")
Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 16
Synovitis, Acne, Pustulosis, Hyperostosis and Osteitis (SAPHO) Syndrome with Henoch–Schönlein Purpura: A Case Report
Authors Wang R, Li Y, Liu Y, Hou X, Li C
Received 18 October 2022
Accepted for publication 20 March 2023
Published 24 April 2023 Volume 2023:16 Pages 1089—1094
DOI https://doi.org/10.2147/CCID.S392909
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jeffrey Weinberg
Ruoyi Wang,1,2,* Yingzi Li,1,2,* Yuyue Liu,3 Xiujuan Hou,1,2,* Chen Li4,*
1Beijing University of Chinese Medicine, Beijing, People’s Republic of China; 2Department of Rheumatology, Dongfang Hospital Beijing University of Chinese Medicine, Beijing, People’s Republic of China; 3Department of Pathology, Dongfang Hospital Beijing University of Chinese Medicine, Beijing, People’s Republic of China; 4Department of Rheumatology, Fangshan Hospital Beijing University of Chinese Medicine, Beijing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xiujuan Hou; Chen Li, Email [email protected]; [email protected]
Introduction: SAPHO (synovitis, acne, pustulosis, hyperostosis, and osteitis) syndrome is a rare disease clinically characterized by a wide range of cutaneous and osteoarticular manifestations, involving multiple system impairments. Vasculitis is a rare comorbidity of SAPHO. Henoch–Schönlein purpura (HSP) is a vasculitis involving the capillaries and arterioles mediated by IgA immune complex. No case report of SAPHO syndrome with HSP was ever found.
Case: Here we reported a case of SAPHO syndrome complicated with HSP and was successfully treated by methylprednisolone and tofacitinib.
Discussion: Although the treat-to-target management of HSP and the first-line clinical medication have given some advices on the treatment. A precise treatment was still needed based on the pathogenesis of the comorbidity. The mechanism of the co-occurrence includes innate immunity and adapted immunity. Considering the active inflammatory reaction and the rapid disease progression, methylprednisolone and tofacitinib were prescribed.
Conclusion: HSP is a new comorbidity of SAPHO. The spectrum of cutaneous small-vessel vasculitis in SAPHO syndrome was enriched. A new treatment approach for SAPHO with HSP was provided.
Keywords: Henoch–Schönlein purpura, SAPHO syndrome, tofacitinib, vasculitis
Introduction
Synovitis, acne, pustulosis, hyperostosis, and osteitis syndrome (SAPHO) syndrome is a rare disease first reported by Chamot et al in 1987.1 As an auto-inflammatory system disease, SAPHO may involve multiple system impairments. It has been reported that SAPHO could cause vasculitis such as pulmonary vasculitis,2 retinal vasculitis,3 and Behçet’s disease (BD).4 So it was obvious that there are some relationships between SAPHO and vasculitis. Henoch–Schönlein Purpura (HSP) is also called immunoglobulin A (IgA) vasculitis. It is a vasculitis involving the capillaries and arterioles mediated by IgA immune complex. HSP was also one of the comorbidities of rheumatoid arthritis5 and ankylosing spondylitis.6 So HSP may have similar pathogenesis with auto-inflammation diseases. However, no case report of SAPHO syndrome with HSP was ever found. The etiology is still unknown. There are several similar pathogeneses of the two diseases. Here we described a Chinese young woman with SAPHO syndrome and HSP. However, the treat-to-target management of HSP and the first-line clinical medication have given some advices on the treatment. A more precisely treatment was still needed based on the pathogenesis of the comorbidity. Finally, this case was successfully treated by methylprednisolone and tofacitinib, which provided a valuable asset for the treatment.
Case Report
A 25-year-old Chinese woman presented with palm erythematous in which pinhead-sized intraepidermal pustules formed over 3 years. She was diagnosed as palmoplantar pustulosis (PPP). In the past 3 years, the painful midshaft of the left clavicle and the sternoclavicular joint could be eased with oral diclofenac sodium. However, there existed problems of recurrence. The maculopapular rash in dark purple appeared on the lower extremities (Figure 1A). No indication of fading was observed when pressed. The maculopapular rash rapidly progressed to a wide range. The lesion in the pretibial skin rapidly progressed into a 1-centimeter-sized ulcer (Figure 1B). The patient presented with no swelling or the painful of knee and ankle joints. She also denied a history of allergies and other diseases.
