleavage of the Tyr(P) P−O bond, and second, Asp181 participates in positioning and activating the water molecule that splits the cysteinyl–phosphate bond in the enzyme–phosphate intermediate. At both stages, closure of the WPD loop is essential in bringing Asp181 close to the phosphate group. Our results showed that dipeptides were able to interact with the Asp181 from the WPD loop of PTP1B phosphatase.
It was found that the capacity of compounds to inhibit PTP1B depended on their nature, position, and number of substituents in the inhibitor structure, eg, the presence of some specific substituents increases the ability of flavonoids to inhibit PTP1B.37 In the near future, we plan to study the effect on PTPs of more peptides with different side groups, since they appear to lead to promising inhibitory compounds.
Conclusion
Here, we showed that the dipeptides and tripeptides tested were able to bind and interact with allosteric sites of PTP1B and SHP2 phosphatases. The compounds tested decreased the enzymatic activity of phosphatases PTP1B and SHP2, with IC50 values in micromolar ranges, as well as the viability of MCF7 breast cancer cells. Our studies show that small peptide compounds can be considered a promising base for design of studies on specific inhibitors of PTPs.
Abbreviations
PTP, protein tyrosine phosphatase; HER1, epidermal growth factor receptor; JAK, Janus kinase; STAT, signal transducer and activator of transcription protein; Src, proto-oncogene tyrosine-protein kinase.
Acknowledgments
We acknowledge financial support from the project IP2015 038,774 from the Polish Ministry of Science and Higher Education. JAT acknowledges funding from NSERC (Canada) and the Allard Foundation.
Disclosure
The authors report no conflicts of interest in this work.
References
Nunes-Xavier CE, Martín-Pérez J, Elson A, Pulido R. Protein tyrosine phosphatases as novel targets in breast cancer therapy. Biochim Biophys Acta. 2013;1836(2):211–226. | ||
den Hertog J, Groen A, van der Wijk T. Redox regulation of protein-tyrosine phosphatases. Arch Biochem Biophys. 2005;434(1):11–15. | ||
Östman A, Hellberg C, Böhmer FD. Protein-tyrosine phosphatases and cancer. Nat Rev Cancer. 2006;6(4):307–320. | ||
Balavenkatraman KK, Aceto N, Britschgi A, Mueller U, Bence KK, Neel BG, Bentires-Alj M. Epithelial protein-tyrosine phosphatase 1B contributes to the induction of mammary tumors by HER2/Neu but is not essential for tumor maintenance. Mol Cancer Res. 2011;9(10):1377–1384. | ||
Aceto N, Bentires-Alj M. Targeting protein-tyrosine phosphatases in breast cancer. Oncotarget. 2012;3(5):514–515. | ||
LaRochelle JR, Fodor M, Ellegast JM, et al. Identification of an allosteric benzothiazolopyrimidone inhibitor of the oncogenic protein tyrosine phosphatase SHP2. Bioorg Med Chem. 2017;25(24):6479–6485. | ||
Scott LM, Lawrence HR, Sebti SM, Lawrence NJ, Wu J. Targeting protein tyrosine phosphatases for anticancer drug discovery. Curr Pharm Des. 2010;16(16):1843–1862. | ||
Liao H, Pei D. Cell-permeable bicyclic peptidyl inhibitors against T-cell protein tyrosine phosphatase from a combinatorial library. Org Biomol Chem. 2017;15(45):9595–9598. | ||
Martin KR, Narang P, Xu Y, et al. Identification of small molecule inhibitors of PTPσ through an integrative virtual and biochemical approach. PLoS One. 2012;7(11):e50217. | ||
Swingle M, Ni L, Honkanen RE. Small-molecule inhibitors of ser/thr protein phosphatases: specificity, use and common forms of abuse. Methods Mol Biol. 2007;365:23–38. | ||
Yu ZH, Chen L, Wu L, Liu S, Wang L, Zhang ZY. Small molecule inhibitors of SHP2 tyrosine phosphatase discovered by virtual screening. Bioorg Med Chem Lett. 2011;21(14):4238–4242. | ||
Garcia Fortanet J, Chen CH, Chen YN, et al. Allosteric Inhibition of SHP2: Identification of a Potent, Selective, and Orally Efficacious Phosphatase Inhibitor. J Med Chem. 2016;59(17):7773–7782. | ||
Ganou CA, Eleftheriou PT, Theodosis-Nobelos P, Fesatidou M, Geronikaki AA, Lialiaris T, Rekka EA. Docking analysis targeted to the whole enzyme: an application to the prediction of inhibition of PTP1B by thiomorpholine and thiazolyl derivatives. SAR QSAR Environ Res. 2018;29(2):133–149. | ||
