")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Synthesis and in vivo magnetic resonance imaging evaluation of biocompatible branched copolymer nanocontrast agents
Authors Jackson A, Chandrasekharan P, Shi J, Rannard SP, Liu Q, Yang CT, He T
Received 18 May 2015
Accepted for publication 30 June 2015
Published 18 September 2015 Volume 2015:10(1) Pages 5895—5907
DOI https://doi.org/10.2147/IJN.S88764
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Dr Thomas Webster
Alexander W Jackson,1,* Prashant Chandrasekharan,2,* Jian Shi,3 Steven P Rannard,4 Quan Liu,5 Chang-Tong Yang,6 Tao He1,7
1Institute of Chemical and Engineering Sciences (ICES), 2Laboratory of Molecular Imaging, Singapore Bioimaging Consortium, Agency for Science Technology and Research (A* STAR), 3Department of Biological Science, National University of Singapore, Singapore; 4Department of Chemistry, University of Liverpool, Liverpool, United Kingdom; 5School of Chemical and Biomedical Engineering, 6Lee Kong Chian School of Medicine, Nanyang Technological University, Singapore; 7School of Chemistry and Chemical Engineering, HeFei University of Technology, Anhui, People’s Republic of China
*These authors contributed equally to this work
Abstract: Branched copolymer nanoparticles (Dh =20–35 nm) possessing 1,4,7, 10-tetraazacyclododecane-N,N',N",N'"-tetraacetic acid macrocycles within their cores have been synthesized and applied as magnetic resonance imaging (MRI) nanosized contrast agents in vivo. These nanoparticles have been generated from novel functional monomers via reversible addition–fragmentation chain transfer polymerization. The process is very robust and synthetically straightforward. Chelation with gadolinium and preliminary in vivo experiments have demonstrated promising characteristics as MRI contrast agents with prolonged blood retention time, good biocompatibility, and an intravascular distribution. The ability of these nanoparticles to perfuse and passively target tumor cells through the enhanced permeability and retention effect is also demonstrated. These novel highly functional nanoparticle platforms have succinimidyl ester-activated benzoate functionalities within their corona, which make them suitable for future peptide conjugation and subsequent active cell-targeted MRI or the conjugation of fluorophores for bimodal imaging. We have also demonstrated that these branched copolymer nanoparticles are able to noncovalently encapsulate hydrophobic guest molecules, which could allow simultaneous bioimaging and drug delivery.
Keywords: branched copolymer nanoparticles, gadolinium chelate, MRI, RAFT polymerization
Introduction
Recently, there has been an increased interest in the development of nanomaterials for the diagnosis and treatment of different diseases.1–5 A guiding principle in this rapidly growing area is the ability of nanoparticles to mimic the high affinity and multivalent interactions that exist in cellular systems offering the potential for highly efficient synthetic diagnostic and therapeutic systems.6–12 One clear advantage of nanomaterials lies in their potential to be used as noninvasive diagnostic tools with the capacity to combine multiple modalities on one probe and the longer term opportunities for simultaneous application as drug-delivery systems. Therefore, the development of a new generation of nano-sized theranostics is a very appealing goal for contemporary polymer scientists. Nanomaterials employing a wide range of substances, both organic (dendrimers, liposomes, and natural and synthetic polymers) and inorganic materials (quantum dots, metallic nanostructures, and metal oxides), can be used as diagnostic and therapeutic tools. When decorated with special functionalities, nanomaterials may also be applied in a wide range of different molecular imaging techniques, such as single-photon emission tomography, positron emission tomography, optical imaging, and magnetic resonance imaging (MRI).13
MRI is an attractive imaging technique with several advantages such as high soft-tissue resolution and discrimination in any imaging plane, high spatial resolution tomography, and lack of radioactivity. To improve contrast during MRI, exogenous contrast agents can be employed aiding the distinction between diseased and normal tissues by altering the longitudinal and transverse relaxation times (T1 and T2) of water protons. Although various paramagnetic compounds have been evaluated for application within MRI, gadolinium(III) (Gd(III)) chelates continue to be the most wildly used T1-weighted contrast agents. To date, most Gd(III) contrast agents are small molecular chelates such as the clinically approved Gd(III)-diethylene diamine penta-acetic acid (Magnevist®) and Gd(III)-N, N′, N″, N″′-tetracarboxymethyl-1,4,7,10-tetraazacyclododecane (Gd[DOTA]) (Dotarem®).14,15 However, small molecular Gd(III) contrast agents usually suffer from transient tissue retention, appreciable toxicities, poor sensitivity, rapid renal clearance, and unfavorable pharmacokinetic profiles, which collectively results in a short imaging time window, low contrast, and limited clinical applications.
To address these issues, there has been an increased interest in developing polymeric nanocontrast agents for MRI. To date, most polymer nanocontrast agents focus on Gd(III) chelates conjugated onto dendrimers,16–22 liposomes,23–30 micelles,31–35 core cross-linked star polymers,36,37 and hyperbranched polymers.36–38 Polymer-based nanoparticle MRI contrast agents have demonstrated optimal pharmacokinetics and circulation half-life, increased tolerance to enzymatic degradation, as well as greater physical and chemical stability which improve their therapeutic value.16–38 For example, Liu et al have recently reported38 that Gd(III) chelates have been conjugated onto hyperbranched polyprodrug amphiphiles which demonstrate prolonged blood circulation with a half-life up to ~9.8 hours.
However, the synthesis of dendrimers often suffers from challenging multistep synthesis and purification. Indirect assembly strategies utilizing amphiphilic block copolymers can be problematic as micelles are not very stable on dilution, leading to uncertainty during in vivo studies; the need for additional micelle cross-linking increases the inherent complexity. Therefore, we see the need for the development of more versatile and efficient synthetic routes to polymer nanoparticle MRI contrast agents. The groups Li et al have made significant advancements with their preparation36,37 of core cross-linked star and hyperbranched polymer nanoparticles. They have elegantly demonstrated37 that the precise molecular location of gadolinium has a significant influence on relaxivity. One of the key advantages of core cross-linked star and classic hyperbranched polymer nanoparticles is the inclusion of cross-linking during the polymerization process, a clear benefit when compared to the traditional micellar approach. Unlike micellar assemblies, these polymer nanoparticles do not possess large hydrophobic domains which are required to permit the loading of hydrophobic guest molecules. Our previous research39–42 has focused on the development of branched copolymer nanoparticles through a similar synthetic approach to the synthesis of core cross-linked star polymers via the “arm-first” method. In this present work, we have shown that it is possible to synthesize structures which exist between micelles and core cross-linked star polymers. During the cross-linking polymerization stage, we include a significant amount of a hydrophobic comonomer. The resulting branched copolymer nanoparticles comprise a hydrophilic corona and a hydrophobic covalently cross-linked core; this synthetic approach does not require post-polymerization modification to incorporate macrocycle functionalities which is a common additional synthetic step in other similar systems. We believe that these structures will yield a great advancement in the field of bioimaging as there is the possibility for simultaneous imaging and delivery of hydrophobic pharmaceuticals.
Herein, we present the direct synthesis of stable Gd(III)- chelated branched copolymer nanoparticles, synthesized from novel monomers via reversible addition–fragmentation chain transfer (RAFT) polymerization without the need for self-assembly techniques (Figure 1). We have demonstrated that these nanoparticles are promising MRI contrast agents and are able to perfuse and passively target tumor cells, presumably by means of the enhanced permeation and retention (EPR) effect. The hydrophobic core is able to simultaneously chelate gadolinium and noncovalently encapsulate hydrophobic guest molecules, hence combining the properties of micellar assemblies with the synthetic advantages of core cross-linked star polymers. We have also shown that it is possible to adorn these nanoparticles with succinimidyl ester-activated benzoate functionalities, which may be used in future studies for conjugation of various targeting moieties to achieve active bioimaging or to introduce small molecule fluorescent probes to produce multimodality bioimaging nanoparticles.