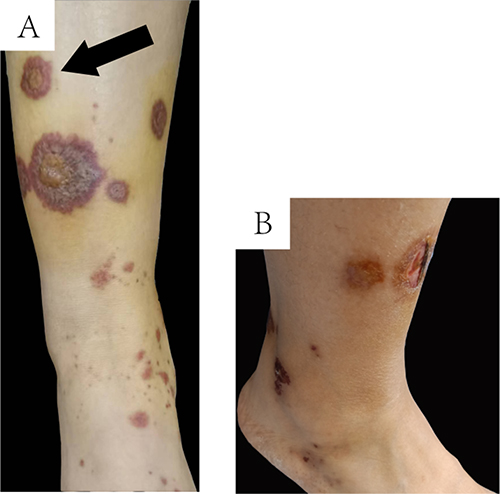 |
Figure 1 The cutaneous manifestation. (A) The maculopapular rash in dark purple appeared on the lower extremities; (B) The lesion in the pretibial skin rapidly progressed into 1-centimeter-sized ulcer. |
The laboratory results showed that the amount of platelets was 323×105/L (range, 98–300.2×105/L). The concentration of IgA was 4.93g/L (range, 0.7–4 g/L). The erythrocyte sedimentation rate (ESR) was up to 108 mm/h (range, 0–20mm/h). The functions of liver and kidney were normal. Rheumatoid factor, anti-streptolysin O, anti-cyclic citrullinated peptide antibody, human leukocyte antigen B27, antinuclear antibody, and parathyroid hormone were all negative. 99mTc-MDP bone scintigraphy showed an abnormal nuclide concentration in the left clavicle region accompanied by left clavicle deformation (Figure 2).
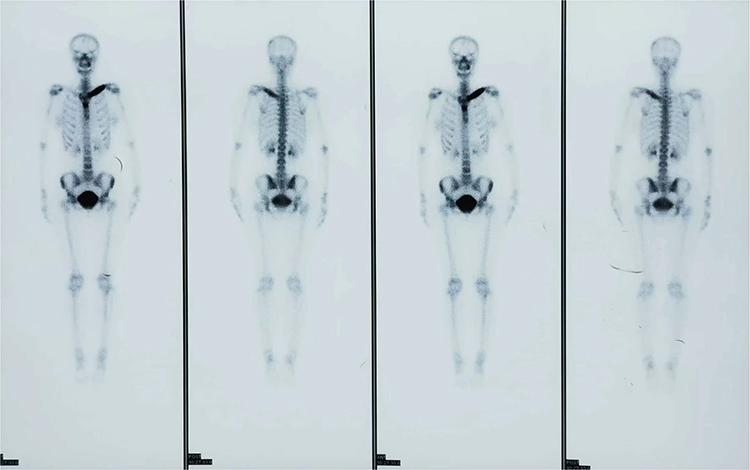 |
Figure 2 99mTc-MDP bone scintigraphy showed an abnormal nuclide concentration in left clavicle region accompanied by left clavicle deformation. |
Histopathological examination of a biopsy specimen from a lesion on the right anterior tibial skin showed that granulosa cell hyperplasia of epidermal small focus, and vascular cluster hyperplasia visible in the superficial layer of the dermis, with fibrin-like degeneration of vascular wall, nucleate dust and red blood cell spillover, which conform to the morphological manifestation of vasculitis (Figure 3A and B). Direct immunofluorescence showed deposition of IgA and C3 in the vessel walls (Figure 3C and D).
