")
Back to Journals » International Journal of Nanomedicine » Volume 15
Synergistic Treatment of Tumor by Targeted Biotherapy and Chemotherapy via Site-Specific Anchoring of Aptamers on DNA Nanotubes
Authors Chen R, Sun P, Chu X, Pu X, Yang Y, Zhang N, Zhao Y
Received 28 July 2019
Accepted for publication 1 February 2020
Published 27 February 2020 Volume 2020:15 Pages 1309—1320
DOI https://doi.org/10.2147/IJN.S225142
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Run Chen,1,* Pengchao Sun,2,* Xiao Chu,1 Xiaorong Pu,1 Yang Yang,1 Nan Zhang,1,3,4 Yongxing Zhao1,3,4
1Department of Pharmaceutics, School of Pharmaceutical Sciences, Zhengzhou University, Zhengzhou, Henan 450001, People’s Republic of China; 2Institute for Biological Interfaces 1, Karlsruhe Institute of Technology, Karlsruhe 76344, Germany; 3HeNan Key Laboratory of Targeting Therapy and Diagnosis for Critical Diseases, Zhengzhou, Henan 450001, People’s Republic of China; 4Key Laboratory of Advanced Pharmaceutical Technology, Ministry of Education of China, Zhengzhou, Henan 450001, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yongxing Zhao; Nan Zhang
School of Pharmaceutical Sciences, Zhengzhou University, No. 100 KeXue Ave, Zhengzhou, HeNan 450001, People’s Republic of China
Tel +86 371 67739165
Fax +86 371 67739546
Email [email protected]; [email protected]
Background: Aptamers have been widely used as targeted therapeutic agents due to its relatively small physical size, flexible structure, high specificity, and selectivity. Aptamers functionalized nanomaterials, not only enhance the targeting of nanomaterials, but can also improve the stability of the aptamers. We developed aptamer C2NP (Apt) conjugated straight DNA nanotubes (S-DNT-Apt) and twisted DNA nanotubes (T-DNT-Apt) as nanocarriers for doxorubicin (DOX).
Methods: The twisted DNA nanotubes (T-DNT) and straight DNA nanotubes (S-DNT) were assembled with a scaffold and hundreds of staples. Apt was site-specifically anchored on DNA nanotubes with either different spatial distribution (3 or 6 nm) or varied stoichiometry (15Apt or 30Apt). The developed nanocarriers were characterized with agarose gel electrophoresis and transmission electron microscopy. The drug loading and release in vitro were evaluated by measuring the fluorescence intensity of DOX using a microplate reader. The stability of DNT in cell culture medium plus 10% of FBS was evaluated by agarose gel electrophoresis. The cytotoxicity of DNA nanostructures against K299 cells was tested with a standard CCK8 method. Cellular uptake, cell apoptosis, cell cycle and reactive oxygen species level were investigated by flow cytometry. The expression of p53 was examined by Western Blot.
Results: T-DNT-30Apt-6 exhibited the highest cytotoxicity when the concentration of Apt was 120 nM. After intercalation of DOX, the cytotoxicity of DOX@T-DNT-30Apt-6 was further enhanced due to the combination of chemotherapy of DOX and biotherapy of Apt. The enhanced cytotoxicity of DOX@T-DNT-30Apt-6 can be explained by the increase in the cellular uptake, cell apoptosis and intracellular ROS levels. Additionally, the interaction between Apt and its receptor CD30 could upregulate the expression of p53.
Conclusion: These results demonstrate that both stoichiometry and spatial arrangement of Apt on T-DNT-Apt influence the anticancer activity. The developed twisted DNA nanotubes may be a solution for the synergistic treatment of cancer.
Keywords: DNA nanotubes, aptamers, controlled spatial distribution, biotherapy and chemotherapy
Introduction
Nanocarriers have been widely used for cancer therapy.1 These nanocarriers include inorganic materials2 and organic materials.3 Recently, an emerging natural material, DNA, has drawn much attention for drug delivery. Based on the complementary base pairing rule, hundreds of short single-stranded DNA (ssDNA, staples) can hybridize to a long single-stranded DNA (scaffold) and be folded into a two-dimension (2D) or three-dimension (3D) DNA origami nanostructure.4 Doxorubicin (DOX) has been widely used for cancer therapy.5–7 It can noncovalently bind duplex DNA by the interaction between the amino group of DOX and the cytosine of DNA.8 DOX-loaded DNA nanostructures have been used for cancer therapy.9 However, DOX delivered by these nanocarriers can only passively accumulate at the tumor site.
DNA aptamers are short oligonucleotides which can specifically bind to their receptors with high affinity. DNA aptamers immobilized nanocarriers, such as mesoporous silica nanoparticles,10 gold nanoparticles,11 protein nanoparticles,12 polymer nanoparticles,13 etc., enhanced not only the targeting specificity of the nanocarriers but also improved the stability of aptamers. Given these advantages, DNA aptamers modification has been an excellent solution to enhance the targeting specificity of nanocarriers.14 DNA aptamers can be immobilized on DNA nanostructures either by extending staples with the sequence of aptamers15 or by hybridization method.16 DNA aptamer C2NP (Apt) is a 31 nt oligonucleotide with high affinity and specificity to CD30 receptor which is a diagnostic biomarker and overexpressed in tumor cells, such as K299 cells (human anaplastic large cell lymphoma cell line). It was reported that Apt interacts with its receptor and induces cell apoptosis.17 Apt probe had been developed and exhibited high sensitivity and a long-lasting signal in vivo.18 Our previous work also showed that Apt modified rectangular DNA origami nanostructures (RE-4Apt and RE-16Apt) increased the targeting delivery and nanocarriers with a high density of Apt (RE-16Apt) exhibited stronger anti-cancer activity in vitro.19
Extracellular matrix (ECM) plays a vital role in cell adhesion, spreading, and migration. It was reported that the distance and amount of ECM-protein affected cell binding and spreading. ECM-protein micropatterned substrate can guide the shape of cells.20 Besides the ECM-protein, ligands can also influence cell behavior. Nanoscale arrangements of ephrin-A5 can affect EphA2 receptor activation in human breast cancer cells.21 Inspired by these findings, we would like to find out how Apt with different spatial distribution and stoichiometry impacts the cell activities.
