Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Synthesis, characterization, and evaluation of poly (D,L-lactide-co-glycolide)-based nanoformulation of miRNA-150: potential implications for pancreatic cancer therapy
Authors Arora S, Swaminathan S, Kirtane A, Srivastava S ![]() , Bhardwaj A, Singh S, Panyam J, Singh A
, Bhardwaj A, Singh S, Panyam J, Singh A
Received 5 February 2014
Accepted for publication 1 April 2014
Published 18 June 2014 Volume 2014:9(1) Pages 2933—2942
DOI https://doi.org/10.2147/IJN.S61949
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Sumit Arora,1 Suresh K Swaminathan,2 Ameya Kirtane,2 Sanjeev K Srivastava,1 Arun Bhardwaj,1 Seema Singh,1 Jayanth Panyam,2 Ajay P Singh1,3
1Department of Oncologic Sciences, Mitchell Cancer Institute, University of South Alabama, Mobile, Alabama, USA; 2Department of Pharmaceutics, The University of Minnesota, Minneapolis, USA; 3Department of Biochemistry and Molecular Biology, College of Medicine, University of South Alabama, Mobile, Alabama, USA
Abstract: MicroRNAs are small (18–22 nucleotide long) noncoding RNAs that play important roles in biological processes through posttranscriptional regulation of gene expression. Their aberrant expression and functional significance are reported in several human malignancies, including pancreatic cancer. Recently, we identified miR-150 as a novel tumor suppressor microRNA in pancreatic cancer. Furthermore, expression of miR-150 was downregulated in the majority of tumor cases, suggesting that its restoration could serve as an effective approach for pancreatic cancer therapy. In the present study, we developed a nanoparticle-based miR-150 delivery system and tested its therapeutic efficacy in vitro. Using double emulsion solvent evaporation method, we developed a poly (D,L-lactide-co-glycolide) (PLGA)-based nanoformulation of miR-150 (miR-150-NF). Polyethyleneimine (a cationic polymer) was incorporated in PLGA matrix to increase the encapsulation of miR-150. Physical characterization of miR-150-NF demonstrated that these nanoparticles had high encapsulation efficiency (~78%) and exhibited sustained release profile. Treatment of pancreatic cancer cells with miR-150-NF led to efficient intracellular delivery of miR-150 mimics and caused significant downregulation of its target gene (MUC4) expression. Inhibition of MUC4 correlated with a concomitant decrease in the expression of its interacting partner, HER2, and repression of its downstream signaling. Furthermore, treatment of pancreatic cancer cells with miR-150-NF suppressed their growth, clonogenicity, motility, and invasion. Together, these findings suggest that PLGA-based nanoformulation could potentially serve as a safe and effective nanovector platform for
miR-150 delivery to pancreatic tumor cells.
Keywords: PLGA nanoparticles, miR-150, MUC4, invasion, migration
Introduction
Pancreatic cancer remains one of the most lethal malignancies in the United States.1,2 The overall median survival after diagnosis is 2–8 months, and the 5-year survival rate is only 3%–6%, indicating the ineffectiveness of current treatment approaches.1–3 It is estimated that nearly 45,220 Americans were diagnosed with pancreatic malignancy, and over 38,460 died from this disease during the past year, marking it as the fourth leading cause of cancer-related death.2,3 This grim scenario has placed a major emphasis on the development of novel therapeutic tools for effective management of this devastating malignancy. In this regard, microRNAs (miRNAs or miRs) have emerged as a novel class of biomolecules that can be exploited clinically, not only as diagnostic and prognostic biomarkers, but also as novel targets for pancreatic cancer therapy.
miRNAs are small noncoding gene-regulatory RNAs that exhibit aberrant expression in several human malignancies, including pancreatic cancer. miRNAs play important roles in cancer initiation, progression, metastasis, and therapeutic resistance through downregulation of pathologically relevant gene targets.4–7 Hence, inhibition of overexpressed oncogenic miRNAs or restitution of downregulated tumor-suppressor miRNAs in cancer cells could provide new therapeutic avenues to treat cancer. Recently, we identified miR-150 as a novel, tumor suppressor miRNA in pancreatic cancer.8 Expression of miR-150 was downregulated in the majority of pancreatic tumor cases, suggesting its restoration could serve as an effective approach for therapy. However, the physicochemical characteristics of miRNAs – namely anionic charge, hydrophilic nature, sensitivity to nuclease degradation in plasma, and inefficient uptake by tumor cells – are major hurdles in achieving their cellular restoration by exogenous means.9,10 Therefore, the application of miRNAs as potential therapeutic agents requires delivery approaches that protect the miRNAs from nucleases and enhance uptake of the miRNAs by the tumor cells.
Two types of gene delivery systems, viral and nonviral, have been developed.11,12 The advantage of using viral vectors is their high transduction efficiency resulting in efficient delivery of the genetic material into the target cells.13 However, the use of viral gene delivery systems is limited by their inherent immunogenicity, low loading capacity, and other quality control issues.11,14,15 On the other hand, nonviral vectors (liposomes, polymer-based systems, and organic or inorganic nanoparticles) are safer for human use, albeit with lower delivery effectiveness.16–19 In this study, we investigated poly (D,L-lactide-co-glycolide) (PLGA)-based nanoformulation for the delivery of miR-150 to pancreatic cancer cells in vitro. PLGA is a biodegradable and biocompatible polymer and has already been approved by United States Food and Drug Administration for human use.20,21 Our studies show that miR-150-loaded nanoformulation (miR-150-NF) resulted in efficient intracellular delivery of miR-150, potent inhibition of its target gene (MUC4), and associated changes in signaling pathways. Lastly, miR-150-NF treatment led to inhibition of pancreatic tumor cell growth and clonogenicity, and suppressed their malignant behavioral properties. Together, our study characterizes a PLGA-based nanovector platform that could potentially be useful for efficient delivery of miR-150 to the pancreatic tumor cells and, thus, pave the way for novel and effective miRNA-based cancer therapeutics.
Materials and methods
Cell culture
The human pancreatic cancer cell lines, Colo-357 and HPAF, were maintained in a humidified atmosphere of 5% CO2 at 37°C as monolayer cultures in Roswell Park Memorial Institute (RPMI) 1640 media (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS) (Atlanta Biologicals, Lawrence Ville, GA, USA), penicillin (100 units/mL), and streptomycin (100 μg/mL) (Thermo Fisher Scientific). Validation of cell lines was done by short tandem repeats genotyping and/or expression of defined molecular markers (MUC1, MUC4, vimentin, and DPC4). Moreover, cells were continuously monitored for their typical morphology and intermittently tested for mycoplasma using MycoSensor PCR assay kit (Stratagene, La Jolla, CA, USA) as per manufacturer’s protocol.