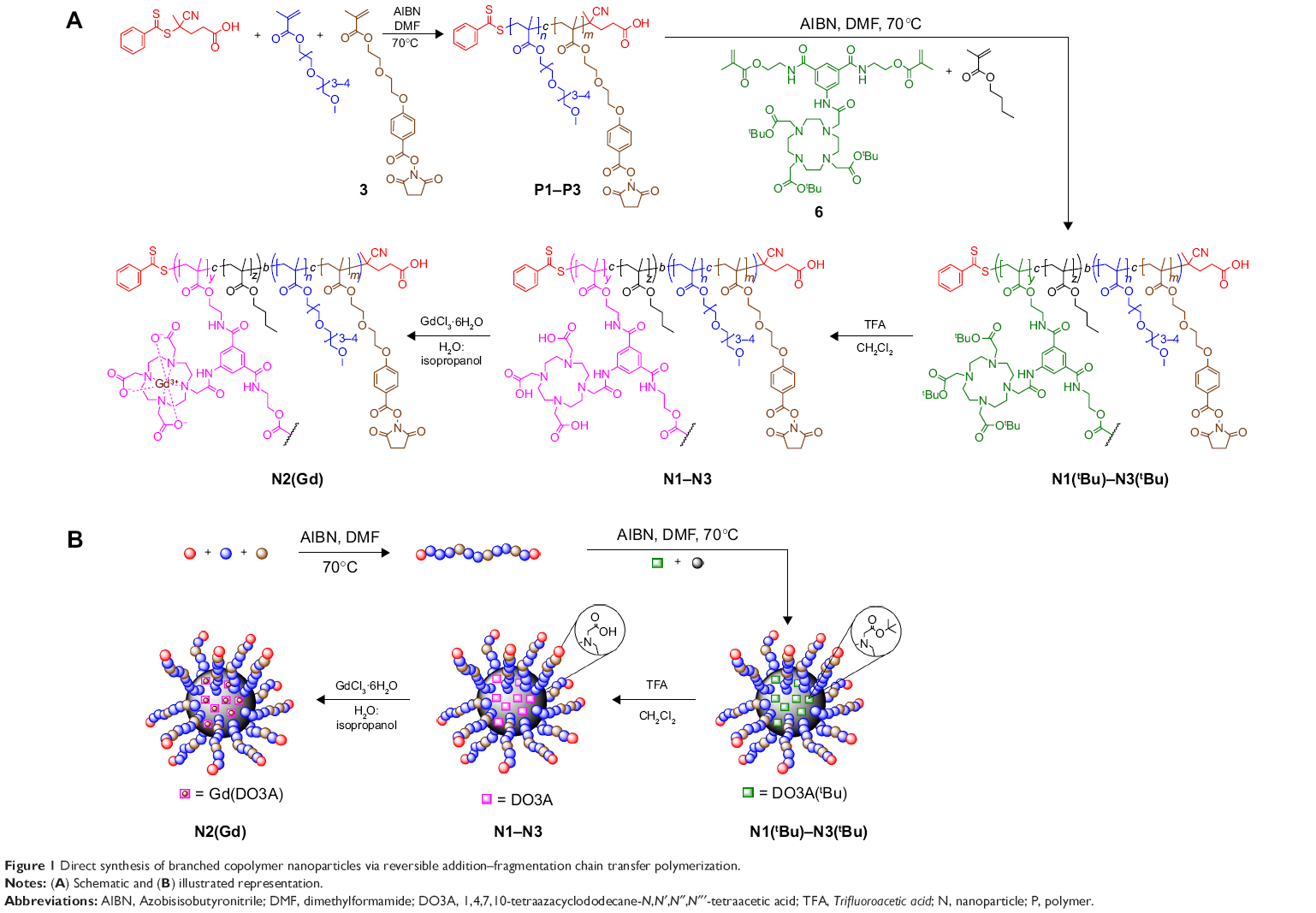 | Figure 1 Direct synthesis of branched copolymer nanoparticles via reversible addition–fragmentation chain transfer polymerization. |
Methods
Relaxivity measurements
Longitudinal (r1) and transverse (r2) relaxivities were measured using a 7 T Bruker ClinScan (Bruker Biospin GmbH, Karlsruhe, Germany) with a Siemens interface; a 72 mm volume coil was used. T1 was calculated using an inversion-recovery sequence; repetition time (TR)/echo time (TE) =5,000 milliseconds/7 milliseconds; average =3; flip angle =90°; and inversion times =31, 150, 300, 700, 1,200, 1,800, 2,400, 3,000, 3,600, 4,000, 4,500, and 4900 milliseconds. T2 was measured using a multiple spin echo sequence; TR =5,000 milliseconds; TE =12.9 milliseconds; number of echoes (NE) =14. The sample was maintained at 36°C using a circulated water bath. T2 maps were obtained by the inline acquisition sequence program. T1 was calculated from the inversion-recovery images, by fitting for the decay curve. The values r1 and r2 were reported for 1.0 mmol concentration of the contrast agent (slope of the linear fit between 1/T1 or 1/T2 versus concentration of contrast agent in mmol). The relaxivity is represented as mM−1s−1 ± standard deviation (n=3).
HK-2 cell viability assay
Ethical approval for the use of commercial cell line of human origin was sought from National University of Singapore – Institutional Review Board (NUS-IRB). HK-2 proximal tubule epithelial cells (ATCC, CRL-2190) were cultured in keratinocyte serum-free media (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with prequalified human recombinant epidermal growth factor, bovine pituitary extract, and 1% penicillin–streptomycin solution at 37°C in a humidified incubator with 5% CO2. For the cell viability assay, the cell passage number was kept within 3–5. In general, 5,000 viable cells/100 μL media were added in each well of a 96-well plate. The cells were allowed to attach for 24 hours. N2(Gd) nanoparticles (contrast agent) were prepared at different concentrations using sterile 1X phosphate-buffered saline (PBS). The prepared solution was further sterilized under UV light for 45–60 minutes. After 24 hours’ cell attachment, 10 μL of the contrast agent at different concentration was added (six wells for one concentration). To another six wells, 10 μL of sterile 1X PBS was added; these wells were used as positive control. Each concentration of the contrast agent was added to one well containing no cells but 100 μL of the culture media alone. The cells were incubated with the contrast agent at different time points. Four hours before each time point, 10 μL of Cell Counting Kit-8 (Sigma-Aldrich Co., St Louis, MO, USA) was added into each of the wells. After 4 hours’ incubation, the absorbance of each well was read at 450 nm (reference wavelength of 650 nm) using a well plate reader (SpectraMax 340PC; Molecular Devices LLC, Sunnyvale, CA, USA). The cell viability was calculated after correction for absorbance with the control wells. The data is represented as concentration versus percent cell viability ± standard error and:
| (1) |
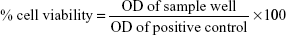
Xenograft model and in vivo MRI study
All animal work was done under Institutional Animal Care and Use Committee ethical policies and guidelines #100591. U87MG human glioma xenograft was grown on SCID mice (6 weeks old, female). In brief, the cells grown in Dulbecco’s Modified Eagle’s Medium with 10% fetal bovine serum and 1% penicillin–streptomycin were prepared at concentrations of 1×106–3×106 viable cells in volumes of approximately 100–150 μL 1X PBS (with 20% v/v Matrigel to get a slightly viscous solution). Using a 30G needle, the cells were slowly injected under the skin on the right flank of the animal. Tumors were periodically measured and animals were scanned when the tumor size reached 150–200 mm3. N2 nanoparticles were injected with a dose of 0.045 mmol of Gd(III)/kg of animal. MRI was done using a 7 T Bruker ClinScan, with a mouse transmit and receive body coil. The first phase tracer kinetics were studied using a FLASH sequence with a temporal resolution of 10 seconds in the coronal plane; TR/TE =10.62/1.32 milliseconds, flip angle =20°. Angiographic image was obtained by subtraction of pre- and post-contrast images and generating a maximum intensity projection in the 2D-plane. T1-weighted image was obtained using a gradient echo sequence in the axial plane with TR/TE =400/2.6 milliseconds and flip angle =90°; the image acquisition was gated with the respiration with the animal in 1.5% (v/v) isoflurane with 80 beats per minute respiration rate. T1 maps were obtained using a turbo-spin echo inversion recovery sequence with respiration gating, with TR/TE =5,000/7.7 milliseconds and flip angle =180°, and fat saturation and turbo factor of 3. Inversion times =41, 500, 1,000, 1,500, 2,000, 3,000, and 4,000 milliseconds. T1 maps were obtained with the in-house Matlab program.