To this end, we used twisted DNA nanotubes (T-DNT) as carriers of Apt. We modified the T-DNT with either 15 or 30 of Apt which were defined as DOX@T-DNT-15Apt and DOX@T-DNT-30Apt. Given that the linear distance between the binding sites of CD30 is 5.7 nm,22 we designed Apt modified nanotubes with the spatial distance of 3 or 6 nm which were donated as DOX@T-DNT-15Apt-3, DOX@T-DNT-30Apt-3, DOX@T-DNT-15Apt-6, DOX@T-DNT-30Apt-6 respectively. The spatial distribution of Apt was firstly designed on straight DNT (S-DNT) map, and Apt was then modified at the same positions on T-DNT. The design and the illustration of each DNT can be found in Figure S2. We firstly compared the biotherapy of Apt modified S-DNT (S-DNT-30Apt-3, S-DNT-30Apt-6) and T-DNT (T-DNT-30Apt-3, T-DNT-30Apt-6) to confirm that the spatial distribution of Apt on T-DNT remained the same as designed in S-DNT. An anti-cancer drug, doxorubicin (DOX), was then loaded to the DNA nanotubes for the chemotherapy. In our present study, the effects of spatial distribution and stoichiometry of Apt on cell behaviors were investigated. Both bioactivities of Apt and chemotherapy of DOX were studied. Furthermore, the anti-tumor mechanism of Apt anchored T-DNT was explored. We demonstrated that Apt anchored T-DNT with different spatial distribution and stoichiometry showed various anti-cancer activities. DOX@T-DNT-30Apt-6 increased the biotherapy of Apt and chemotherapy of DOX. Western blot results showed that Apt induces cell apoptosis by upregulation of p53 expression in K299 cells. Our results indicate that Apt modified T-DNT is not only a promising strategy for targeting delivery but also an efficient solution for the synergistic treatment of cancer.
Materials and Methods
Materials
Scaffolds (7560 and 8634) were derived from M13mp18 which was donated by Prof. Dr. B. Högberg (Karolinska Institutet, Sweden). C2NP and single-stranded DNA was synthesized by Bioneer (Daejeon, Korea). K299 cells (human anaplastic large cell lymphoma cell line) were obtained from CoBioer (Nanjing, China). Agarose was supplied by Biowest (Nuaill´e, France). Fetal bovine serum (FBS) was purchased from BI (Kibbutz, Israel). Cell apoptosis assay kit, cell cycle kit, DCFH-DA cellular reactive oxygen species assay kit, and DOX hydrochloride (DOX∙HCl) were obtained from Solarbio (Beijing, China). Dihydrochloride (DAPI), ethidium bromide (EB), 1Kbp DNA Marker, DNA loading buffer, 4% paraformaldehyde, phosphate-buffered solution (PBS) powder, and Difco Skim Milk were supplied by Solarbio (Beijing, China). The following antibodies were used: anti-p53 (catalog number: WL01333; Wanleibio). Rabbit monoclonal anti-β-actin (1:1000, catalog number WL01774; Wanleibio, China) was used as a housekeeping protein. All other chemicals were obtained from Aladdin (Shanghai, China). All commercial chemicals and agents were used without any purification and modification.
Preparation of DNT
Twisted and straight DNT (T-DNT and S-DNT) were assembled following Rothmund’s method.23 Briefly, scaffold (100 nM, 2 µL) and staples (200 nM, 5 µL) were mixed in TEM buffer (Tris 5 mM, EDTA 1 mM, MgCl2 5 mM, pH 7.8) with a final volume of 40 µL. Folding was carried out by rapid heating from 25 to 80°C (4°C/s, maintained at 80°C for 5 min), followed by slow cooling from 80 to 60°C (0.004°C/s, maintained at 60°C for 10 min) and from 60 to 24°C (0.001°C/s). For the second slow cooling step, the temperature was maintained for 400 s after every one-degree drop. Aptamer C2NP anchored DNT (T-DNT-15Apt-3, T-DNT-15Apt-6, T-DNT-30Apt-3, T-DNT-30Apt-6, S-DNT-30Apt-3, S-DNT-30Apt-6) were constructed by the same procedure with prolonged staples. Microscope visible carriers, FAM-T-DNT-30Apt-6, were prepared by FAM-labeled staples. The obtained DNT was stored at 4°C for further study.
Characterization of DNT
DNT were characterized by agarose gel electrophoresis and transmission electron microscopy (TEM). For the agarose gel electrophoresis, 1.3 μL of EB (10 μg/mL) was added for gel imaging. Samples mixed with 6×loading buffer (1:0.2, v/v) were loaded to the pockets of agarose gel (1.5%, in 1×TBE buffer) which was later run at 70 V in an ice bath. After 4 h, the gel was imaged by a gel imaging system (Biorad, USA). The morphology of DNT was further imaged with TEM (FEI, America). DNT (2 μL, 4 nM) was spotted on a glow-discharged, carbon-coated Formvar grid, and incubated for 20 s, followed by staining with uranyl formate (2%, w/v, in water). The excess solution was removed by filter paper, followed by observation with TEM.
DOX Loading and Release
A calibration curve of DOX, ranging from 4 to 256 µM, was firstly built by a microplate reader for the quantification of DOX (BioTek, USA; λex=485 nm, λem=591 nm). DOX@T-DNT-30Apt was obtained by incubating DOX∙HCl solution and T-DNT-30Apt-6 at room temperature in the dark. The concentration of DOX was optimized to get the maximum loading capacity. Firstly, various amounts of DOX (20 μL; 16, 32, 64, 128, 256, 512, and 1024 μM) were incubated with T-DNT-30Apt-6 (20 μL, 4 nM). After 3 h, DOX@T-DNT-30Apt-6 was centrifuged down (15,000 rpm, 10 min) and the fluorescence (F) of free DOX in the supernatant was measured by the microplate reader and the concentration (C) was calculated by the calibration curve. The drug-loading capacity of DOX@T-DNT-30Apt-6 was determined by equation (1):
(1)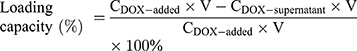
The UV-vis spectra of the resulting nanocarriers were recorded by a UV-vis spectrophotometer (Agilent, USA) to confirm the successful encapsulation of DOX. The zeta potential of the nanocarriers was measured by zeta potential analyzer using a Zetasier Nano-ZS (Malvern, UK).