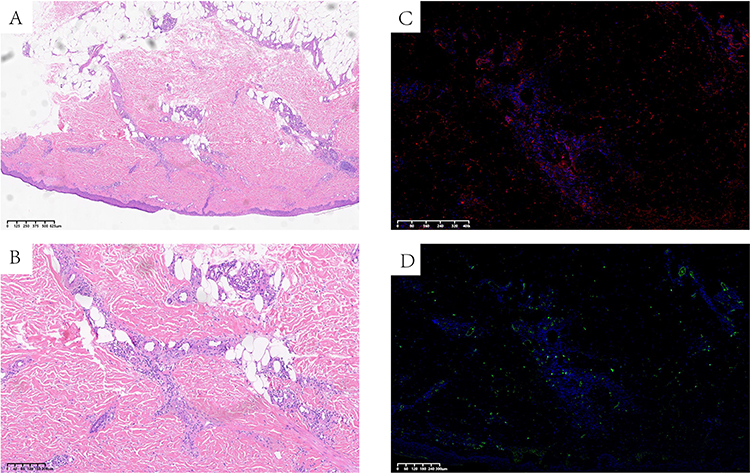 |
Figure 3 HE staining: (A and B) Granulosa cell hyperplasia of epidermal small focus, and vascular cluster wall, nucleate dust and red blood cell spillover, which conform to the morphological manifestation of vasculitis. (C and D) Direct immunofluorescence showed deposition of IgA and C3 in the vessel walls of the lesion biopsy. |
The co-occurrence of PPP and characteristic osteoarticular findings in the midshaft of the left clavicle and the sternoclavicular joint suggested the diagnosis of SAPHO syndrome, according to the diagnostic criteria by Kahn in 2003.7 The skin lesion and the associated histopathological examination suggested the diagnosis of HSP based on the diagnostic criteria by EULAR/PRINTO/PRES in 2010.8 Oral methylprednisolone (20mg per day) was prescribed in light of the hyperinflammatory state and the severe symptoms. Skin ulcer began to heal and the rest of the skin lesions gradually scabbed and subsided 4 days later (Figure 4).
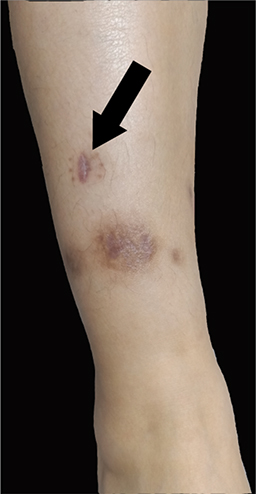 |
Figure 4 After the treatment of methylprednisolone and tofacitinib, the ulcer was healed and other lesions were subsided. |
All the aforementioned symptoms were markedly alleviated, and the ESR obviously reduced to 10mm/h (0–20) after 1 month of treatment. However, the long-term treatment of corticosteroids could lead to adverse effects. Our previous study indicated that tofacitinib could significantly decrease the inflammatory marker and improve the symptoms.9 Thus, the administration was changed from methylprednisolone (20mg per day) to tofacitinib (5mg twice a day). The ulcer was completely healed without further joint inflammation and new skin lesions after tofacitinib treatment for 2 months.
Discussions
SAPHO syndrome is a rare auto-inflammatory disorder that usually involves both osteoarticular and cutaneous. It has many cutaneous manifestations. The most common manifestations are PPP, acne conglobata, and hidradenitis suppurativa.10 HSP is an auto-inflammatory disease with a mechanism of the abnormal IgA deposits in vessel walls.
In this case, a definite diagnosis of both SAPHO syndrome and HSP was made according to their criteria for typical manifestations. Although adults are less susceptible to HSP than children, they often suffered severe clinical cutaneous involvement such as hemorrhagic blister and necrotic skin lesions.11 According to the treat-to-target management of HSP, the extensive rapidly progressing rash may be an indication to use a short course of steroids.12 The first-line clinical treatments of SAPHO syndrome are mainly nonsteroidal anti-inflammatory drugs (NSAIDs) and corticosteroids. The patient was suffering from high ESR and the rapidly progressing disease process. In order to alleviate the inflammatory reaction and slow down the disease process, a treatment of potent steroids was needed. The anti-inflammatory effect of methylprednisolone is three times of prednisolone and oral methylprednisolone was therefore prescribed. Finally, the disease was effectively controlled. Considering her active inflammatory reaction and a long-term corticosteroids therapy could lead to side effects, a proper immunoregulatory prescription was thus needed.