Preparation of miR-150-loaded PLGA/Polyethylenimine nanoparticles
Ester terminated PLGA (molecular weight: 40 kDa) and PEI (molecular weight: 25 kDa) were purchased from LACTEL Absorbable Polymers (Birmingham, AL, USA). The ratio of lactic acid to glycolic acid in PLGA is 50:50. PLGA (32 mg) and PEI (100 μg) were dissolved in 1 mL of chloroform.A mixture of acetylated bovine serum albumin (4 mg) and miR-150 (283.5 μg) in 0.2 mL of 1X-Tris-ethylenediaminetetraacetic acid (EDTA) buffer was added into organic phase (the molar ratio of PEI and miRNA was ~1:8), vortexed briefly, and then sonicated using a probe sonicator (~3 W, Sonicator XL; Misonix, Inc., Farmingdale, NY, USA) for 30 seconds over an ice bath to form water-in-oil emulsion. The water-in-oil emulsion was then added into 6 mL of 2.5% w/v polyvinyl alcohol solution prepared with 1X Tris-EDTA buffer and sonicated at 9 W for 3 minutes over ice bath. The emulsion was further stirred for 18 hours at ambient conditions, and then for 2 hours under vacuum to remove chloroform. Nanoparticles were recovered by ultracentrifugation (35,000 rpm for 35 minutes at 4°C, Optima TM, LE-80 K; Beckman Coulter Inc., Brea, CA, USA), washed three times (first wash using 1X Tris-EDTA buffer and the next two washes with sterile deionized water), and then lyophilized (FreeZone 4.5; Labconco, Kansas City, MO, USA).
Encapsulation efficiency
The amount of miR-150-loaded in nanoparticles was determined as described previously.22 In brief, the amount of un-entrapped miR-150 mimics in the nanoparticle wash solutions obtained during the formulation step was quantified using the Quant-iT™ PicoGreen Assay kit (Thermo Fisher Scientific, Waltham, MA, USA). Fluorescence resulting from the binding of PicoGreen reagent to miR-150 was measured in a black-bottomed 96-well plate using FLx-800 microplate reader (BIO-TEK Instruments, Winooski, VT, USA). miR-150 loading in nanoparticles was determined by subtracting the total amount of miR-150 recovered in the wash solutions (miR-150w) from the initial amount of miR-150 (miR-150i) added. Encapsulation efficiency was calculated as:
(miR-150i–miR-150w/miR-150i) × 100 |
Physicochemical characterization
The surface morphology and size of nanoparticles were characterized by transmission electron microscopy using Technai™ G2 Spirit Bio TWIN (FEI Company, Hillsboro, OR, USA). Hydrodynamic diameter and zeta potential of nanoparticles were measured in distilled water using Delsa™ Nano C (Beckman Coulter Inc.). Zeta potential values were calculated from measured velocities using Smoluchowski equation. Mean hydrodynamic diameters were calculated for size distribution by weight with the assumption of a lognormal distribution.
In vitro release assay
About 1 mg of miR-150-loaded nanoparticles were incubated with 1 mL of release medium (Tris–EDTA buffer with or without 10% FBS, pH 7.4) in a rotary shaker at 100 rpm at 37°C. At predetermined intervals (after every 24 hours for 14 days), the nanoparticle suspension was centrifuged at 7,500 rpm at 4°C for 10 min. Supernatant (1 μL) was analyzed using the nucleic acid measurement application of NanoDrop ND-1,000 spectrophotometer (Thermo Fisher Scientific), and the amount of miRNA was reported as μg/mL. Release medium was used as a control. Samples were taken in triplicate for each time point.
Measurement of delivery efficacy
Pancreatic cancer cells were seeded in six-well plates and grown until they reached 60%–70% confluence. Cells were treated with miR-150 mimics (Ambion, Austin, TX, USA) (with or without Lipofectamine [Thermo Fisher Scientific]), miR-150-NF (0.05 mg, 0.1 mg, 0.2 mg), or blank (unloaded, 0.2 mg) PLGA nanoparticles. After 16 hours of transfection/treatment, media was replaced with complete media and cells were further cultured for 48 hours. Expression level of mature miR-150 was examined using quantitative reverse-transcription polymerase chain reaction (PCR) assay as described in the following section.
RNA isolation and quantitative reverse-transcription PCR assay
TRIzol reagent (Thermo Fisher Scientific) was used to extract total RNA. Complementary DNA was synthesized using the High Capacity complementary DNA Reverse Transcription Kit (Thermo Fisher Scientific) following manufacturer’s instruction. PCR was performed in 96-well plates using SYBRGreen Master Mix (Thermo Fisher Scientific) on an iCycler system (Bio-Rad Laboratories Inc., Hercules, CA, USA). U6 small nuclear RNA was used as an internal control. Relative amounts of miRNA were normalized against U6 small nuclear RNA, and fold change for miRNA was calculated by the 2–ΔΔCt method. All the primers used in this study are described earlier.8 The thermal conditions used for the real-time PCR were as follows: cycle 1, 95°C for 10 minutes; cycle 2 (×40), 95°C for 10 seconds and 58°C for 45 seconds.
Western blot analysis
Cells were processed for protein extraction and western blotting as described earlier.23 Immunodetection was carried out using specific antibodies against MUC4 (8G7), pY1248-HER2 (mouse monoclonal) (Santa Cruz Biotechnology Inc., Dallas, TX, USA), pHER2, pERK1/2 (mouse monoclonal), ERK1/2, FAK and pFAK (rabbit monoclonal) (Epitomics, Burlingame, CA, USA), and β-actin (mouse monoclonal) (Sigma-Aldrich Co., St Louis, MO, USA). All secondary antibodies (Santa Cruz Biotechnology Inc.) were used at 1:2,000 dilutions. Proteins were visualized with the Super Signal West Femto Maximum sensitivity substrate kit (Thermo Fisher Scientific) and the signal was detected using an LAS-3,000 image analyzer (Fuji Photo Film Co., Tokyo, Japan).