Release property of DOX was studied by a dialysis method.15 Typically, DOX solution, DOX@S-DNT-30Apt-6 or DOX@T-DNT-30Apt-6 (4 nM, 20 μL) was enclosed in Slide-A-Lyzer™ MINI Dialysis Devices (Thermo Fisher, Germany) which were floated in PBS (pH7.4, 37°C). At the set time points (0.5, 1, 2, 4, 8, 12, 24, and 48 h), three dialysis tubes of each sample were taken out and the concentration of DOX remained inside the tube was quantified. The cumulative release was calculated by equation (2):
(2)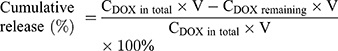
Stability Evaluation
The stability of both DOX and T-DNT were investigated by monitoring the fluorescence intensity of DOX and migration of T-DNT in agarose gel, respectively. To this end, free DOX (64 μM) and DOX@T-DNT-30Apt-6 (4 nM) were incubated with cell culture medium with 10% of FBS in the dark at 37°C. Samples (5 μL) were collected at set time points (0, 2, 4, 6, 8, 12, 24 and 48 h). The fluorescence intensity (F) of DOX was determined by a microplate reader. Typically, samples were mixed with DMSO (2 μL) and PBS (43 μL, pH7.4). The mixtures were shaken at 100 rpm for 3 min before measurement. The relative stability of DOX was calculated via equation (3):
(3)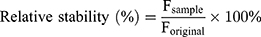
The migration of T-DNT was analyzed by agarose gel electrophoresis as the same procedure for the characterization of T-DNT.
Cell Culture
K299 cells were cultivated in RPMI 1640 with 10% of FBS under standard conditions (CO2: 5%, temperature: 37°C, humidity: 100%). Cells were split every two or three days. Only cells in the exponential phase were used for the following experiments.
In vitro Anticancer Activity
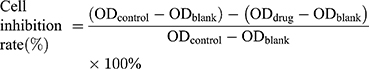
Anticancer activity of the developed nanocarriers was tested against K299 cell line by a standard cell counting-8 kit (CCK8) assay.15 K229 cells were seeded in 96 well plates at a density of 1×104 cells/well and incubated under standard cell culture conditions for 12 h before each experiment. Firstly, various concentrations of T-DNT (1, 2, 3, 4 and 5 nM) and MgCl2 (0.625, 1.25, 2.5 and 6.25 mM) were treated with K299 cells for 48 h to test the cytotoxicity of T-DNT and MgCl2. Secondly, cell culture medium with different concentration of T-DNT-30Apt-3, T-DNT-30Apt-6, S-DNT-30Apt-3 and S-DNT-30Apt-6 (CDNT: 2, 4 and 6 nM) were added to each well and incubated for 48 h to investigate the differences of anti-cancer activities of each group. Bioactivity of Apt was evaluated by incubation of Apt, T-DNT-15Apt-3, T-DNT-15Apt-6, T-DNT-30Apt-3 and T-DNT-30Apt-6 with cells at different concentrations (CApt: 15, 30, 60 and 120 nM) for 48 h. Chemotherapy of free DOX, DOX@T-DNT, and DOX@T-DNT-30Apt-6 was determined by incubation with cells for 48 h at different concentrations of DOX (CDOX: 0.5, 1 and 5 µM; CDNT: 4 nM). Bioactivity and chemotherapy of the developed nanocarriers were studied by treatment cells with DOX, DOX@T-DNT and DOX@T-DNT-30Apt-6 at different concentrations (CDNT: 2, 4 and 6 nM; CDOX: 1 µM) for 48 h. After each incubation, CCK8 assay was carried out following the manufacturer’s instruction. The Uv-vis absorbance (OD) of the medium at 450 nm was measured by a microreader and the inhibition rate was calculated by equation (4):
(4)
Cellular Uptake
Cellular uptake of DOX was imaged by confocal laser scanning microscopy (CLSM, Olympus, Tokyo, Japan) and quantified by flow cytometric analysis (BD FACSCanto II, USA). K299 cells were seeded in 12-well plates for 12 h before each experiment.
For the CLSM imaging, cells were treated with DOX, DOX@T-DNT and DOX@T-DNT-30Apt-6 (CDNT: 4 nM, CDOX: 1 μM) for different times. At set time points (0.5, 1, 2 and 4 h), the uptake process was terminated by adding ice-cold PBS. The excess DOX or DOX-loaded nanocarriers were removed by another 3 times washing steps with PBS. Subsequently, the cells were fixed with 4% paraformaldehyde and cell nuclei were stained with DAPI (10 μg/mL, 15 min), followed by CLSM imaging. Intracellular location of T-DNT-30Apt-6 was also investigated by CLSM. In brief, K299 cells were incubated with FAM-T-DNT-30Apt-6 (CDNT: 4 nM). After 4 h, cellular uptake was stopped by ice-cold PBS. Cells were further fixed, stained and imaged as the same procedure.
With respect to intracellular DOX quantification, cells were incubated with DOX, DOX@T-DNT and DOX@T-DNT-30Apt-6 (CDNT: 4 nM, CDOX: 1 μM) for 4h. The cellular uptake was terminated by ice-cold PBS and the excess DOX or DOX-loaded carriers were removed by washing cells with PBS for 3 times. 10,000 cells were automatically collected and intracellular signal of DOX was quantified by flow cytometric analysis (BD FACSCanto II, USA).
Cell Apoptosis
Apoptotic cells were determined by dual staining with an Annexin V-FITC and propidium iodide (PI) kit. Typically, K299 cells (2×105 cells/well) were seeded in 12-well plates and incubated at the standard cell culture conditions for 12 h. The cells were treated with DOX, DOX@T-DNT and DOX@T-DNT-30Apt-6 (CDNT: 4 nM, CDOX: 1 μM). After 48 h, cells were collected and washed thrice with PBS. The cells were resuspended in 1 mL of binding buffer followed by treatment with FITC-Annexin V (5 μL, 20 min) and PI (5 μL, 10 min) in turn. The cell suspension was incubated for 15 min in the dark before flow cytometric analysis.