The pathogenesis of the two diseases still remains unclear; however, there may be some immune and inflammatory association between them. The immune mechanism association between SAPHO syndrome and HSP contains innate immunity and adapted immunity. From the innate immunity perspective, enhanced migration and adhesion of peripheral blood (PB) neutrophils from SAPHO patients were observed.13 It was anticipated that IgA complexes could activate neutrophils via the IgA Fc receptor FcalphaRI (CD89), thereby inducing neutrophil migration and activation in HSP.14 The reduction of NK cells15,16 and the proliferation of Th17 cells17,18 in PB were observed in both SAPHO syndrome and in HSP. Meanwhile, IL-6 and IL-8 levels in PB were increased in the two diseases as well. These abnormal changes were associated with joint inflammation and cutaneous lesions.10,19 IL-18 was related to the incidence risk of PPP in SAPHO syndrome.10 In the meanwhile, IL-18 gene polymorphisms also played an important role in the pathogenesis of HSP.20
Tofacitinib is a new small-molecule inhibitor of the JAK signaling pathway, which mainly acts on JAK1 and JAK3. It could modulate the Th17/Treg balance.21 JAK/STAT is the downstream core signaling molecules of the IL-6 signaling pathway. NF-κB is the significant signaling molecule in the downstream of IL-8 and IL-18 signaling pathways. It has been proven that the cross-talk of JAK/STAT signaling pathway and NF-κB signaling pathway may accelerate disease progression.22 Tofacitinib can alleviate the disease activity by inhibiting the combining of JAK with STAT. It also has a convinced effect in the treatment of psoriatic arthritis,23 ankylosing spondylitis,24 and SAPHO syndrome.9,25,26 Although tofacitinib was reported to treat refractory cutaneous leukocytoclastic vasculitis, it had not been reported in HSP treatment. Based on the inference of the co-occurrence mechanism of the two diseases and our previous experiences, tofacitinib was prescribed. The disease activity was controlled and no rashes reoccurred.
At present, among all the cutaneous small-vessel vasculitis, only BD was considered as a comorbidity of SAPHO syndrome. Both BD and HSP are all ascribed as cutaneous small-vessel vasculitis which was triggered by the deposition of the immune complex. In our point of view, HSP is also a new comorbidity of SAPHO syndrome as discussed above, which enriched the spectrum of cutaneous small-vessel vasculitis in SAPHO syndrome. Methylprednisolone and tofacitinib successfully treated SAPHO with HSP, which provided a new therapeutic approach.
Ethics Statement
This case has obtained the institutional ethical approval from Ethics Committee of Fangshan Hospital of Beijing University of Chinese Medicine (Approval #FZY JS-2021-002).
Consent
Written informed consent for publication of their details was obtained from the patient.
Author Contributions
All authors made a significant contribution to the work reported. All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
This research was funded by the National Natural Science Foundation of China (No. 82074246) for CL and Construction Project of Clinical Key Specialty in Fengtai District of Beijing for XH.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Chamot AM, Benhamou CL, Kahn MF, et al. Acne-pustulosis-hyperostosis-osteitis syndrome. Results of a national survey. 85 cases. Rev Rhum Mal Osteoartic. 1987;54(3):187–196. French.
2. Norikane T, Yamamoto Y, Arai-Okuda H, et al. Pulmonary vasculitis on dual-phase 18F-FDG PET/CT in SAPHO syndrome. Clin Nucl Med. 2022;47(5):e411–e413. doi:10.1097/RLU.0000000000004104
3. Shanmugam VK, Phillpotts M, Brady T, et al. Retinal vasculitis with chronic recurrent multifocal osteomyelitis: a case report and review of the literature. BMC Rheumatol. 2019;3:29. doi:10.1186/s41927-019-0076-5
4. Yabe H, Takano Y, Nomura E, et al. Two cases of SAPHO syndrome accompanied by classic features of Behcet’s disease and review of the literature. Clin Rheumatol. 2008;27(1):133–135. doi:10.1007/s10067-007-0697-8
5. Nishiya K, Oosaki F, Nakamura T, et al. Rheumatoid arthritis associated with Henoch-Schönlein purpura. Clin Exp Rheumatol. 2000;18(5):653–654.
6. Beauvais C, Kaplan G, Mougenot B, et al. Cutaneous vasculitis and IgA glomerulonephritis in ankylosing spondylitis. Ann Rheum Dis. 1993;52(1):61–62. doi:10.1136/ard.52.1.61
7. Gao S, Deng X, Zhang L, et al. The comparison analysis of clinical and radiological features in SAPHO syndrome. Clin Rheumatol. 2021;40(1):349–357. doi:10.1007/s10067-020-05187-0
8. Sirufo MM, Raggiunti M, Magnanimi LM, et al. Henoch-Schönlein purpura following the first dose of COVID-19 viral vector vaccine: a case report. Vaccines. 2021;9(10):10. doi:10.3390/vaccines9101078
9. Li Y, Huo J, Cao Y, et al. Efficacy of tofacitinib in synovitis, acne, pustulosis, hyperostosis and osteitis syndrome: a pilot study with clinical and MRI evaluation. Ann Rheum Dis. 2020;79(9):1255–1257. doi:10.1136/annrheumdis-2020-217250
10. Przepiera-Będzak H, Brzosko M. SAPHO syndrome: pathogenesis, clinical presentation, imaging, comorbidities and treatment: a review. Adv Dermatol Allergol. 2021;38(6):937–942. doi:10.5114/ada.2020.97394
11. Pillebout E, Sunderkötter C. IgA vasculitis. Semin Immunopathol. 2021;43(5):729–738. doi:10.1007/s00281-021-00874-9
12. Abu-Zaid MH, Salah S, Lotfy HM, et al. Consensus evidence-based recommendations for treat-to-target management of immunoglobulin A vasculitis. Ther Adv Musculoskel. 2021;13:1759720X–2110596X.