Biodistribution study
Six SCID mice with U87MG xenograft tumor were randomly divided into two groups. In each group, three animals were injected with 0.045 mmol of gadolinium/kg/100 μL of nanoparticles and one mouse in each group was injected with 100 μL of 0.9% normal saline. Of two groups, one group was sacrificed at 2 hours and the other at 4 hours postinjection of nanoparticles. The animals were exsanguinated by cardiac puncture under 2% isoflurane. The blood sample was heparinized and stored at 4°C. Organs were snap frozen in liquid nitrogen and freeze-dried for 2 days at −75°C and 0.011 mbar (Christ Alpha 2–4 LD freeze dryer). Blood samples were analyzed using inductively coupled plasma mass spectrometry (ICP-MS). A known volume (100 μL) of blood was treated with 200 μL of concentrated nitric acid. The solution was heated to 80°C for 1 hour. The digested samples were made to 20 mL using ultrapure water (Veolia Water, Paris, France). A known volume of 1X PBS (100 μL) with 1% heparin was used as a control. Freeze-dried organs were weighed and homogenized using a mortar and pestle. From the homogenized organs, a known amount was taken in clean vials and digested using 200 μL of concentrated nitric acid at 80°C for 1 hour. The digested samples were made to 20 mL using ultrapure water (ELGA LabWater system). All samples were filtered through 0.45 μm syringe filter before analysis using ICP-MS.
Results
Monomer synthesis and characterization
Our first objective was to synthesize the succinimidyl ester benzoate functional diethylene glycol methacrylate monomer 3 (Figure 2A). Initially compound 1 was prepared via ether formation between 4-hydroxybenzoic acid and 2-(2-chloroethoxy)ethanol which were refluxed in the presence of potassium hydroxide (1H NMR spectrum can be found in Figure S1). Compound 2 (1H NMR, Figure S2) was then synthesized via esterification between compound 1 and methacrylic acid in the presence of p-toluenesulfonic acid. Compound 2 was then activated with N-hydroxysuccinimide in the presence of dicyclohexylcarbodiimide to produce monomer 3 (1H NMR, Figure S3). The dimethacrylate 1,4,7,10-tetraazacyclododecane-N, N′, N″, N″′-tetraacetic acid (DO3A) functional monomer 6 (Figure 2B) was synthesized through reaction of 5-aminoisophthalic acid with an excess of 2-aminoethyl methacrylate in the presence of the coupling agent dicyclohexylcarbodiimide, and the resulting compound 4 (1H NMR, Figure S4) was subsequently reacted with bromoacetyl bromide to produce compound 5 (1H NMR, Figure S5). The dimethacrylate DO3A bifunctional monomer 6 was isolated after coupling compound 5 with tri-tert-butyl 1,4,7,10-tetraazacyclododecane-1, 4,7-triacetate in the presence of potassium carbonate. Compound 6 (1H NMR, Figure S6) was found to be very pure and highly stable.
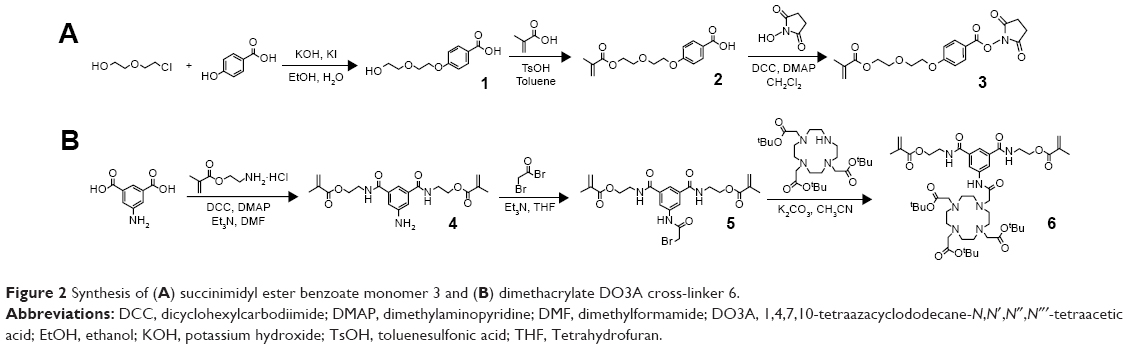 | Figure 2 Synthesis of (A) succinimidyl ester benzoate monomer 3 and (B) dimethacrylate DO3A cross-linker 6. |
Synthesis of branched copolymer nanoparticles via RAFT polymerization
The overall synthesis of branched copolymer nanoparticles via RAFT polymerization is illustrated in Figure 1A and B. Initially, linear oligoethylene glycol-based copolymers (P1–P3) of varying lengths were synthesized via RAFT polymerization. It is well understood that nanoparticles are prone to sequestration by the mononuclear phagocyte system, which is triggered by the adsorption of plasma proteins onto nanoparticles.43 We chose to employ poly(ethylene glycol)-based monomers within the nanoparticle corona generating “stealth-like” nanoparticles to minimize protein adsorption and avoid mononuclear phagocyte system clearance. Due to the controlled nature of the RAFT technique, these linear copolymer building blocks retain their terminal dithioester functional groups allowing subsequent polymerization. Copolymerization in the presence of the novel DO3A containing dimethacrylate cross-linker 6 and n-butyl ethacrylate was then performed leading to highly branched but soluble macromolecules (N1(tBu)–N3(tBu)). This process is in part similar to our previous reports on the synthesis of anisotropic branched copolymer nanoparticles where atom transfer radical polymerization was applied.39,41 However, atom transfer radical polymerization has disadvantages such as the employment of copper catalysts which may lead to residual toxicity and there is often compatibility issues with some monomer functionalities (eg, –COOH). RAFT polymerization has been beneficial here in the synthesis of branched copolymer nanoparticles. Recently, RAFT has been applied in synthesis of classic hyperbranched or core cross-linked star polymer nanocontrast agents, which has demonstrated good control of polymer structure.36–38,44,45
The novel dimethacrylate cross-linker 6 employed incorporates the DO3A macrocycles simultaneously during the branching process ensuring the DO3A macrocycles are predominantly linked to the branched copolymers through two methacrylate functionalities, increasing their stability to undesirable ester hydrolysis and holding them within the nanoparticle core. The tert-butyl protecting groups within the DO3A macrocycles were converted to the corresponding carboxylic acids (N1–N3) and gadolinium was chelated within the core of N2 furnishing N2(Gd) which was applied in vivo as a nanoparticle MRI contrast agent. We have shown that the succinimidyl ester benzoate acid functionalities present within the nanoparticle corona have survived the chelation process making these nanoparticles versatile platforms for the conjugation of other actives through amide bond formation.
The RAFT chain transfer agent 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid was used to mediate the copolymerization of various mixtures of monomer 3 and oligo(ethylene glycol) methyl ether methacrylate (OEGMA) monomethyl ether at 70°C in dimethylformamide (DMF) to produce the linear copolymer building blocks P1–P3 (Figure 1). 1H NMR analysis (Figure S7) indicated the copolymers prepared possessed compositions of the two monomers identical to the feed ratios and displayed a peak at δ =2.89 ppm, confirming the presence of the pendant succinimidyl ester benzoate functional groups. Gel permeation chromatography analysis (Table 1; Figure S8) of P1–P3 confirmed relatively dispersities and suggested good control over molecular weights.