Reactive Oxygen Species (ROS)
Reactive oxygen species assay kit was used to analyze ROS levels in K299 cells. Briefly, K299 cells (2×105 cells/well) were seeded in a 12-well plate. After incubation for 12 h, cells were treated with DOX, DOX@T-DNT and DOX@T-DNT-30Apt-6 (CDNT: 4 nM, CDOX: 1 μM) and further incubated at standard conditions. After 48 h, the cells were collected and washed with PBS for 3 times. The cells were resuspended in 500 μL of cell culture medium containing 2,7-Dichlorofluorescin diacetate (DCFH-DA; 10 μM, 500 μL). All samples were incubated under standard conditions for another 25 min and washed with cell culture medium (without FBS) and PBS for several times before flow cytometric analysis.
Cell Cycle
DNA content quantitation assay kits were used for cell cycle stages measurement. K299 cells were seeded at a density of 2×105 cells/well in a 12-well plate. After incubation for 12 h, cells were treated with the fresh medium which contained DOX, DOX@T-DNT, and DOX@T-DNT-30Apt-6 (CDNT: 4 nM, CDOX: 1 μM). After cultivation for 48 h, cells were collected (1000 rpm, 5 min) and washed twice with PBS. The pellets were redispersed in 500 µL of ethanol (70%) and incubated at 4°C overnight for fixation. Cells were washed with PBS twice to remove any ethanol and treated with RNase (100 µL) at 37°C for 30 min, and stained with PI (200 µL, 30 min) for flow cytometric analysis.
Western Blot
K299 cells were seeded in a 6-well plate at a density of 5×105 cells/well. After treatment with DOX, DOX@T-DNT and DOX@T-DNT-30Apt-6 (CDNT: 4 nM, CDOX: 1 μM) for 48 h, cells were collected and washed twice with ice-cold PBS, followed by lysis in RIPA tissue/cell lysis buffer (radio immunoprecipitation assay; 1 mM, 50 µL; Solarbio Science) for 30 min on an ice bath. The cells were mixed every 10 min during the lysis. Samples were collected by centrifugation (14,000×g, 10 min, 4°C) and quantified by a BCA kit (Bradford Protein Assays, Thermo Fisher Scientific, Germany).24 Subsequently, the samples (20 µg) were separated on a sodium dodecyl sulfate-polyacrylamide gel (SDS-PAGE; 12%, 120 V, 1 h) and transferred onto a polyvinylidene fluoride (PVDF) membrane via wet transfer (200 mA, 1 h). After being blocked with nonfat milk at room temperature (5%, in PBST (0.1% of Tween 20 in PBS); 3 h, 100 rpm), the membrane was incubated with primary antibody (1:1000; PBST; overnight, 4°C). After being washed with PBST (3 times, 30 min), the membrane was further incubated with the secondary antibody (1:10,000, PBST; 2 h, 100 rpm). Protein signals were developed with an enhanced chemiluminescence kit (Merck Millipore, Darmstadt, Germany).
Statistical Analysis
All data were expressed as the mean ± SD. The graphs were performed by Origin 8.0. Statistical analyses were performed using SPSS. All data were compared with a single factor analysis of variance (ANONA). The results of flow cytometric analysis were performed by Flow Jo software. All experiments in this study were conducted at least three times.
Results and Discussion
Construction and Characterization of DNA Nanotubes
The scaffold DNA was extracted from M13mp18, as shown in Supporting Information Figure S1. Scaffolds can be folded into S-DNT and T-DNT with the help of staples (Tables S1 and S2) via DNA origami nanotechnology.25 Extending the sequence at the end of staples is a simple method to immobilize Apt on DNA nanostructures.15 These staples provide possibilities for the modification of Apt at different positions to achieve the controllable quantity and spatial distribution of Apt on the DNA nanotubes. In the present study, scaffolds with 8634 or 7560 bases were folded into T-DNT and S-DNT by one step annealing respectively.26 By extending staple with a sequence of Apt, DNT-Apt with different spatial distribution (3 or 6 nm) and stoichiometry (15 or 30 Apt) were constructed (Scheme 1, Figure S2) for precise treatment of cancer. The optimized conditions for the synthesis of DNA nanotubes are as follows: the concentration of Mg2+ is 5 mM, and the molar ratio of staples to scaffold is 5:1, as shown in Figure S3.
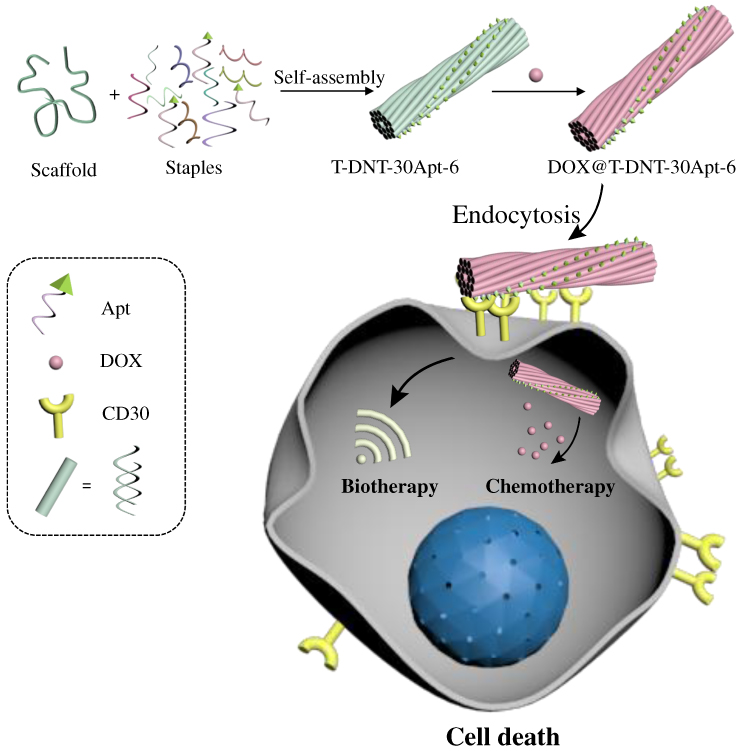 |
Scheme 1 Schematic illustration of Apt (C2NP) anchoring DNA nanotubes (DNT) for biotherapy and chemotherapy. On the one hand, the developed nanocarriers could effectively get access to cells by CD30 receptor moderated endocytosis which can increase the chemotherapy of doxorubicin (DOX). On the other hand, Apt modified DNT with different spatial distribution (3 or 6 nm) and stoichiometry (15Apt or 30Apt) could activate cellular signaling which induces cell apoptosis, resulting in different anticancer activities. |
DNA nanotubes were characterized by TEM and agarose gel electrophoresis. Figure 1A and B showed the TEM images of T-DNT-30Apt-6 and S-DNT-30Apt-6, respectively. Both DNA nanotubes are uniform with nanorod-like morphology. The size is about 140 nm × 14 nm. Given that the length of Apt is only 31 nt, they are too small to be imaged by TEM. Migration of scaffold, T-DNT, T-DNT-30Apt-6, and T-DNT-15Apt-6 in an agarose gel is shown in Figure 1C. The slight retardation difference among scaffold, T-DNT, T-DNT-30Apt-6 and T-DNT-15Apt-6 suggested the different molecular weights of the structures. Slower migrations are observed in Apt loaded T-DNT.