Cell growth and clonogenicity assays
For cell growth assay, cells (2×103 cells per well) were treated with miR-150-NF or blank PLGA nanoparticles (0.05 mg each), as described above, and cultured for 5 days. Cell growth was then monitored using WST-1 assay kit (Hoffman-La Roche Ltd., Basel, Switzerland) as per manufacturer’s instructions. The absorbance was measured at a wavelength of 450 nm using a Bio-Rad Benchmark microplate reader (Bio-Rad Laboratories Inc.).For clonogenicity assay, cells were cultured in six-well plates until they reached 60%–70% confluence, and were subsequently treated with miR-150-NF or blank PLGA nanoparticles (0.05 mg each). Following 48 hours transfection, cells were trypsinized and plated in six-well plates at a density of 1×103 cells per well in a regular media for colony formation. After 2 weeks, colonies were fixed with methanol, stained with crystal violet, photographed, and counted using image analysis software (Gene Tools; Syngene, Frederick, MD, USA).
Motility and invasion assays
Cells were treated with miR-150-NF or blank PLGA nanoparticles (0.05 mg each) for 48 hours, trypsinized, and subjected to motility and invasion assays. For motility assay, cells (1×106) were plated in the top chamber of mono-coated polyethylene terephthalate membrane (six-well insert, pore size 8 μm; BD Biosciences, San Jose, CA, USA). For the invasion assay, 2.5×105 cells were plated in the top chamber of the transwell with a Matrigel-coated polycarbonate membrane (24 wells, 0.8 μm; BD Biosciences). Respective media with 10% FBS was added to the lower chamber as a chemoattractant. After 16 hours of incubation, cells remaining on the upper surface of the insert membrane were removed by cotton swab. Cells that had migrated or invaded through the membrane/Matrigel to the bottom of the insert were fixed, stained with Diff-Quick cell staining kit (Siemens Healthcare Diagnostics Inc., Newark, DE, USA), mounted on a slide, photographed using inverted phase contrast microscope, and counted in ten random view fields.
Statistical analysis
Each experiment was performed independently at least three times. All the values were expressed as mean ± standard deviation. Wherever appropriate, the data were also subjected to unpaired two-tailed Student’s t-test. A value of P<0.05 was considered as significant.
Results
Characterization of miR-150-NF
Nanoparticles were characterized for physicochemical properties, namely morphology, size, zeta potential, loading efficiency, and in vitro release profile. Transmission electron microscopy studies revealed that nanoparticles were spherical in shape with a smooth surface and possessed an average particle size of ~168 nm (for blank nanoparticles, Figure 1A) and ~183 nm (for miR-150-NF, Figure 1B). Dynamic light scattering studies demonstrated an average hydrodynamic diameter of ~307 nm and a polydispersity of 0.229 for blank nanoparticles, whereas miR-150-NF exhibited average diameter of 399 nm and a polydispersity of 0.177 (data not shown). Nanoparticles had a net negative surface charge and exhibited a zeta potential of −20.22 mV for blank nanoparticles and −20.63 mV for miR-150-NF. Furthermore, our data demonstrate efficient encapsulation of miR-150 mimics (miRNA loading efficiency =78.3%) in PLGA nanoparticles. Additionally, in vitro release profile of miR-150-NF indicated an initial burst release of 9.4% and 21% of the encapsulated miR-150 in serum deprived TE-buffer and serum (10% FBS) containing TE-buffer, respectively, in the first 24 hours followed by a more sustained release over the next 14 days (Figure 2). These studies also show that miRNA release is faster in the presence of serum (>62% release vs <30% in the absence of serum).
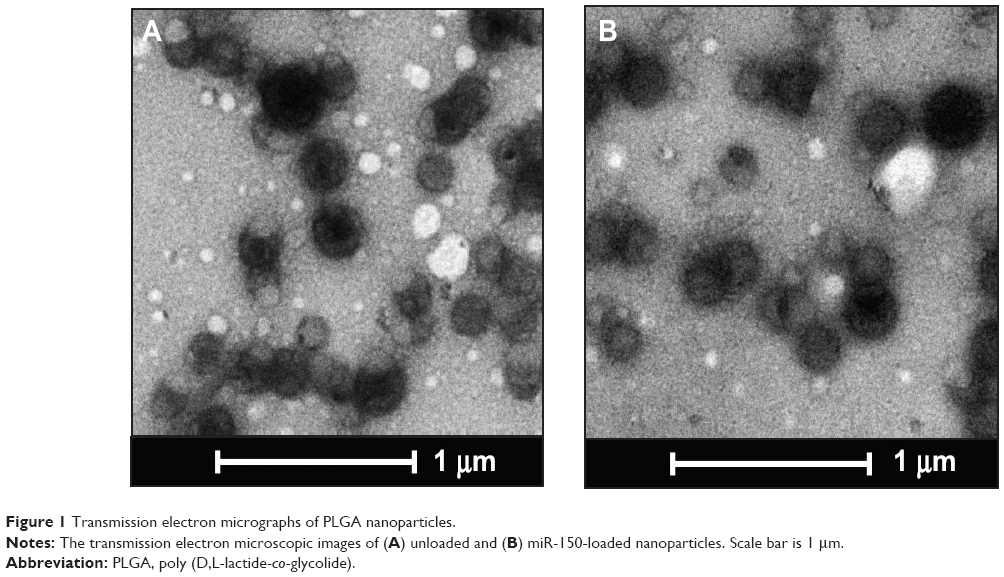 | Figure 1 Transmission electron micrographs of PLGA nanoparticles. |
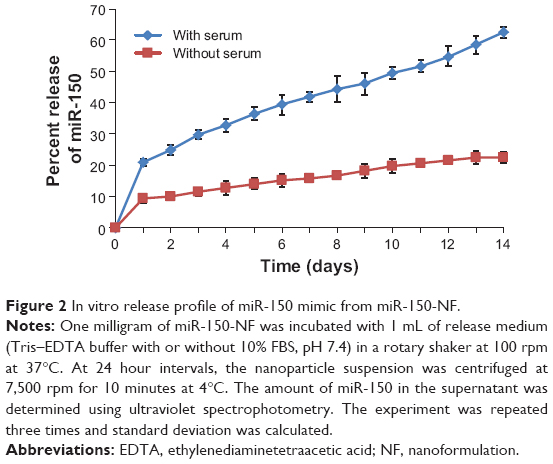 | Figure 2 In vitro release profile of miR-150 mimic from miR-150-NF. |
miR-150-NF efficiently delivers miR-150 mimics to pancreatic cancer cells
An effective delivery system should possess high transfection efficiency in order to deliver oligonucleotides proficiently into the cells. We evaluated whether the miR-150-NF is able to increase the level of miR-150 in pancreatic cancer cells. Our data revealed that miR-150 expression was significantly increased (~28-fold in Colo-357 and ~26-fold in HPAF cells) in 0.1 mg miR-150-NF (containing 0.69 μg miR-150)-treated cells as compared to those transfected with miR-150– Lipofectamine (containing 0.708 μg miR-150) (Figure 3).Moreover, miR-150 expression was further increased in a dose-dependent manner (0.05 mg to 0.2 mg) in both the cell lines after treatment with miR-150-NF. Our data suggest that miR-150-NF is highly efficient in delivering miR-150 to pancreatic cancer cells.