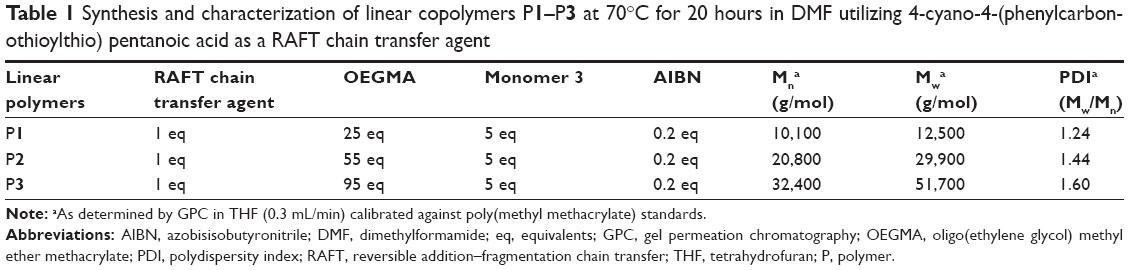 | Table 1 Synthesis and characterization of linear copolymers P1–P3 at 70°C for 20 hours in DMF utilizing 4-cyano-4-(phenylcarbonothioylthio) pentanoic acid as a RAFT chain transfer agent |
The linear copolymers P1–P3 were chain extended (Figure 1) with n-butyl methacrylate in the presence of the dimethacrylate DO3A functional cross-linker 6 at 70°C in DMF to produce the branched copolymers N1(tBu)–N3(tBu) (Table 2) composed of covalently cross-linked copolymer chains. The ratio of the branching dimethacrylate monomer 6 to the growing polymer chain is restricted to three monomer units per chain and the reaction is carried out under relatively dilute conditions; thus, the formation of a macromolecular gel is inhibited and discrete soluble molecular species are formed. 1H NMR analysis (Figure 3A; Figure S9) of N1(tBu)–N3(tBu) displayed a peak at δ =2.89 ppm confirming the continued presence of the succinimidyl ester benzoate functionalities. The linear polymerizations of P1–P3 and the subsequent chain extensions to prepare N1(tBu)–N3(tBu) were monitored by 1H NMR spectroscopy which confirmed the polymerizations reached conversions of 95%–98%. Therefore, the average number of each component monomer and their respective ratios are equal to the feed equivalents displayed in Tables 1 and 2.
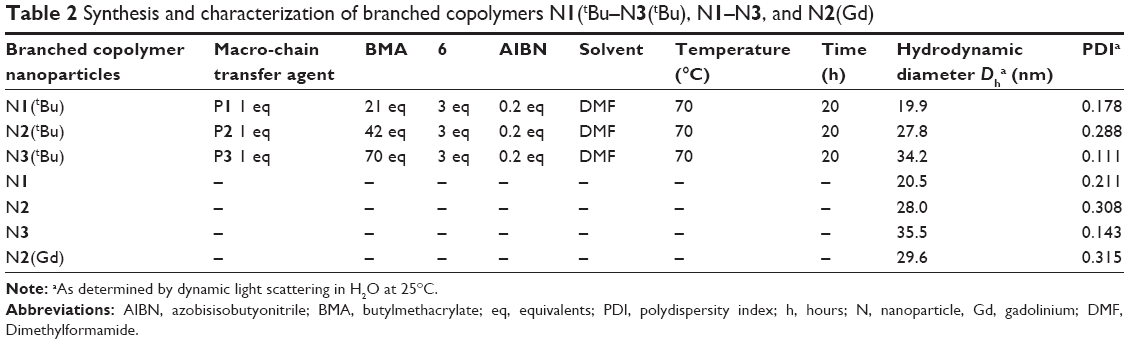 | Table 2 Synthesis and characterization of branched copolymers N1(tBu–N3(tBu), N1–N3, and N2(Gd) |
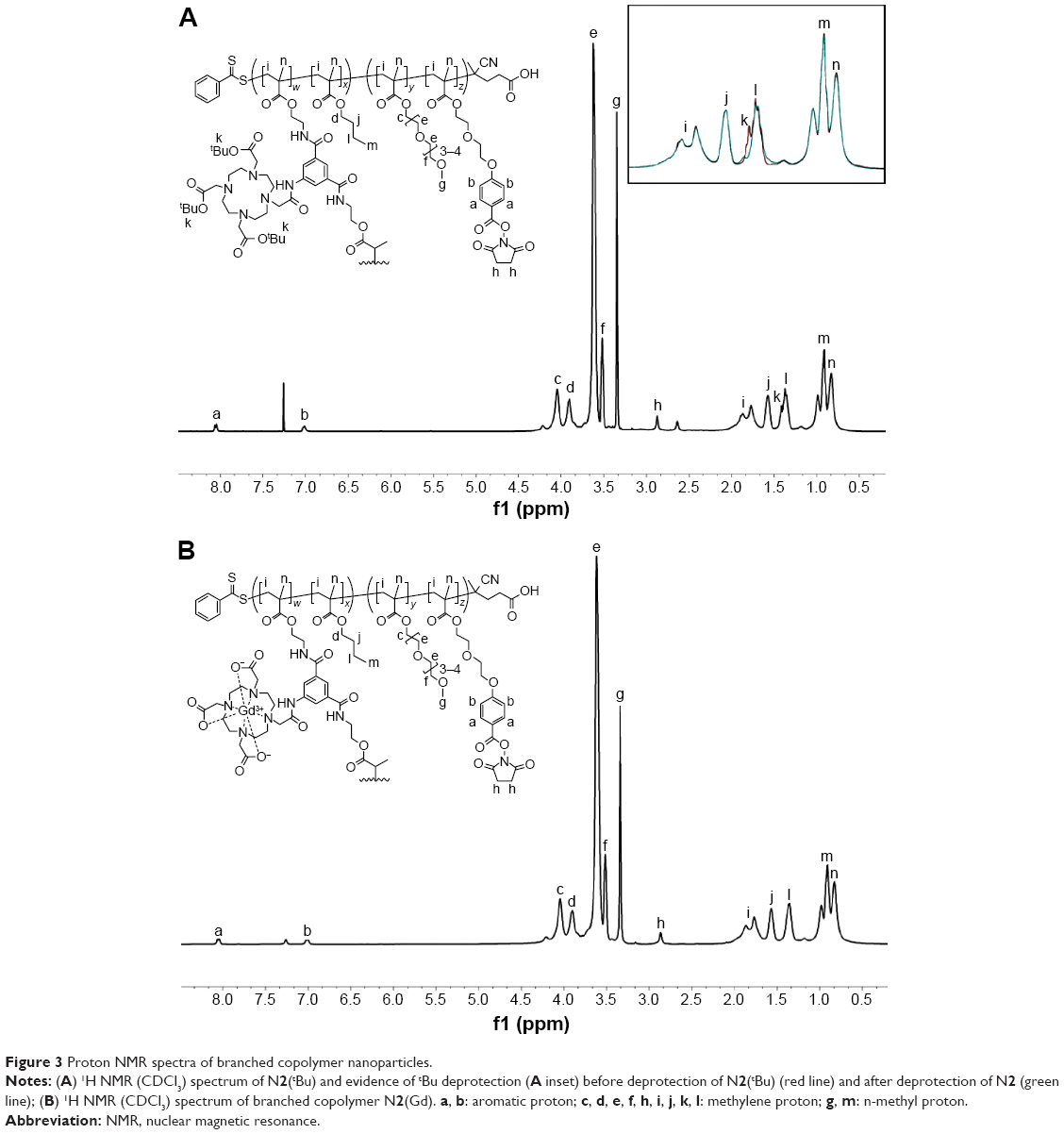 | Figure 3 Proton NMR spectra of branched copolymer nanoparticles. |
To prepare aqueous solutions, N1(tBu)–N3(tBu) were dissolved in acetone followed by dropwise addition of water. After evaporation of acetone, clear aqueous nanoparticle solutions were obtained. Dynamic light scattering (DLS) analysis (Table 2; Figure S10) the nanoparticles are prepared in various sizes in the range of 20–35 nm which is appropriate for in vivo experiments. These results clearly show the ease in which branched copolymer nanoparticles with different diameters can be prepared. To permit chelation of gadolinium, the tert-butyl groups present within the DO3A macrocycles had to be deprotected to the corresponding carboxylic acids. This was achieved by treating the branched copolymers N1(tBu)–N3(tBu) with Trifluoroacetic acid (TFA) in CH2Cl2 (Figure 1); the resulting branched copolymers N1–N3 were characterized by 1H NMR spectroscopy (Figure S11) which confirmed, when overlapped with the 1H NMR spectra of N1(tBu)–N3(tBu) (Figure 3A inset), that deprotection had successfully occurred. 1H NMR analysis (Figure S11) also displayed a peak at δ =2.89 ppm, confirming the conditions had not affected the succinimidyl ester benzoate functionalities. DLS analysis in water of N1–N3 (Figure 4A) prepared from corresponding branched copolymers after an identical evaporation process displayed near-identical sizes (Table 2; Figure S10) when compared to N1(tBu)–N3(tBu), respectively, confirming that the deprotection process does not result in any undesirable nanoparticle decomposition. The nanoparticles N1–N3 were further analyzed by cryotransmission electron microscopy (cryo-TEM), which displayed uniform spherical nanoparticles of sizes similar to those determined by DLS (Figure 4B–D).