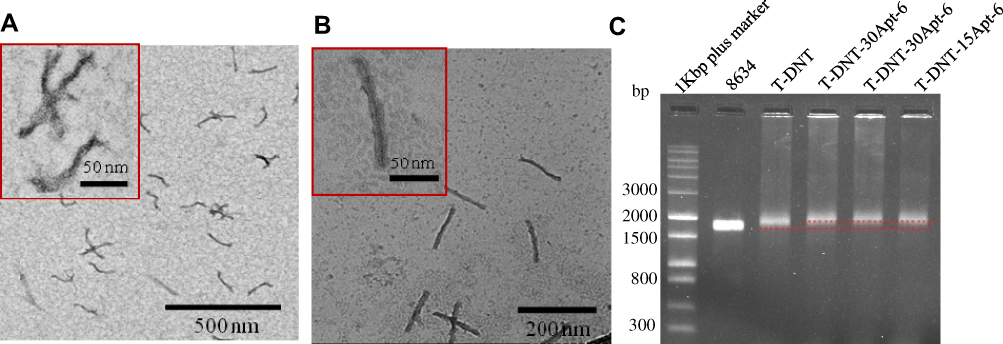 |
Figure 1 Characterization of DNA nanostructures. (A) TEM image of T-DNT-30Apt-6. (B) TEM image of S-DNT-30Apt-6. (C) Agarose gel electrophoresis analysis of scaffold, T-DNT, T-DNT-30Apt-6 and T-DNT-15Apt-6. |
Drug Loading, Stability and Release
DOX was loaded to T-DNT for chemotherapy of lymphoma in vitro by the hydrogen bond between the amino group of DOX and cytosine of DNA.8 After incubation T-DNT (4 nM, 20 μL) with DOX (256 μM, 20 μL) in the dark for 3 h, ~5 molecules of DOX were inserted to each base pair, which was the maximum loading capacity. The successful encapsulation of DOX can be proved either by the zeta potential shift from −25.6 mV to −17.6 mV (Figure S4) or by the UV-vis absorbance of DOX@S-DNT-30Apt-6 at 486 nm (Figure S5). The stability of T-DNT and DOX were investigated in cell culture medium with 10% of FBS at 37°C before anti-cancer studies. As shown in Figure 2A, T-DNT was stable in cell culture medium at least for 8 h and some T-DNT had been degraded at 12 h. Compared with free DOX, the fluorescence of DOX@T-DNT-30Apt-6 was much higher after incubation in cell culture medium for 48 h (Figure 2B). These results demonstrated that T-DNT-30Apt-6 could protect DOX from directly interacting with cell culture medium, resulting in the improvement of the structural stability of DOX in cell culture medium.26
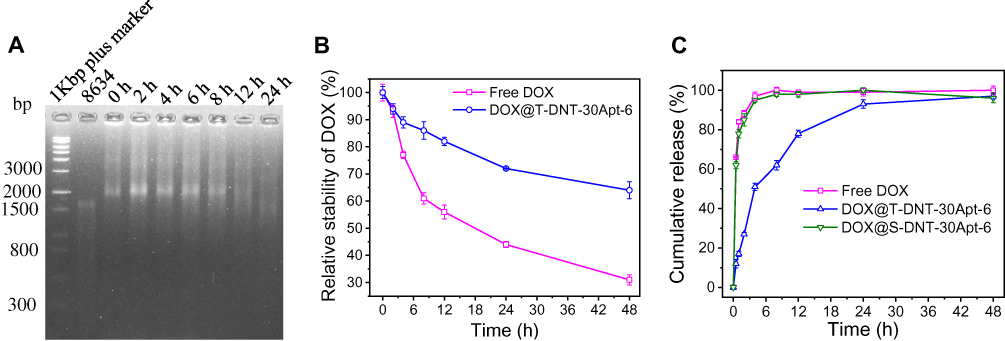 |
Figure 2 Time-dependent stability of T-DNT and DOX, and the release study of DOX from DNA nanotubes in Vitro. (A) Agarose gel analysis of T-DNT after incubation with cell medium plus 10% of FBS for different periods of time. (B) Comparation of the relative stability of free DOX and DOX@T-DNT-30Apt-6 in cell medium plus 10% of FBS. (C) Release curves of DOX from DOX@T-DNT-30Apt-6 and DOX@S-DNT-30Apt-6 in PBS. |
Figure 2C showed the release property of DOX from S-DNT-30Apt-6 and T-DNT-30Apt-6. DOX@S-DNT-30Apt-6 and free DOX exhibited similar release properties. However, the release of DOX from T-DNT-30Apt-6 was much slower in 24 h, which may be due to the more compact structure of T-DNT.26 Given that T-DNT-30Apt-6 showed a controllable release of DOX, T-DNT was selected for further study.
In vitro Anticancer Activity
In vitro anti-tumor activity of DOX@T-DNT-Apt and DOX@S-DNT-Apt were studied on K299 cells with a standard CCK-8 assay. Given that Apt can activate CD30 receptors which could induce cell apoptosis17 and DOX can kill cancer cells, both chemotherapy and biotherapy were investigated in the present study.
Figure S6 confirms that both origami buffer and T-DNT were not cytotoxic at the concentrations which were used for the following studies. Human gastric epithelial cell line (Ges-1) which has low expression of CD30 receptors was used as a negative control experiment. As shown in Figure S7, Apt and Apt modified DNT showed no significant effects on Ges-1 cell activities even with a high concentration of Apt (120 nM).