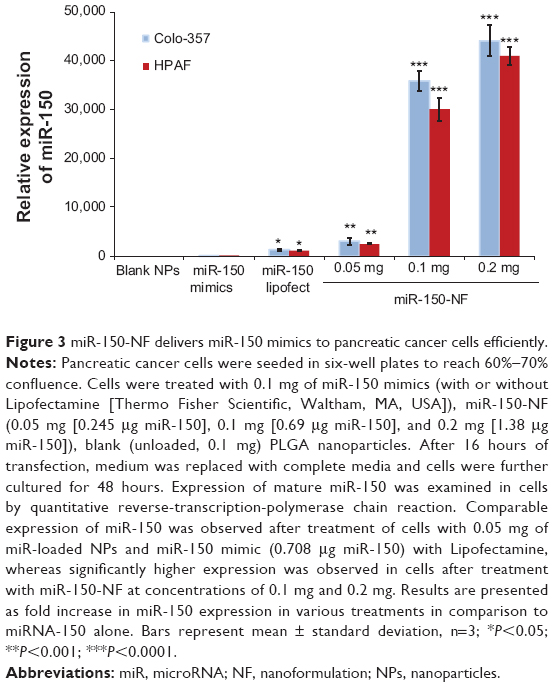 | Figure 3 miR-150-NF delivers miR-150 mimics to pancreatic cancer cells efficiently. |
Treatment of miR-150-NF represses the expression of MUC4 and its interacting partner, HER2, and impacts downstream signaling
We have shown earlier that MUC4 is a direct target of miR-150.8 Therefore, to delineate the functional efficacy of miR-150-NF, we examined its effect on the expression of MUC4. For this, we treated Colo-357 and HPAF cells with blank nanoparticles (0.1 mg), miR-150-NF (0.05 mg or 0.1 mg), or miR-150 mimics with Lipofectamine for 48 hours and examined the expression of MUC4. Our data reveal that both miR-150-NF and nonformulated miR-150 are able to repress MUC4 expression in Colo-357 and HPAF cells.Interestingly, the data demonstrate that the effect of MUC4 inhibition is more potent when cells are treated with miR-150-NF as compared to treatment with Lipofectamine– miR-150 complex (Figure 4). We next examined the effect of miR-150-NF treatment on the expression of HER2 as well as its downstream signaling. HER2 has previously been shown by us to physically interact with MUC4, where the latter causes its stabilization.24 The data from immunoblot analysis reveal that the expression of HER2 and its phosphorylated form (pY1248-HER2) is reduced following treatment with miR-150-NF in both the cell lines (Figure 4). Furthermore, we observed a decreased phosphorylation of ERK and FAK (downstream effectors of HER2) in miR-150-NF-treated cells as compared to blank nanoparticles. Altogether, our data suggest that miR-150-NF represses MUC4 and HER2, leading to attenuation of its downstream signaling in pancreatic cancer cells, thus confirming the functional efficacy of miR-150-NF.
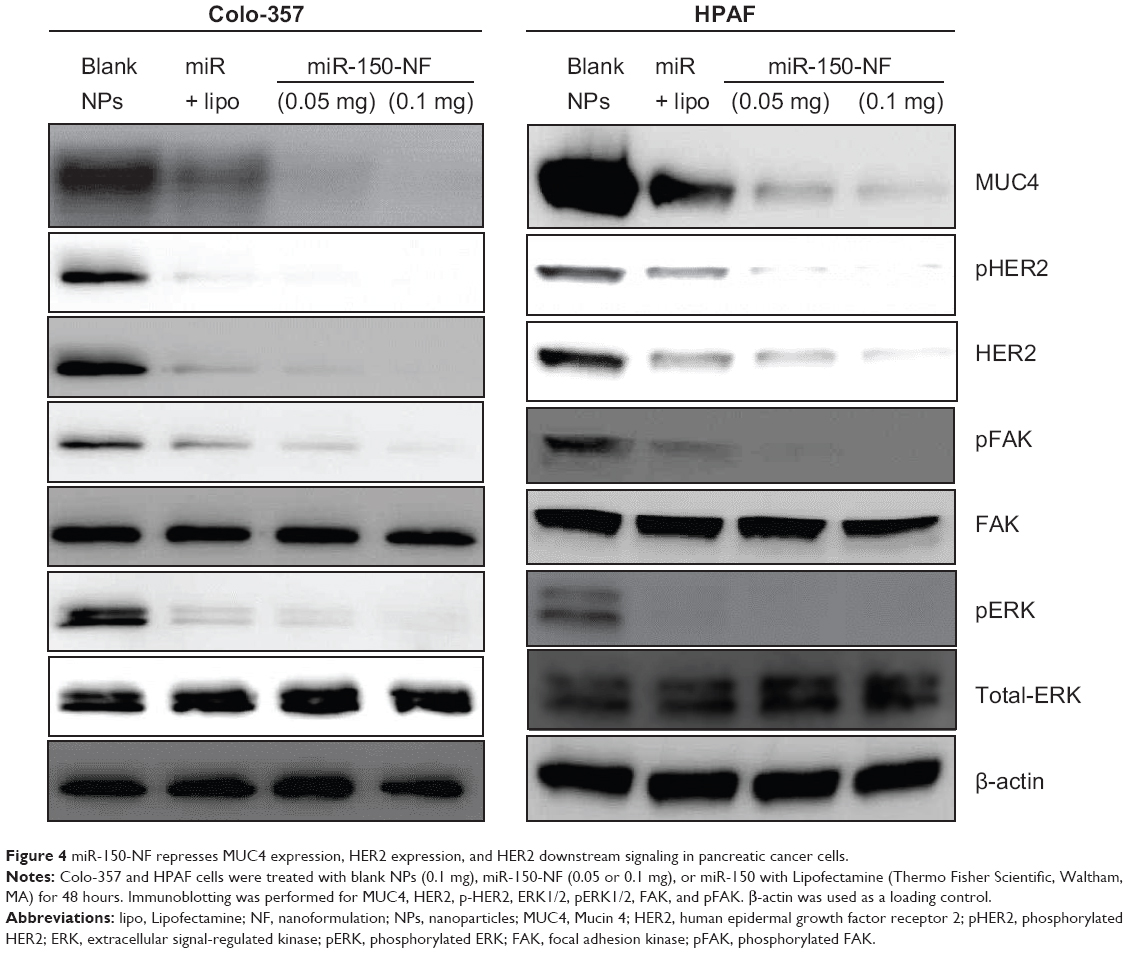 | Figure 4 miR-150-NF represses MUC4 expression, HER2 expression, and HER2 downstream signaling in pancreatic cancer cells. |