13. Sun Y, Li C, Zhu M, et al. Enhanced migration and adhesion of peripheral blood neutrophils from SAPHO patients revealed by RNA-Seq. Orphanet J Rare Dis. 2019;14(1). doi:10.1186/s13023-019-1169-3
14. Heineke MH, Ballering AV, Jamin A, et al. New insights in the pathogenesis of immunoglobulin A vasculitis (Henoch-Schonlein purpura). Autoimmun Rev. 2017;16(12):1246–1253. doi:10.1016/j.autrev.2017.10.009
15. Ding Y, Zhou Y, Li H-R, Xiong Y-H, Yin W, Zhao L. Characteristics of immune function in the acute phase of Henoch-Schönlein purpura. Clin Rheumatol. 2021;40(9):3711–3716. doi:10.1007/s10067-021-05707-6
16. Xu D, Liu X, Lu C. Reduction of peripheral natural killer cells in patients with SAPHO syndrome. Clin Exp Rheumatol. 2019;37(1):12–18.
17. Jen HY, Chuang YH, Lin SC, et al. Increased serum interleukin-17 and peripheral Th17 cells in children with acute Henoch-Schönlein purpura. Pediatr Allergy Immunol. 2011;22(8):862–868. doi:10.1111/j.1399-3038.2011.01198.x
18. Firinu D, Barca MP, Lorrai MM, et al. TH17 cells are increased in the peripheral blood of patients with SAPHO syndrome. Autoimmunity. 2014;47(6):389–394. doi:10.3109/08916934.2014.906582
19. Kimura S, Takeuchi S, Soma Y, et al. Raised serum levels of interleukins 6 and 8 and antiphospholipid antibodies in an adult patient with Henoch-Schönlein purpura. Clin Exp Dermatol. 2013;38(7):730–736. doi:10.1111/ced.12089
20. Torres O, Palomino-Morales R, Miranda-Filloy JA, et al. IL-18 gene polymorphisms in Henoch-Schonlein purpura. Clin Exp Rheumatol. 2010;28(1 Suppl 57):114.
21. Zhou Y, Leng X, Luo S, et al. Tolerogenic dendritic cells generated with tofacitinib ameliorate experimental autoimmune encephalomyelitis through modulation of Th17/Treg balance. J Immunol Res. 2016;2016:5021537. doi:10.1155/2016/5021537
22. Ibrahim S, Salama MA, Selima E, et al. Sitagliptin and tofacitinib ameliorate adjuvant induced arthritis via modulating the cross talk between JAK/STAT and TLR-4/NF-κB signaling pathways. Life Sci. 2020;260:118261. doi:10.1016/j.lfs.2020.118261
23. Gratacós Masmitjà J, González Fernández CM, Gómez Castro S, et al. Efficacy of tofacitinib in the treatment of psoriatic arthritis: a systematic review. Adv Ther. 2021;38(2):868–884. doi:10.1007/s12325-020-01585-7
24. Deodhar A, Sliwinska-Stanczyk P, Xu H, et al. Tofacitinib for the treatment of ankylosing spondylitis: a Phase III, randomised, double-blind, placebo-controlled study. Ann Rheum Dis. 2021;80(8):1004–1013. doi:10.1136/annrheumdis-2020-219601
25. Yang Q, Zhao Y, Li C, et al. Case report: successful treatment of refractory SAPHO syndrome with the JAK inhibitor tofacitinib. Medicine. 2018;97(25):e11149. doi:10.1097/MD.0000000000011149
26. Yuan F, Luo J, Yang Q. SAPHO syndrome complicated by ankylosing spondylitis successfully treated with tofacitinib: a case report. Front Immunol. 2022;13:911922. doi:10.3389/fimmu.2022.911922
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.