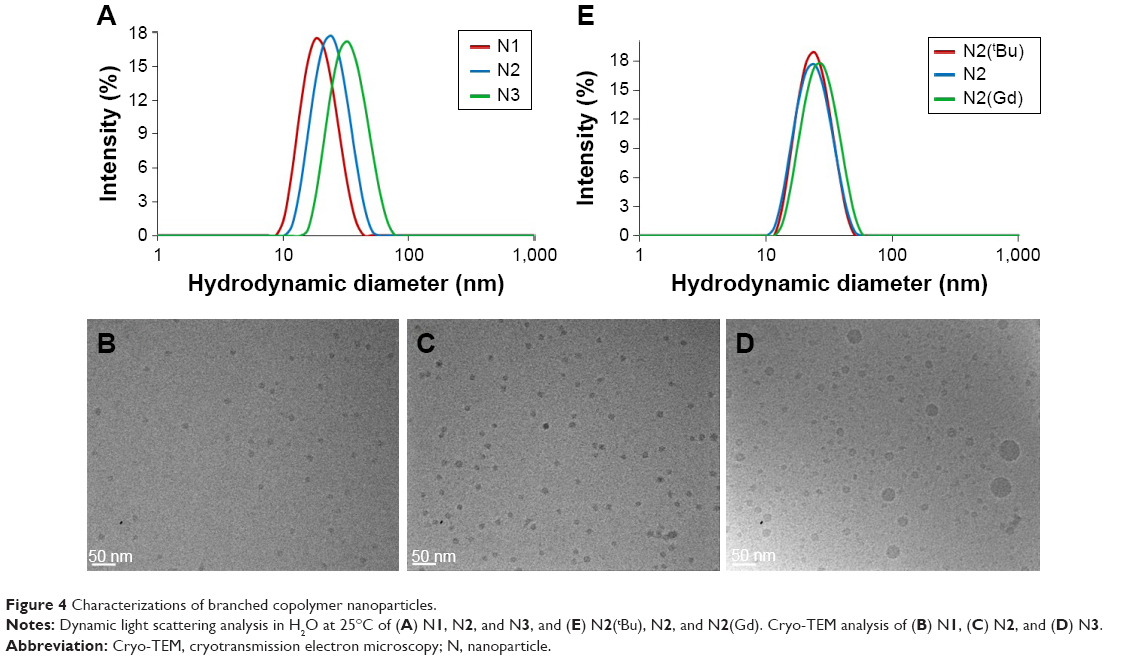 | Figure 4 Characterizations of branched copolymer nanoparticles. |
Preparation of branched copolymer nanoparticle MRI contrast agent
We decided to choose N2 (Dh =28.0 nm) for the following in vivo MRI investigation. An in vivo study investigating the relationship between variations in nanoparticle size and biodistribution is beyond the scope of this initial study. N2 was treated with a large excess of GdCl3·6H2O in a mixture of isopropanol:H2O (30:70) at 55°C for 24 hours to generate N2(Gd) (Figure 1). The solution obtained was extensively dialyzed (molecular weight cutoff: 7 kDa) against water for 48 hours to completely remove the excess Gd, and the aqueous solution obtained was evaporated to dryness. N2(Gd) was then isolated by precipitation from minimal Tetrahydrofuran (THF) into ice-cold hexane. 1H NMR analysis of N2(Gd) (Figure 3B) confirmed no decomposition, which was further verified by DLS analysis of N2(Gd) which displayed an almost identical size to nanoparticles N2(tBu) and N2 (Figure 4E; Table 2). Once again, 1H NMR results (Figure 3B) displayed a peak at δ =2.89 ppm, confirming that the succinimidyl ester benzoate functionalities had survived all reactions and processing that the branched copolymers had undergone and no undesirable hydrolysis had occurred during the chelation process.
ICP-MS was used to analyze the concentration of gadolinium within a known quantity of N2(Gd) which confirmed a successful chelation. N2(Gd) was then applied as an MRI contrast agent. The Gd(III)-chelated DO3A macrocycle units in the nanoparticle improve the hydrophilicity of the nanoparticle core, thus ensuring the ability of surrounding water molecules to approach the core and directly exchange with the coordinated water in the Gd(DO3A) macrocycles. Relaxivity measurements in deionized water (Figure 5A and B) using a 7 T Bruker ClinScan determined the r1 to be 14.16 mM−1s−1 and r2 of 54.59 mM−1s−1, with a r2/r1 ratio of 3.86. Most of the small molecular gadolinium contrast agents can be classified as extracellular fluid contrast agents, including Gd(DOTA), Gd(III)-diethylenediaminepentaacetic acid, and their derivatives. These contrast agents possess r1 relaxivity values of 3–6 mM−1s−1 at higher magnetic field.46,47 Small molecular contrast agents which were protein bounded showed intravascular characteristics and have a r1 relaxivity of 6 mM−1s−1 at higher a magnetic field.48 In our study of N2(Gd), r1 relaxivity (14.16 mM−1s−1) is almost three-times the value of commercial small molecular contrast agents. The relatively large r1 value for N2(Gd) nanoparticles can be beneficial to acquire MRI signals at lower concentrations of nanoparticles compared to small-molecule MRI gadolinium contrast agents. In order to optimize acquisition parameters for the in vivo study, phantoms were run using a FLASH sequence (Figure S12) with varying TR and flip angle, with a minimum TE. The signal from the phantom was calculated and normalized with respect to that of the background. It was observed that a flip angle of 20° provided a higher signal-to-noise ratio (SNR) with TRs of 10, 20, and 50 milliseconds. The proceeding in vivo experiment thus employed a TR of 10 milliseconds and flip angle of 20° to acquire images with a temporal resolution of 10 seconds per acquisition.
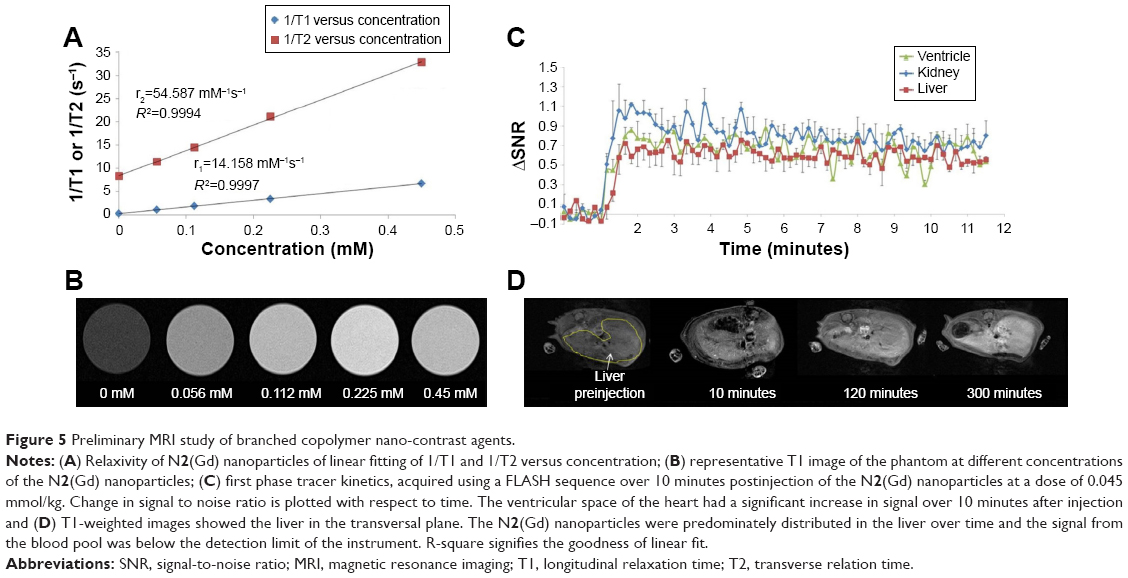 | Figure 5 Preliminary MRI study of branched copolymer nano-contrast agents. |
In vitro cytotoxicity study
Since most gadolinium-based contrast agents are cleared through the kidney, it is essential to study the in vitro toxicity on HK-2 cell lines associated with the gadolinium-chelated N2(Gd) nanoparticles (Figure S13). The half maximal inhibitory concentration (IC10) values for N2(Gd) nanoparticles are 0.33 mM, 0.45 mM, and 0.49 mM at 24, 48, and 72 hours’ incubation, respectively. The low IC10 value at 24 hours suggests that the cells were burdened immediately after the exposure to the nanoparticles; consequently, the IC10 values increased, suggesting a reduced toxic effect of the nanoparticles with time. The low IC10 values indicated that N2(Gd) nanoparticles have negligible toxic effect at three-times the clinically administered dose of gadolinium. Free gadolinium ions from the dissociation of chelates can cause nephrogenic systemic fibrosis in patients with insufficient renal clearance. The use of macrocycles present within biocompatible and kinetically stable polymer nanoparticles could prevent gadolinium-related toxicity.