miR-150-NF treatment inhibits growth and clonogenicity of pancreatic cancer cells
In order to examine the effects of miR-150-NF on pancreatic cancer cell phenotype, we performed a series of in vitro assays. First, we studied the effect of miR-150-NF on the cellular morphology and growth of pancreatic cancer cells. Cells treated with miR-150-NF (0.05 mg and 0.1 mg) showed alterations in morphology as compared to untreated cells (Figure 5). At 0.1 mg concentration of miR-150-NF, remarkable changes in the morphology of pancreatic cancer cells (Colo-357 as well as HPAF) were observed as illustrated by rounded shape, smaller size, reduced spreading over the culture dish, and detachment from the substratum as compared to the control cells (Figure 5). Furthermore, the growth kinetics study demonstrated significant growth inhibition in miR-150-NF-treated pancreatic cancer cells (~66% in Colo-357 and ~59% in HPAF) on day 5 as compared to their respective controls (Figure 6A). We next examined the effect of miR-150-NF on the anchorage-dependent clonogenic ability of the pancreatic cancer cells. The data reveal that miR-150-NF decreases the clonogenic ability of Colo-357 and HPAF cells by 78.4% and 69.3%, respectively, as compared to that observed in blank nanoparticle-treated cells(Figure 6B). Thus, our findings suggest a significant antitumor efficacy of miR-150-NF against pancreatic cancer cells.
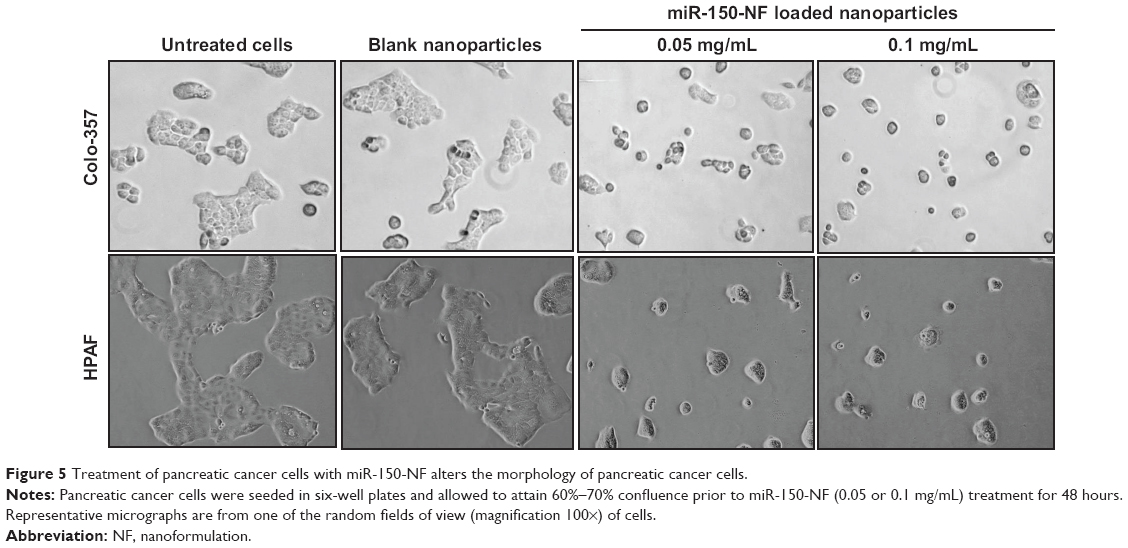 | Figure 5 Treatment of pancreatic cancer cells with miR-150-NF alters the morphology of pancreatic cancer cells. |
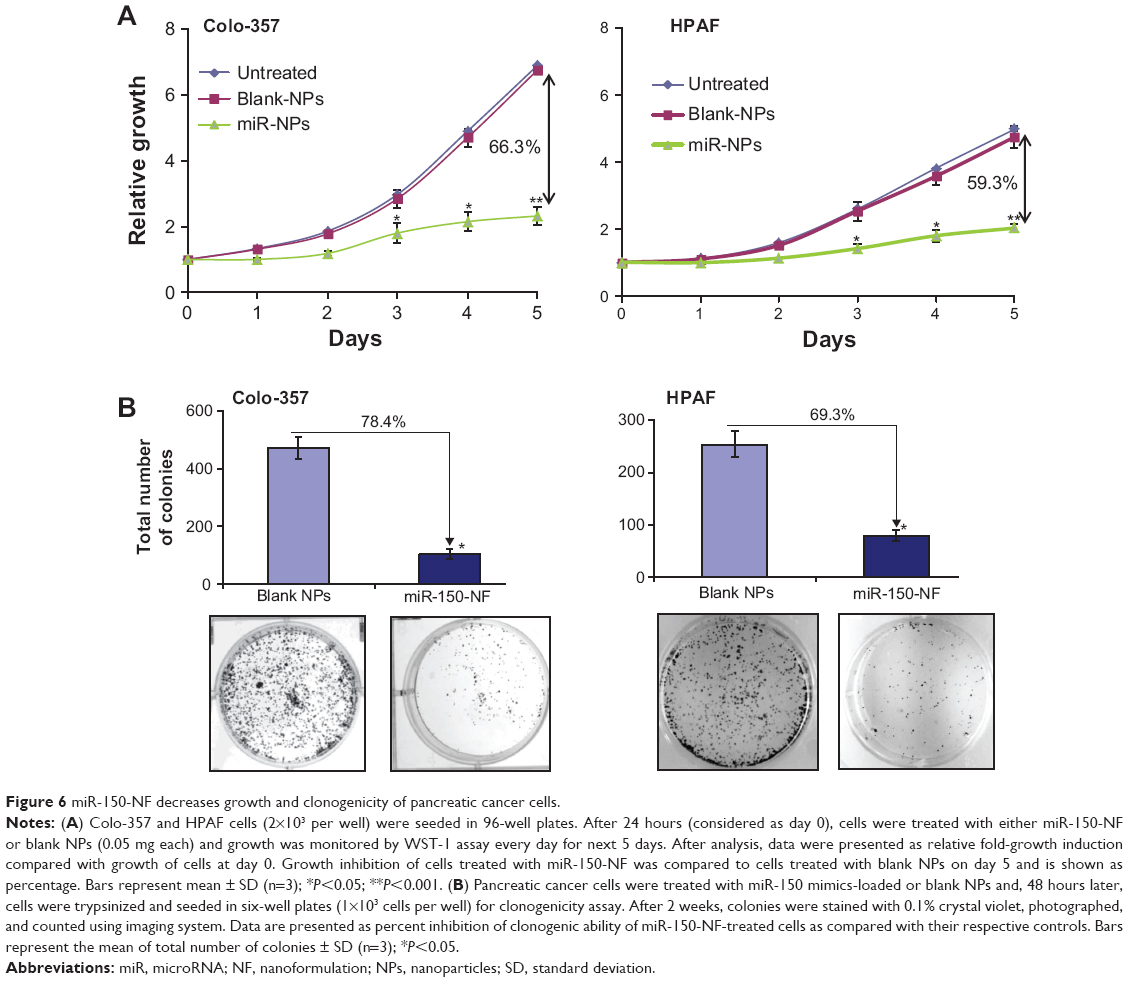 | Figure 6 miR-150-NF decreases growth and clonogenicity of pancreatic cancer cells. |
Treatment with miR-150-NF results in the suppression of malignant behavior of pancreatic cancer cells