We firstly investigated biotherapy of S-DNT and T-DNT to confirm that the spatial distribution of Apt on T-DNT remains the same as designed in S-DNT. As shown in Figure 3A, at 2 nM of DNT (CApt: 60 or 30 nM), S-DNT-30Apt-3, T-DNT-30Apt-3, S-DNT-15Apt-6 and T-DNT-15Apt-6 showed similar cytotoxicity on K299 cells due to the low concentration of Apt. At 4 nM (CApt: 120 or 60 nM) and 6 nM (CApt: 180 or 90 nM), cell inhibition rates of S-DNT-30Apt-6 and T-DNT-30Apt-6 (p>0.05) showed no significant difference (p>0.05), which is similar between S-DNT-30Apt-3 and T-DNT-30Apt-3. These results indicate that the spatial distribution of Apt in T-DNT is the same as S-DNT. Furthermore, T-DNT-30Apt-6 and S-DNT-30Apt-6 demonstrated significantly higher cell inhibition rate than T-DNT-30Apt-3 and S-DNT-30Apt-3 (p<0.001). These results can be explained by the fact that the distance between the binding site of CD30 on the cell membrane is about 5.7 nm22 and S-DNT-30Apt-6 and T-DNT-30Apt-6 activated more CD30 receptors which induced more cell apoptosis.17,22 Besides, T-DNT could control the release of DOX, and further studies were carried out on T-DNT.
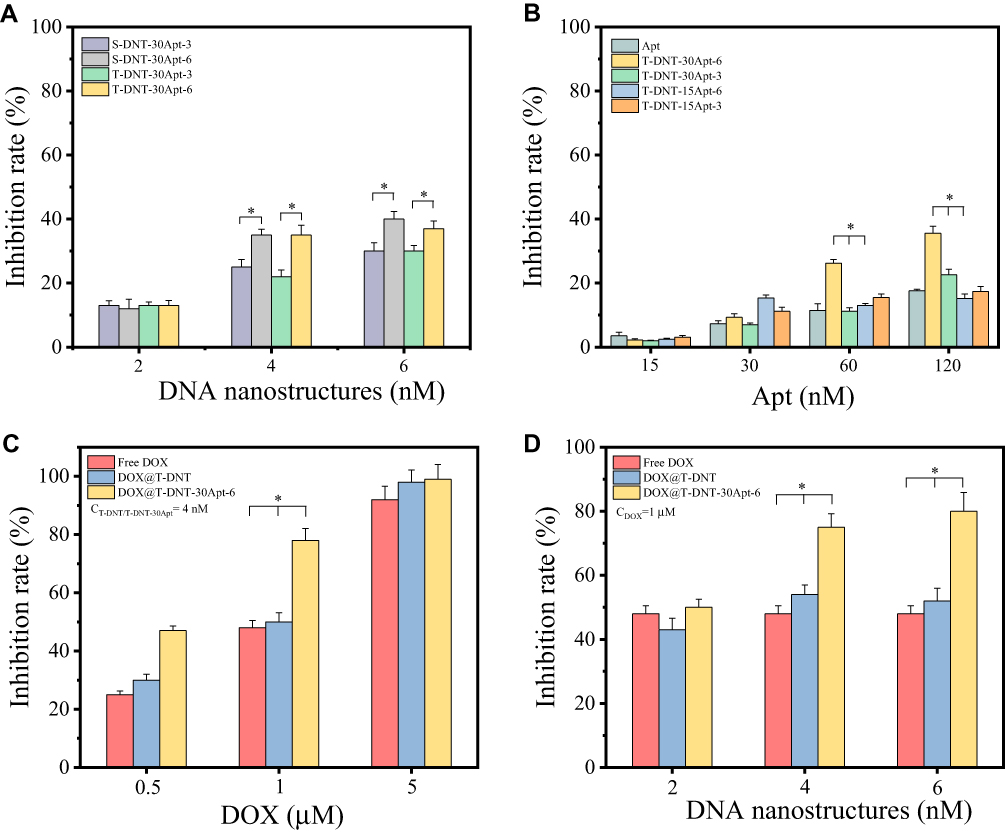 |
Figure 3 In Vitro anti-cancer activity of DNA nanostructures against K299 cells. (A) Bioactivity of S-DNT-30Apt and T-DNT-30Apt with different spatial distributions (3 or 6 nm) (*P<0.001). (B) Optimization of concentration and spatial distribution of Apt on DNA nanostructures (*P<0.001, T-DNT-30Apt-6 vs T-DNT-30Apt-3 and T-DNT-15Apt-6). (C) Synergetic bioactivity and chemotherapy of DOX, DOX@T-DNT and DOX@T-DNT-30Apt-6 against K299 cells with various concentrations of DOX (*P<0.001, DOX@T-DNT-30Apt-6 vs Free DOX and DOX@T-DNT). (D) Synergetic bioactivity and chemotherapy of DOX, DOX@T-DNT and DOX@T-DNT-30Apt-6 against K299 cells with various concentrations of DNA nanostructures (*P<0.001, DOX@T-DNT-30Apt-6 vs free DOX and DOX@T-DNT). |
We then optimized the spatial distribution and concentration of Apt for biotherapy. Figure 3B shows the biotherapy of T-DNT with different spatial distribution and stoichiometry of Apt. Similarly, at the lower concentration of Apt (15 nM and 30 nM), there were no significant differences in cell inhibition rates between each group. However, with the increase of concentration of Apt (60 nM and 120 nM), the anti-cancer activity varies. T-DNT-30Apt-6 exhibited excellent anti-cancer activities, and the inhibition rate was the highest when the concentration of Apt was 120 nM. What’s more, anti-cancer activity of DNA nanotubes with 6 nm between Apt was superior to that of 3 nm. These results indicated that a high concentration of Apt with 6 nm of spatial distribution indeed increased cell apoptosis. Therefore, a higher concentration of T-DNT-30Apt-6 (CDNT=4 nM, CApt=120 nM) was used for the study of chemotherapy of DOX.