In vivo MRI studies
Initial in vivo experiments were carried out to evaluate the application of the N2(Gd) nanoparticles as nano-sized MRI contrast agents. With small size and poly(ethylene glycol)-based coronas which are known to prevent opsonization and uptake by the reticuloendothelial system,49,50 it was hypothesized that these nanoparticles would have a long blood circulation time. The first phase tracer kinetics (Figure 5C) showed a considerable signal enhancement in the ventricle of the heart, kidney, and liver. Signal remained steady in the ventricular space of the heart over 10 minutes (Figure S14). However, no extracellular uptake of the N2(Gd) nanoparticles was observed in the kidney. The N2(Gd) nanoparticles mainly distributed in the blood pool. Typically, the anti-biofouling nature of poly(ethylene glycol) prevents opsonization, which is the primary immune response by macrophages that leads to an increased accumulation of contrast agents in the reticuloendothelial system.51–53 The signal increases in the liver up to 5 hours postinjection (Figure 5D) indicating a long blood retention time, which could be promising for tumor detection.
After the initial imaging study was carried out, we decided to investigate the dynamic contrast imaging of N2(Gd) nanoparticles on SCID mice bearing U87MG human glioma xenograft tumor, with the aim to investigate the potential for passive tumor detection. MRI scans were carried out when the tumor size reached 150–200 mm3 in volume. The maximum intensity projection image acquired at 10 minutes post-administration of N2(Gd) nanoparticles (Figure 6A) showed considerable signal enhancement at the ventricle of the heart, blood vessels in the kidney, liver, and tumor. The SNR plot versus time comparing tumor and muscle (Figure 6B) demonstrated a signal enhancement in the tumor compared to the muscle. This is probably due to polymer nanoparticles penetrating “leaky” blood vessels, known as passive targeting via the EPR effect at the tumor during the process of angiogenesis.54,55 The T1 value decreased over time (Figure 6C and D) and reached a steady low value. The long blood circulation time of N2(Gd) nanoparticles together with the EPR effect provides an advantage for tumor detection by passive targeting.
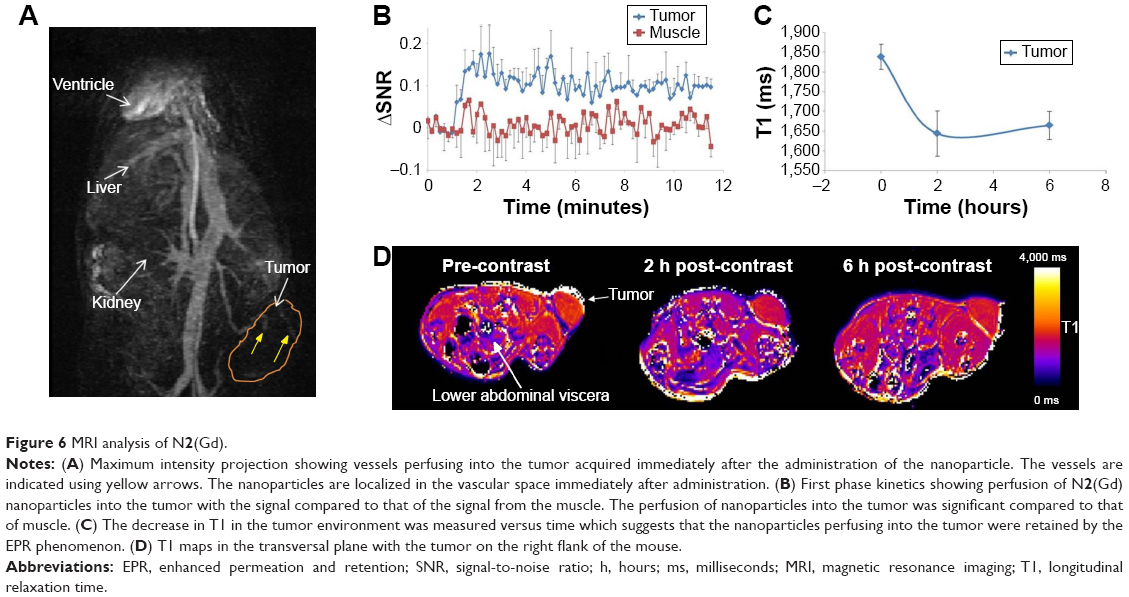 | Figure 6 MRI analysis of N2(Gd). |
Biodistribution study
A biodistribution study was then performed on the same tumor-bearing mice to determine the percentage injected dose per gram (ID/g) of organs/blood at 2 and 4 hours postinjection of N2(Gd); analysis was performed using ICP-MS (Figure 7). The blood Gd content was as high as 14% ID/g at 2 hours postinjection and decreased to 8% ID/g at 4 hours postinjection. This quantified blood profile confirms the long blood circulation properties of the nanoparticles. The reticuloendothelial system such as the liver and spleen received most N2(Gd) at approximately 0.15% ID/g at 2 and 4 hours. The kidney had relatively low uptake of Gd: 0.02% ID/g. Although during the first phase of N2(Gd) nanoparticle administration the kidney had the highest SNR (Figure 5C), the uptake of gadolinium in the kidney was low at 2 and 4 hours postinjection, indicating that the amount of Gd received from the kidney during the first phase is being excreted together with renal clearance of the nanoparticles. The tumor received 0.004% ID/g of N2(Gd) at 2 and 4 hours postinjection, possibly from the uptake as a result of “leaky” blood vessels. The accumulation of nanoparticles in the brain was negligible, indicating that N2(Gd) nanoparticles do not cross the blood–brain barrier. These results confirm that N2(Gd) remains in the blood for long periods and may be able to actively target cells once adorned with targeting moieties via amide bond formation with the succinimidyl ester benzoic acid functionalities. With a high relaxivity and a long blood retention time, N2(Gd) nanoparticles could potentially be considered for application in magnetic resonance angiography at a low dose of gadolinium.56,57
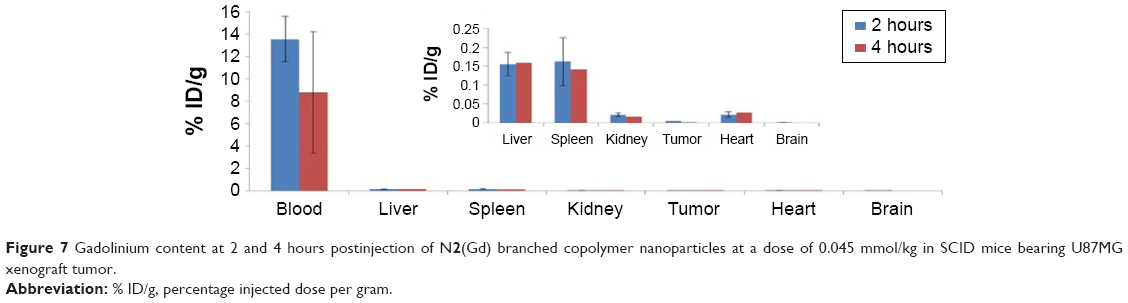 | Figure 7 Gadolinium content at 2 and 4 hours postinjection of N2(Gd) branched copolymer nanoparticles at a dose of 0.045 mmol/kg in SCID mice bearing U87MG xenograft tumor. |
Hydrophobic guest encapsulation
To demonstrate the structural advantages of our branched copolymer nanoparticles, the hydrophobic dye Nile red was encapsulated within the hydrophobic core of N2(Gd). Excess Nile red and N2(Gd) were dissolved in acetone, and H2O was added dropwise. The solution was left to stir at room temperature and acetone allowed to evaporated. The resulting aqueous solution was filtered and the loading capacity determined at approximately 10 percentage by weight of Nile red with respect to N2(Gd). This loading capacity is typical for similar branched copolymer nanoparticles prepared in our laboratory. The solution obtained displayed an intense red/purple color indicating a successful encapsulation (Figure 8). Relaxivity measurements determined the r1 to be 12.75 mM−1s−1, and only slightly lower when compared to pure N2(Gd) (r1=14.16 mM−1s−1), confirming that the presence of the hydrophobic guest has little effect on relaxivity and MRI capabilities. It is hypothesized that the presence of the DO3A macrocycles within the nanoparticle core improve the hydrophilicity to the extent that surrounding water molecules are able to approach the core and exchange with the coordinated water in the Gd(DO3A) macrocycles. This experiment further demonstrates the utility of these branched copolymer nanoparticle platforms. We have observed that these branched copolymer nanoparticles are highly stable when chelated with Gd and loading with Nile red. After storing for 3 months, no precipitation was observed and the samples still displayed high relaxivity values.