In our previous studies, we have shown an important role of MUC4 in potentiating the aggressive malignant behavior of pancreatic tumor cells.8 Therefore, we investigated the effect of miR-150-NF treatment on motility and invasive potential of Colo-357 and HPAF cells. Cell motility assay was performed by following the migration of tumor cells under chemotactic drive (FBS) using a Boyden’s chamber. Data show a significant decrease (~3.1- and 2.8-fold in Colo-357 and HPAF cells, respectively) in the number of migrated cells following miR-150-NF treatment as compared to their respective controls (Figure 7). Furthermore, Matrigel invasion assay was performed to determine the effect of miR-150-NF treatment on invasiveness of pancreatic cancer cells. The data demonstrate that, following treatment with miR-150-NF, invasiveness of the Colo-357 and HPAF cells was decreased ~4.0- and 3.3-fold, respectively, as compared to the cells treated with blank nanoparticles (Figure 7). Altogether, the data suggest a potent effect of miR-150-NF treatment on attenuating the malignant potential of pancreatic cancer cells.
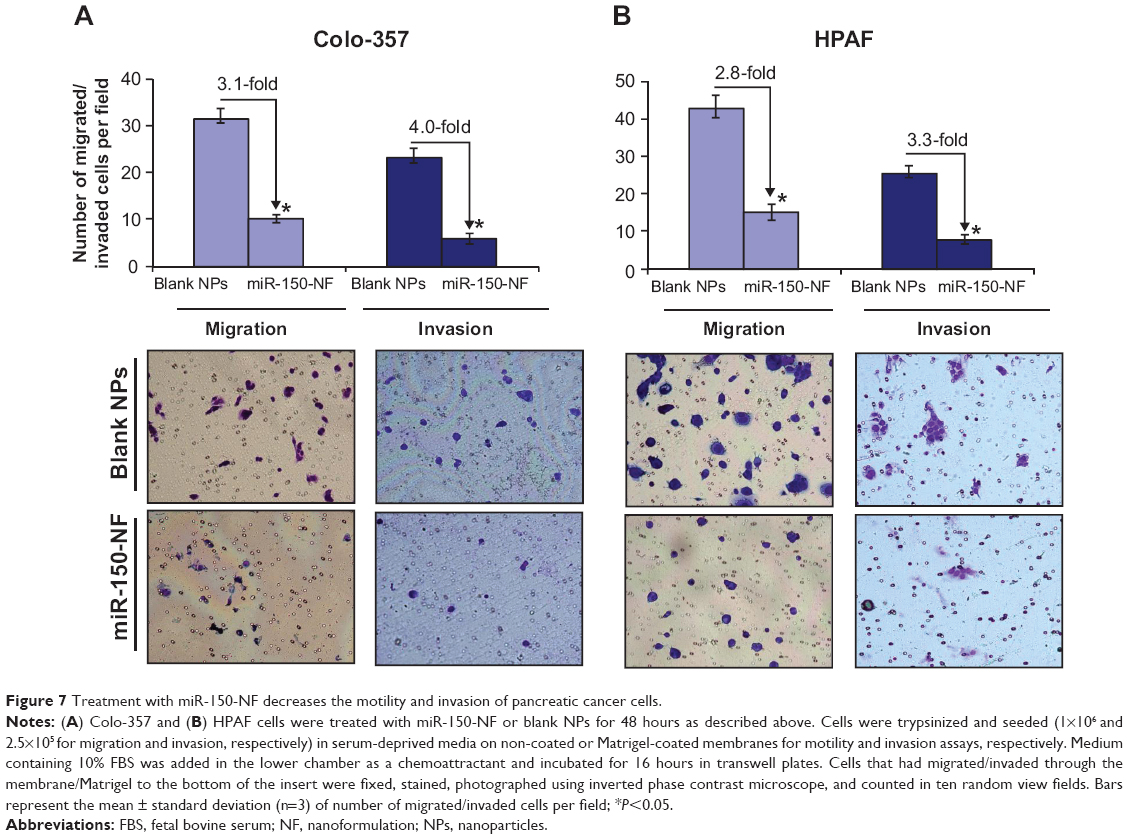 | Figure 7 Treatment with miR-150-NF decreases the motility and invasion of pancreatic cancer cells. |
Discussion
miRNA delivery approach, also known as miRNA replacement therapy, is focused on reinstating functional miRNAs that are lost in the diseased cells. In the present study, we generated PLGA-based miR-150-NF to facilitate the delivery of miR-150 into pancreatic cancer cells exhibiting its downregulated expression. Our studies show that miR-150-NF was able to efficiently deliver miR-150 mimics to pancreatic cancer cells. Treatment with miR-150-NF led to the repression of miR-150 gene target, MUC4, and also caused downregulation of its interacting partner, HER2. We also observed suppression of HER2 and its downstream signaling upon treatment with miR-150-NF, which resulted in significant inhibition of pancreatic cancer cell growth, clonogenicity, motility, and invasion.