We further optimized the concentration of DOX to obtain the maximum chemotherapy of the nanocarriers. As shown in Figure 3C, at the same level of DOX, the inhibition rate of DOX@T-DNT was slightly higher than free DOX, but much lower than DOX@T-DNT-30Apt-6. DOX@T-DNT-30Apt-6 exhibited the highest inhibition rate at 0.5 µM and 1 µM of DOX. These results revealed that T-DNT increased the anti-cancer activity of DOX and Apt promoted DNA nanocarriers to get access to tumor cells. However, 5 µM of DOX was so high that the anti-cancer activity in free DOX, DOX@T-DNT, and DOX@T-DNT-30Apt-6 all showed high cell inhibition rates. Hence, 1 µM of DOX was used for the study of the combination of chemotherapy and biotherapy.
Figure 3D shows the combination of chemotherapy and biotherapy of DOX@T-DNT-30Apt-6. Compared to free DOX and DOX@T-DNT, DOX@T-DNT-30Apt-6 exhibited a higher anti-cancer activity at different concentrations. The anti-cancer activity of DOX@T-DNT-30Apt-6 increased along with the increase of concentration. The inhibition rate of DOX@T-DNT-30Apt-6 (CDNT=4 nM, CDOX=1 µM) was much higher than that of either Apt (Figure 3B) or free DOX at the same conditions. These findings revealed that DOX@T-DNT-30Apt-6 exhibited both chemotherapy and biotherapy. Chemotherapy of DOX and biotherapy of Apt could be further enhanced after combination.
Cellular Uptake
Cellular uptake of DOX, DOX@T-DNT, and DOX@T-DNT-30Apt-6 was also studied. As shown in Figure 4A and Figure S8, cells which were treated with DOX@T-DNT-30Apt-6 showed the strongest fluorescence signal of DOX. Quantitative analysis of intracellular DOX by flow cytometry (Figure 4B) further revealed that cells treated with DOX@T-DNT-30Apt-6 showed the highest (99.9%) DOX level. These results demonstrate that Apt improved the cellular uptake of DOX by CD30 receptor moderated endocytosis.27 Thereby, chemotherapy of DOX can be significantly enhanced.
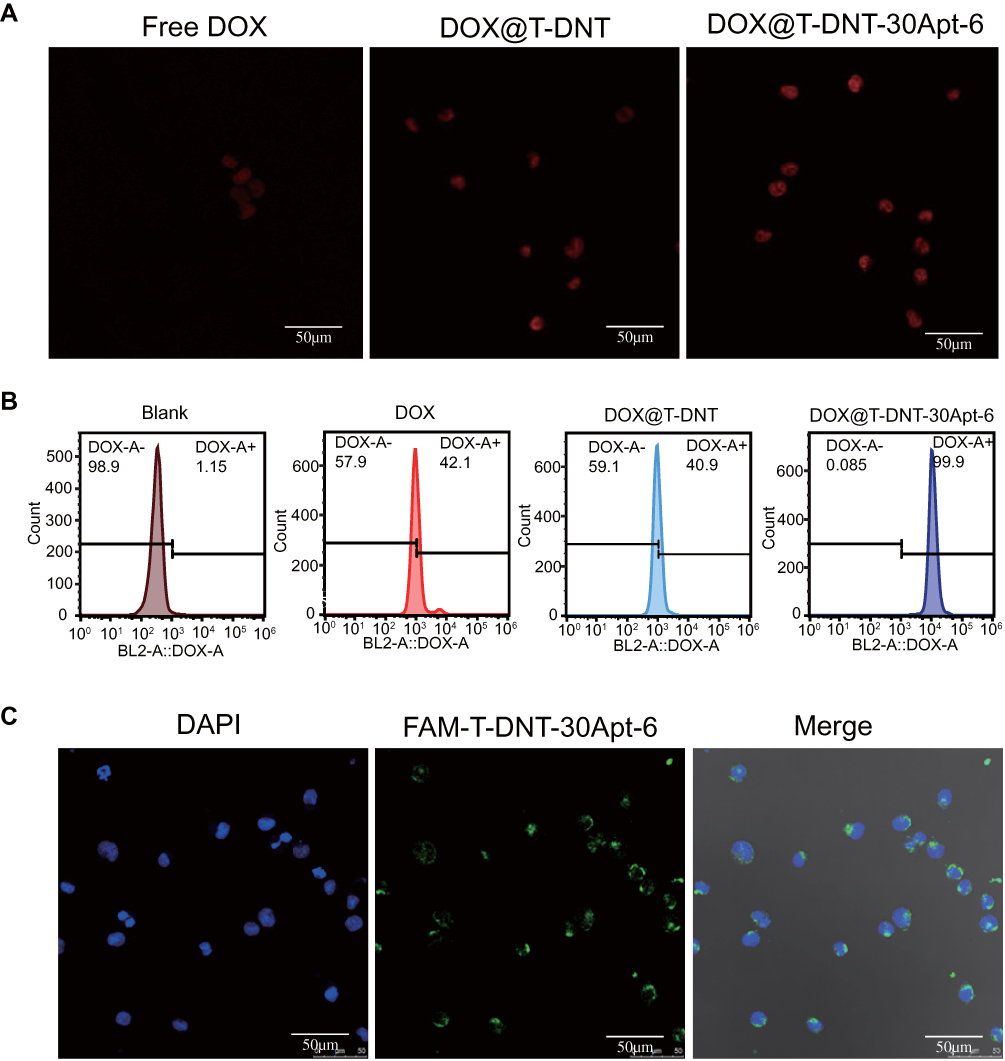 |
Figure 4 Intracellular uptake of DOX, DOX@T-DNT and DOX@T-DNT-30Apt-6. (A) CLSM images of K299 cells after incubation with DOX, DOX@T-DNT and DOX@T-DNT-30Apt-6 for 4h. (B) Flow cytometry histogram profiles of intracellular uptake of DOX after incubation with DOX, DOX@T-DNT and DOX@T-DNT-30Apt-6 for 4 h. (C) CLSM images of K299 cells after incubation with FAM-T-DNT-30Apt-6 for 4 h. |
Intracellular location of FAM-T-DNT-30Apt-6 was investigated by CLSM (Figure 4C). After incubation with K299 cells for 4 h, FAM-T-DNT-30Apt-6 was mainly accumulated around the cell nucleus and no signal of FAM was detected inside the cell nucleus. This experiment demonstrated that FAM-T-DNT-30Apt-6 can be taken up by cells and cannot be digested into small fractions which can get access to the cell nucleus within 4 h.