 | Figure 8 Encapsulation of Nile red within N2(Gd). |
Conclusion
Stable branched copolymer nanoparticles of varying sizes (Dh =20–35 nm) have been developed and employed as MRI nanosized contrast agents. RAFT polymerization has been employed to prepare these novel nanoparticles possessing DO3A macrocycles within their cores and succinimidyl ester benzoate functionalities within their coronas. It has been demonstrated that these nanoparticles can chelate gadolinium, and in vitro cytotoxicity studies using HK-2 cells established their negligible toxicity profile. In vivo MRI experiments showed that these nanoparticles have a high relaxivity and a long blood retention time. Xenograft experiments further illustrated the ability of these nanoparticles to perfuse and passively accumulate in tumor cells, presumably through the enhanced EPR effect. The presence of the succinimidyl ester benzoate functionalities within the nanoparticle coronas will permit future surface modification with fluorophores or targeting moieties to generate nanoparticles to study opportunities for bimodal imaging nanoprobes or active cell-targeting contrast agents. The chelation with different metal ions (111In(III) and 68Ga(III)) may also afford various single-photon emission tomography/positron emission tomography nanoprobe contrast agents. We have also shown that it is possible to load hydrophobic guest molecules (Nile red) within the nanoparticle cores, without any significant loss in relaxivity, which demonstrates the potential for simultaneous bioimaging and drug delivery.
Acknowledgments
This research was supported by the JCO Grant (1131CFG002) of A*STAR (Agency for Science, Technology and Research), and the Tier 1 grant (RG38/14) funded by the Ministry of Education, Singapore.
Disclosure
The authors report no conflicts of interest in this work.
References
Kumar C, editor. Nanotechnologies for the Life Sciences. Weinheim: Wiley; 2006. | ||
De M, Ghosh PS, Rotello VM. Applications of nanoparticles in biology. Adv Mater. 2008;20:4225–4241. | ||
Allen TM, Cullis PR. Drug delivery systems: entering the mainstream. Science. 2004;303:1818–1822. | ||
Riehemann K, Schneider SW, Luger TA, Godin B, Ferrari M, Fuchs H. Nanomedicine – challenge and perspectives. Angew Chem Int Ed Engl. 2009;48:872–897. | ||
Ferrari M. Cancer nanotechnology: opportunities and challenges. Nat Rev Cancer. 2005;5:161–171. | ||
Kumar AM, Sivakova S, Fox JD, Green JE, Marchant RE, Rowan SJ. Molecular engineering of new supramolecular scaffold coatings that can reduce static platelet adhesion. J Am Chem Soc. 2008;130:1466–1476. | ||
Sudeep PK, Emrick T. Functional Si and CdSe quantum dots: synthesis, conjugate formation, and photoluminescence quenching by surface interactions. ACS Nano. 2009;3:4105–4109. | ||
Xu H, Minoia A, Tomović Z, et al. A multivalent hexapod: conformational dynamics of six-legged molecules in self-assembled monolayers at a solid-liquid interface. ACS Nano. 2009;3:1016–1024. | ||
Zareie HM, Boyer C, Bulmus V, Nateghi E, Davis TP. Temperature-responsive self-assembled monolayers of oligo(ethylene glycol): control of biomolecular recognition. ACS Nano. 2008;2:757–765. | ||
Lu W, Arumugam SR, Senapati D, et al. Multifunctional oval-shaped gold-nanoparticle-based selective detection of breast cancer cells using simple colorimetric and highly sensitive two-photon scattering assay. ACS Nano. 2010;4:1739–1749. | ||
Ostaci RV, Damiron D, Grohens Y, Léger L, Drockenmuller E. Click chemistry grafting of to poly(ethylene glycol) brushes to alkyne-functionalized pseudobrushes. Langmuir. 2010;26:1304–1310. | ||
Stukel JM, Li RC, Maynard HD, Caplan MR. Two-step synthesis of multivalent cancer-targeting constructs. Biomacromolecules. 2010;11:160–167. | ||
Cheon J, Lee JH. Synergistically integrated nanoparticles as multimodal probes for nanobiotechnology. Acc Chem Res. 2008;41:1630–1640. | ||
Caravan P, Ellison JJ, McMurry TJ, Lauffer RB. Gadolinium(III) chelates as MRI contrast agents: structure, dynamics, and applications. Chem Rev. 1999;99:2293–2352. | ||
Lauffer RB. Paramagnetic metal complexes as water proton relaxation agents for NMR imaging: theory and design. Chem Rev. 1987;87:901–927. | ||
Wiener EC, Brechbiel MW, Brothers H, et al. Dendrimer-based metal chelates: a new class of magnetic resonance imaging contrast agents. Magn Reson Med. 1994;31:1–8. | ||
Fu Y, Raatschen HJ, Nitecki DE, et al. Cascade polymeric MRI contrast media derived from poly(ethylene glycol) cores: initial syntheses and characterizations. Biomacromolecules. 2007;8:1519–1529. | ||
Langereis S, de Lussanet QG, van Genderen MH, et al. Evaluation of Gd(III)DTPA-terminated poly(propylene imine) dendrimers as contrast agents for MR imaging. NMR Biomed. 2006;19:133–141. | ||
Jászberényi Z, Moriggi L, Schmidt P, et al. Physicochemical and MRI characterization of Gd3+-loaded polyamidoamine and hyperbranched dendrimers. J Biol Inorg Chem. 2007;12:406–420. | ||
Rudovský J, Botta M, Hermann P, Hardcastle KI, Lukes I, Aime S. PAMAM dendrimeric conjugates with a Gd-DOTA phosphinate derivative and their adducts with polyaminoacids: the interplay of global motion, internal rotation, and fast water exchange. Bioconjug Chem. 2006;17:975–987. | ||
Lebdusková P, Sour A, Helm L, et al. Phosphinic derivative of DTPA conjugated to a G5 PAMAM dendrimer: an 17O and 1H relaxation study of its Gd(III) complex. Dalton Trans. 2006;28:3399–3406. | ||
Laus S, Sour A, Ruloff R, Tóth E, Merbach AE. Rotational dynamics account for pH-dependent relaxivities of PAMAM dendrimeric, Gd-based potential MRI contrast agents. Chemistry. 2005;11:3064–3076. | ||
Tilcock C, Unger E, Cullis P, MacDougall P. Liposomal Gd-DTPA: preparation and characterization of relaxivity. Radiology. 1989;171:77–80. | ||
Fossheim SL, Fahlvik AK, Klaveness J, Muller RN. Paramagnetic liposomes as MRI contrast agents: influence of liposomal physicochemical properties on the in vitro relaxivity. Magn Reson Imaging. 1999;17:83–89. | ||
Løkling KE, Fossheim SL, Skurtveit R, Bjørnerud A, Klaveness J. pH-sensitive paramagnetic liposomes as MRI contrast agents: in vitro feasibility studies. Magn Reson Imaging. 2001;19:731–738. | ||