Cicero AFG, Fogacci F, Colletti A. Potential role of bioactive peptides in prevention and treatment of chronic diseases: a narrative review. Br J Pharmacol. 2017;174(11):1378–1394. | ||
Blanco-Míguez A, Gutiérrez-Jácome A, Pérez-Pérez M, et al. From amino acid sequence to bioactivity: the biomedical potential of antitumor peptides. Protein Sci. 2016;25(6):1084–1095. | ||
Boohaker RJ, Lee MW, Vishnubhotla P, Perez JM, Khaled AR. The use of therapeutic peptides to target and to kill cancer cells. Curr Med Chem. 2012;19(22):3794–3804. | ||
McGregor DP. Discovering and improving novel peptide therapeutics. Curr Opin Pharmacol. 2008;8(5):616–619. | ||
Gilad Y, Firer M, Gellerman G. Recent innovations in peptide based targeted drug delivery to cancer cells. Biomedicines. 2016;4(2):11. | ||
Liang JF, Yang VC. Synthesis of doxorubicin-peptide conjugate with multidrug resistant tumor cell killing activity. Bioorg Med Chem Lett. 2005;15(22):5071–5075. | ||
Marqus S, Pirogova E, Piva TJ. Evaluation of the use of therapeutic peptides for cancer treatment. J Biomed Sci. 2017;24(1):21. | ||
Craik DJ, Fairlie DP, Liras S, Price D. The future of peptide-based drugs. Chem Biol Drug Des. 2013;81(1):136–147. | ||
Fodor M, Price E, Wang P, et al. Dual Allosteric Inhibition of SHP2 Phosphatase. ACS Chem Biol. 2018;13(3):647–656. | ||
Wei XQ, Li X, Xin XJ, Tong ZS, Zhang S. Clinical features and survival analysis of very young (age<35) breast cancer patients. Asian Pac J Cancer Prev. 2013;14(10):5949–5952. | ||
Huang WQ, Lin Q, Zhuang X, Cai LL, Ruan RS, Lu ZX, Tzeng CM. Structure, function, and pathogenesis of SHP2 in developmental disorders and tumorigenesis. Curr Cancer Drug Targets. 2014;14(6):567–588. | ||
Chen YN, LaMarche MJ, Chan HM, et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature. 2016;535(7610):148–152. | ||
Xie J, Si X, Gu S, et al. Allosteric Inhibitors of SHP2 with Therapeutic Potential for Cancer Treatment. J Med Chem. 2017;60(24):10205–10219. | ||
Bjorge JD, Pang A, Fujita DJ. Identification of protein-tyrosine phosphatase 1B as the major tyrosine phosphatase activity capable of dephosphorylating and activating c-Src in several human breast cancer cell lines. J Biol Chem. 2000;275(52):41439–41446. | ||
Liao SC, Li JX, Yu L, Sun SR. Protein tyrosine phosphatase 1B expression contributes to the development of breast cancer. J Zhejiang Univ Sci B. 2017;18(4):334–342. | ||
Liu X, Chen Q, Hu XG, et al. PTP1B promotes aggressiveness of breast cancer cells by regulating PTEN but not EMT. Tumour Biol. 2016;37(10):13479–13487. | ||
Wang W, Cao Y, Zhou X, Wei B, Zhang Y, Liu X. PTP1B promotes the malignancy of ovarian cancer cells in a JNK-dependent mechanism. Biochem Biophys Res Commun. 2018;503(2):903–909. | ||
Wang J, Mizui M, Zeng LF, et al. Inhibition of SHP2 ameliorates the pathogenesis of systemic lupus erythematosus. J Clin Invest. 2016;126(6):2077–2092. | ||
An JP, Ha TK, Kim J, Cho TO, Oh WK. Protein Tyrosine Phosphatase 1B Inhibitors from the Stems of Akebia quinata. Molecules. 2016;21(8). | ||
Ramírez-Espinosa JJ, Rios MY, López-Martínez S, et al. Antidiabetic activity of some pentacyclic acid triterpenoids, role of PTP-1B: in vitro, in silico, and in vivo approaches. Eur J Med Chem. 2011;46(6):2243–2251. | ||
Zeng LF, Zhang RY, Yu ZH, et al. Therapeutic potential of targeting the oncogenic SHP2 phosphatase. J Med Chem. 2014;57(15):6594–6609. | ||
Dawson MI, Xia Z, Liu G, et al. An adamantyl-substituted retinoid-derived molecule that inhibits cancer cell growth and angiogenesis by inducing apoptosis and binds to small heterodimer partner nuclear receptor: effects of modifying its carboxylate group on apoptosis, proliferation, and protein-tyrosine phosphatase activity. J Med Chem. 2007;50(11):2622–2639. | ||
Nasiri HR, Mracek P, Grimm SK, Gastaldello J, Kolodzik A, Ullmann D. PAIN-less identification and evaluation of small molecule inhibitors against protein tyrosine phosphatase 1B. Medchemcomm. 2017;8(6):1220–1224. | ||
Proença C, Freitas M, Ribeiro D, et al. Inhibition of protein tyrosine phosphatase 1B by flavonoids: a structure – activity relationship study. Food Chem Toxicol. 2018;111:474–481. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.