Nucleotide delivery requires carriers that are safe, are capable of efficient endocytosis into tumor cells, and possibly possess targeting capabilities for delivery to specific cells.25–28 In the present study, we chose the polymer PLGA for the preparation of nanoparticles to deliver miR-150 due to advantages of PLGA over other polymers. PLGA-based polymer matrices exhibit tremendous versatility that allows for their tailoring to meet the specific need.20,29 Furthermore, PLGA polymers are biodegradable and are approved for human use.20,21 In addition, PLGA-based nanoparticles permit sustained intracellular release of encapsulated agents, leading to potent and prolonged therapeutic efficacy.30–32 However, one of the limitations of the PLGA nanoparticles is their low encapsulation efficiency (<50%) for low molecular weight and hydrophilic molecules.22,29 We were able to overcome this limitation by incorporating PEI, a cationic polymer, in the PLGA matrix. PEI has a high positive charge density and, therefore, helps in enhanced encapsulation of negatively charged moieties such as nucleic acids.33,34 Indeed, addition of cationic polymers, such as PEI or chitosan, has been shown previously to improve encapsulation efficiency of small interfering RNAs.35,36
Additional criteria that affect the efficacy of nanoparticles are their size and zeta potential, which determine the level of cellular and tissue uptake.37–39 Various studies have been conducted to investigate the effect of nanoparticle size on gene delivery efficiency. Smaller size nanoparticles exhibit greater gene delivery efficiency, and some studies suggest 400 nm as the effective upper size limit for passive targeting.37,40–42 Although the average hydrodynamic diameter of our miR-150-loaded nanoparticles was close to this upper limit, they still exhibited efficient intracellular delivery of miRNAs. However, it will be desirable to further refine our formulation methodology to reduce the size of miR-150-NF prior to initiating in vivo studies.
Zeta potential is an indicator of the surface charge of nanoparticles and is a key parameter that determines nanoparticle stability in suspension.37,43 Nanoparticles with high negative or positive zeta potential values are generally more stable in dispersion. Despite the addition of PEI, the net zeta potential of miR-150-NF was –20.63 mV. This could be due to the fact that the amount of PEI in the formulation was very small (100 μg) compared to the amount of PLGA (32 mg). Moreover, the presence of the negatively charged miRNA molecules also would have neutralized some of the cationic charge arising from PEI. In additional studies, we observed a slow and sustained release of miR-150 in vitro, which is highly desirable for sustained therapeutic activity.37,44 Faster miRNA release in the presence of serum (>62% release versus <30% in the absence of serum) might be attributed to a higher percentage of polyanions, mainly due to albumins in serum, that can compete with anionic miRNAs in binding to cationic PEI.
miRNAs play important roles in a wide range of biological processes, such as cell proliferation, apoptosis, differentiation, metabolism, and migration and invasion, through regulation of target gene expression.5,45 To be therapeutically effective, miR-150-NF treatment should be capable of inducing both biological and molecular effects. In that regard, we observed that treatment of pancreatic cancer cells with miR-150-NF caused downregulation of miR-150 target gene (MUC4) expression. Additionally, miR-150-NF also repressed the expression of HER2, an interacting partner of MUC4, leading to attenuation of its downstream signaling (FAK and ERK) in pancreatic cancer cells. These observations correlated with decreased growth and clonogenicity of pancreatic tumor cells and suppression of malignant behavioral properties. These effects were consistent with our previous findings8 and clearly demonstrated the therapeutic efficacy of miR-150-NF in vitro.
In summary, we have successfully developed PLGA-based nanodelivery system of miR-150 that efficiently encapsulates miR-150, demonstrates sustained release, effectively delivers miR-150, and inhibits growth and malignant potential of pancreatic cancer cells. Since PLGA nanoparticles can be easily surface modified,20,25 our formulation also holds immense potential to ensure the targeted delivery of miRNAs to the cancer cells under in vivo setting. Additional studies are, however, needed to further test its effectiveness in vivo. Nonetheless, our data provides proof of the concept for miRNA delivery using PLGA-based nanoformulation.
Acknowledgment
Grant Support: NIH/NCI [CA137513, CA167137, CA175772, CA185490 (to APS) and CA169829, CA186233 (to SS)] and USAMCI.
Disclosure
The authors report no conflicts of interest in this work.
References
Long J, Zhang Y, Yu X, et al. Overcoming drug resistance in pancreatic cancer. Expert Opin Ther Targets. 2011;15:817–828. | ||
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. | ||
Arora S, Bhardwaj A, Singh S, et al. An undesired effect of chemotherapy: gemcitabine promotes pancreatic cancer cell invasiveness through reactive oxygen species-dependent, nuclear factor κB- and hypoxia-inducible factor 1α-mediated up-regulation of CXCR4. J Biol Chem. 2013;19;288:21197–21207. | ||
Alvarez-Garcia I, Miska EA. MicroRNA functions in animal development and human disease. Development. 2005;132:4653–4662. | ||
Bhardwaj A, Singh S, Singh AP. MicroRNA-based Cancer Therapeutics: Big Hope from Small RNAs. Mol Cell Pharmacol. 2010;2:213–219. | ||
Bushati N, Cohen SM. microRNA functions. Annu Rev Cell Dev Biol. 2007;23:175–205. | ||
Srivastava SK, Bhardwaj A, Leavesley SJ, Grizzle WE, Singh S, Singh AP. MicroRNAs as potential clinical biomarkers: emerging approaches for their detection. Biotech Histochem. 2013;88:373–387. | ||
Srivastava SK, Bhardwaj A, Singh S, et al. MicroRNA-150 directly targets MUC4 and suppresses growth and malignant behavior of pancreatic cancer cells. Carcinogenesis. 2011;32:1832–1839. | ||
Kobayashi E, Hornicek FJ, Duan Z. MicroRNA Involvement in Osteosarcoma. Sarcoma. 2012;2012:359739. | ||