Wang T, Hossann M, Reinl HM, et al. In vitro characterization of phosphatidylglyceroglycerol-based thermosensitive liposomes with encapsulated 1H MR T1-shortening gadodiamide. Contrast Media Mol Imaging. 2008;3:19–26. | ||
Kabalka G, Buonocore E, Hubner K, Moss T, Norley N, Huang L. Gadolinium-labeled liposomes: targeted MR contrast agents for the liver and spleen. Radiology. 1987;163:255–258. | ||
Kabalka GW, Buonocore E, Hubner K, Davis M, Huang L. Gadolinium-labeled liposomes containing paramagnetic amphipathic agents: targeted MRI contrast agents for the liver. Magn Reson Med. 1988;8:89–95. | ||
Kabalka GW, Davis MA, Moss TH, et al. Gadolinium-labeled liposomes containing various amphiphilic Gd-DTPA derivatives: targeted MRI contrast enhancement agents for the liver. Magn Reson Med. 1991;19:406–415. | ||
Tilcock C, Ahkong QF, Koenig SH, Brown RD 3rd, Davis M, Kabalka G. The design of liposomal paramagnetic MR agents: effect of vesicle size upon the relaxivity of surface-incorporated lipophilic chelates. Magn Reson Med. 1992;27:44–51. | ||
Zhang G, Zhang R, Wen X, Li L, Li C. Micelles based on biodegradable poly(L-glutamic acid)-b-polylactide with paramagnetic Gd ions chelated to the shell layer as a potential nanoscale MRI-visible delivery system. Biomacromolecules. 2008;9:36–42. | ||
Reynolds CH, Annan N, Beshah K, et al. Gadolinium-loaded nanoparticles: new contrast agents for magnetic resonance imaging. J Am Chem Soc. 2000;122:8940–8945. | ||
Turner JL, Pan D, Plummer R, Chen Z, Whittaker AK, Wooley KL. Synthesis of gadolinium-labeled shell-crosslinked nanoparticles for magnetic resonance imaging applications. Adv Funct Mater. 2005;15:1248–1254. | ||
Gong P, Chen Z, Chen Y, Wang W, Wang X, Hu A. High-relaxivity MRI contrast agents prepared from miniemulsion polymerization using gadolinium(III)-based metallosurfactants. Chem Commun (Camb). 2011;47:4240–4242. | ||
Kaida S, Cabral H, Kumagai M, et al. Visible drug delivery by supramolecular nanocarriers directing to single-platformed diagnosis and therapy of pancreatic tumor model. Cancer Res. 2010;70:7031–7041. | ||
Li Y, Beija M, Laurent S, et al. Macromolecular ligands for gadolinium MRI contrast agents. Macromolecules. 2012;45:4196–4204. | ||
Li Y, Laurent S, Esser L, et al. The precise molecular location of gadolinium atoms has a significant influence on the efficacy of nanoparticulate MRI positive contrast agents. Polym Chem. 2014;5:2592–2601. | ||
Hu X, Liu G, Li Y, Wang X, Liu S. Cell-penetrating hyperbranched polyprodrug amphiphiles for synergistic reductive milieu-triggered drug release and enhanced magnetic resonance signals. J Am Chem Soc. 2015;137:362–368. | ||
He T, Adams DJ, Butler MF, Yeoh CT, Cooper AI, Rannard SP. Direct synthesis of anisotropic polymer nanoparticles. Angew Chem Int Ed Engl. 2007;46:9243–9247. | ||
Hatton FL, Chambon P, McDonald TO, Owen A, Rannard SP. Hyperbranched polydendrons: a new controlled macromolecular architecture with self-assembly in water and organic solvents. Chem Sci. 2014;5:1844–1853. | ||
He T, Adams DJ, Butler MF, Cooper AI, Rannard SP. Polymer nanoparticles: shape-directed monomer-to-particle synthesis. J Am Chem Soc. 2009;131:1495–1501. | ||
Hatton FL, Tatham LM, Tidbury LR, et al. Hyperbranched polydendrons: a new nanomaterials platform with tuneable permeation through model gut epithelium. Chem Sci. 2015;6:326–334. | ||
Hume DA. The mononuclear phagocyte system. Curr Opin Immunol. 2006;18:49–53. | ||
Thurecht KJ, Blakey I, Peng H, et al. Functionalized hyperbranched polymers: towards targeted in vivo 19F magnetic resonance imaging using designed macromlecules. J Am Chem Soc. 2010;132:5336–5337. | ||
Rolfe BE, Blakey I, Squires O, et al. Multimodal Polymer Nanoparticles with Combined 19F Magnetic Resonance and Optical Detection for Tunable, Targeted, Multimodal Imaging in Vivo. J Am Chem Soc. 2014;136:2413–2419. | ||
Chandrasekharan P, Yang CT, Nasrallah FA, Tay HC, Chuang KH, Robins EG. Pharmacokinetics of Gd(DO3A-Lys) and MR imaging studies in an orthotopic U87MG glioma tumor model. Contrast Media Mol Imaging. 2015;10(3):237–244. | ||
Aime S, Caravan P. Biodistribution of gadolinium-based contrast agents, including gadolinium deposition. J Magn Reson Imaging. 2009;30:1259–1267. | ||
Noebauer-Huhmann IM, Kraff O, Juras V, et al. MR Contrast Media at 7Telsa – Preliminary Study on Relaxivities. Poster presented at: Proceedings 16th Scientific Meeting, International Society for Magnetic Resonance in Medicine, Toronto, Ontario, 6 May 2008. | ||
Wu SH, Lin CY, Hung Y, Chen W, Chang C, Mou CY. PEGylated silica nanoparticles encapsulating multiple magnetite nanocrystals for high-performance microscopic magnetic resonance angiography. J Biomed Mater Res B Appl Biomater. 2011;99:81–88. | ||
Fallis S, Beaty-Nosco J, Dorshow RB, Adzamli K. Polyethyleneglycol-stabilized manganese-substituted hydroxylapatite as a potential contrast agent for magnetic resonance imaging: particle stability in biologic fluids. Invest Radiol. 1998;33:847–852. | ||
Ma C, Yang H, Zhou X, Wu B, Zhang G. Polymeric material for anti-biofouling. Colloids Surf B Biointerfaces. 2012;100:31–35. | ||
Yu Q, Zhang Y, Wang H, Brash J, Chen H. Anti-fouling bioactive surfaces. Acta Biomater. 2011;7:1550–1557. | ||
Banerjee I, Pangule RC, Kane RS. Antifouling coatings: recent developments in the design of surfaces that prevent fouling by proteins, bacteria, and marine organisms. Adv Mater. 2011;23:690–718. | ||
Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv Drug Deliv Rev. 2014;66:2–25. | ||
Kwon IK, Lee SC, Han B, Park K. Analysis on the current status of targeted drug delivery to tumors. J Control Release. 2012;164:108–114. | ||
Raman FS, Nacif MS, Cater G, et al. 3.0-T whole-heart coronary magnetic resonance angiography: comparison of gadobenate dimeglumine and gadofosveset trisodium. Int J Cardiovasc Imaging. 2013;29:1085–1094. | ||
Kim CY, Heye T, Bashir MR, Gebhard TA, Merkle EM. Gadofosveset-enhanced magnetic resonance angiography of the thoracic vasculature in the equilibrium phase: feasibility and impact of dose. J Comput Assist Tomogr. 2013;37:732–736. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.