Pecot CV, Calin GA, Coleman RL, Lopez-Berestein G, Sood AK. RNA interference in the clinic: challenges and future directions. Nat Rev Cancer. 2011;11:59–67. | ||
Ibraheem D, Elaissari A, Fessi H. Gene therapy and DNA delivery systems. Int J Pharm. 2014;459:70–83. | ||
Razi SS, Baradaran B, Lotfipour F, Kazemi T, Mohammadnejad L. Gene therapy, early promises, subsequent problems, and recent breakthroughs. Adv Pharm Bull. 2013;3:249–255. | ||
Young LS, Searle PF, Onion D, Mautner V. Viral gene therapy strategies: from basic science to clinical application. J Pathol. 2006;208: 299–318. | ||
Itaka K, Kataoka K. Recent development of nonviral gene delivery systems with virus-like structures and mechanisms. Eur J Pharm Biopharm. 2009;71:475–483. | ||
Schagen FH, Ossevoort M, Toes RE, Hoeben RC. Immune responses against adenoviral vectors and their transgene products: a review of strategies for evasion. Crit Rev Oncol Hematol. 2004;50:51–70. | ||
Aigner A. Nonviral in vivo delivery of therapeutic small interfering RNAs. Curr Opin Mol Ther. 2007;9:345–352. | ||
Merdan T, Kopecek J, Kissel T. Prospects for cationic polymers in gene and oligonucleotide therapy against cancer. Adv Drug Deliv Rev. 2002;54:715–758. | ||
Zhou J, Shum KT, Burnett JC, Rossi JJ. Nanoparticle-Based Delivery of RNAi Therapeutics: Progress and Challenges. Pharmaceuticals (Basel). 2013;6:85–107. | ||
Tyagi N, Rathore SS, Ghosh PC. Enhanced killing of human epidermoid carcinoma (KB) cells by treatment with ricin encapsulated into sterically stabilized liposomes in combination with monensin. Drug Deliv. 2011;18:394–404. | ||
Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Préat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release. 2012;161:505–522. | ||
Langer R, Tirrell DA. Designing materials for biology and medicine. Nature. 2004;428:487–492. | ||
Patil Y, Panyam J. Polymeric nanoparticles for siRNA delivery and gene silencing. Int J Pharm. 2009;367:195–203. | ||
Singh S, Srivastava SK, Bhardwaj A, Owen LB, Singh AP. CXCL12-CXCR4 signalling axis confers gemcitabine resistance to pancreatic cancer cells: a novel target for therapy. Br J Cancer. 2010;103: 1671–1679. | ||
Chaturvedi P, Singh AP, Chakraborty S, et al. MUC4 mucin interacts with and stabilizes the HER2 oncoprotein in human pancreatic cancer cells. Cancer Res. 2008;68:2065–2070. | ||
Jain AK, Das M, Swarnakar NK, Jain S. Engineered PLGA nanoparticles: an emerging delivery tool in cancer therapeutics. Crit Rev Ther Drug Carrier Syst. 2011;28:1–45. | ||
Kesharwani P, Gajbhiye V, Jain NK. A review of nanocarriers for the delivery of small interfering RNA. Biomaterials. 2012;33: 7138–7150. | ||
Panyam J, Labhasetwar V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv Drug Deliv Rev. 2003;55: 329–347. | ||
Wang J, Lu Z, Wientjes MG, Au JL. Delivery of siRNA therapeutics: barriers and carriers. AAPS J. 2010;12:492–503. | ||
Barichello JM, Morishita M, Takayama K, Nagai T. Encapsulation of hydrophilic and lipophilic drugs in PLGA nanoparticles by the nanoprecipitation method. Drug Dev Ind Pharm. 1999;25:471–476. | ||
Cohen H, Levy RJ, Gao J, et al. Sustained delivery and expression of DNA encapsulated in polymeric nanoparticles. Gene Ther. 2000;7: 1896–1905. | ||
Khan A, Benboubetra M, Sayyed PZ, et al. Sustained polymeric delivery of gene silencing antisense ODNs, siRNA, DNAzymes and ribozymes: in vitro and in vivo studies. J Drug Target. 2004;12:393–404. | ||
Panyam J, Zhou WZ, Prabha S, Sahoo SK, Labhasetwar V. Rapid endo-lysosomal escape of poly(DL-lactide-co-glycolide) nanoparticles: implications for drug and gene delivery. FASEB J. 2002;16:1217–1226. | ||
Read ML, Singh S, Ahmed Z, et al. A versatile reducible polycation-based system for efficient delivery of a broad range of nucleic acids. Nucleic Acids Res. 2005;33:e86. | ||
Zhu L, Mahato RI. Lipid and polymeric carrier-mediated nucleic acid delivery. Expert Opin Drug Deliv. 2010;7:1209–1226. | ||
Patil YB, Swaminathan SK, Sadhukha T, Ma L, Panyam J. The use of nanoparticle-mediated targeted gene silencing and drug delivery to overcome tumor drug resistance. Biomaterials. 2010;31:358–365. | ||
Tahara K, Sakai T, Yamamoto H, Takeuchi H, Kawashima Y. Establishing chitosan coated PLGA nanosphere platform loaded with wide variety of nucleic acid by complexation with cationic compound for gene delivery. Int J Pharm. 2008;354:210–216. | ||
Dinarvand R, Sepehri N, Manoochehri S, Rouhani H, Atyabi F. Polylactide-co-glycolide nanoparticles for controlled delivery of anticancer agents. Int J Nanomedicine. 2011;6:877–895. | ||
Gaumet M, Vargas A, Gurny R, Delie F. Nanoparticles for drug delivery: the need for precision in reporting particle size parameters. Eur J Pharm Biopharm. 2008;69:1–9. | ||
Tyagi N, Ghosh PC. Folate receptor mediated targeted delivery of ricin entrapped into sterically stabilized liposomes to human epidermoid carcinoma (KB) cells: effect of monensin intercalated into folate-tagged liposomes. Eur J Pharm Sci. 2011;43:343–353. | ||
Kim JH, Park JS, Yang HN, et al. The use of biodegradable PLGA nanoparticles to mediate SOX9 gene delivery in human mesenchymal stem cells (hMSCs) and induce chondrogenesis. Biomaterials. 2011;32: 268–278. | ||
Prabha S, Zhou WZ, Panyam J, Labhasetwar V. Size-dependency of nanoparticle-mediated gene transfection: studies with fractionated nanoparticles. Int J Pharm. 2002;244:105–115. | ||
Ishida O, Maruyama K, Sasaki K, Iwatsuru M. Size-dependent extravasation and interstitial localization of polyethyleneglycol liposomes in solid tumor-bearing mice. Int J Pharm. 1999;190:49–56. | ||
Hall JB, Dobrovolskaia MA, Patri AK, McNeil SE. Characterization of nanoparticles for therapeutics. Nanomedicine (Lond). 2007;2: 789–803. | ||
Utreja P, Jain S, Tiwary AK. Novel drug delivery systems for sustained and targeted delivery of anti-cancer drugs: current status and future prospects. Curr Drug Deliv. 2010;7:152–161. | ||
Iorio MV, Croce CM. MicroRNAs in cancer: small molecules with a huge impact. J Clin Oncol. 2009;27:5848–5856. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.