Mechanism of Cell Inhibition
The anti-cancer activity of DOX is achieved by inserting DNA base pairs and inhibiting topoisomerase II.8 Previous studies have shown that DOX affects cell apoptosis, cell cycle, and intracellular ROS levels.19 As shown in Figure 5A, free DOX increased early-stage cell apoptosis from 7.2% to 37.4%. After being loaded to T-DNT, cell apoptosis was slightly increased to 41.1%. Due to Apt, cell apoptosis was further increased to 62.3% by DOX@T-DNT-30Apt-6. A similar phenomenon was observed in the ROS analysis (Figure 5B). Intracellular ROS levels were increased sharply in DOX@T-DNT-30Apt-6 (58.7%). Figure 5C confirms that free DOX mainly arrested cell cycle at S stage. And the percentage of cells which were arrested at G2/M was slightly increased to 26.66%. Compared to free DOX, DOX@T-DNT mainly arrested less on G2/M. With the help of Apt, cell cycle arrest in G2/M was further increased to 33.99% in DOX@T-DNT-30Apt-6 group. This result matches our previous study that Apt can slightly arrest cells in G2/M.19
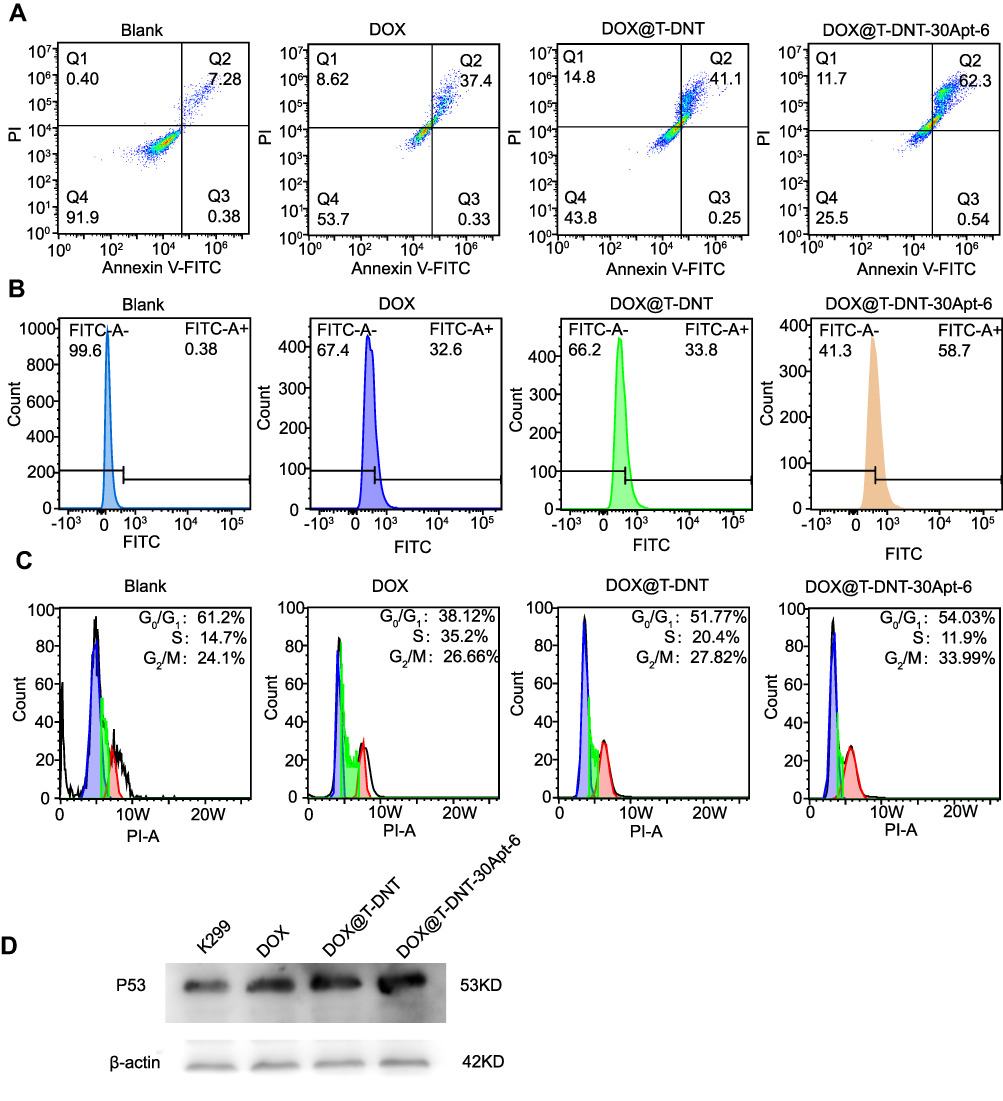 |
Figure 5 Mechanism study of the anti-cancer activity of DOX@T-DNT and DOX@T-DNT-30Apt-6. Representative results of the impacts of free DOX and different DNA origami carriers on (A) cell apoptosis, (B) ROS content (C) cell cycle and (D) p53 expression. |
It was reported that Apt could bind to CD30 receptors and upregulated the downstream signaling, resulting in cell apoptosis.28 Our previous study showed that Apt could result in cell apoptosis, arrest cell cycles in G2/M, and upregulated the intracellular ROS level.19 However, it is still unclear whether Apt has effects on some cell signaling. It is well known that tumor suppressor gene p53 plays a crucial role in cell cycle and cell proliferation.24 We therefore investigated whether DOX@T-DNT-30Apt-6 could induce p53 mediated apoptosis in K299 cells. Western blot analysis showed that both free DOX and DOX@T-DNT could promote the expression of p53 protein (Figure 5D). Moreover, the expression level of p53 was further increased in DOX@T-DNT-30Apt-6 group. These results demonstrated that Apt might induce the apoptosis of K299 cells by inducing the increase of p53 protein expression.
Conclusions
To summarize, we have synthesized tumor-targeting T-DNT nanocarriers by immobilization of Apt with different spatial distribution and stoichiometry. The spatial distribution of Apt which matched the distance of binding sites of CD30 receptors exhibited stronger anti-cancer activity. Furthermore, we investigated the anti-tumor mechanism of Apt. Our results showed that the interaction between C2NP and its receptor CD30 could upregulate the expression of p53, resulting in K299 cells apoptosis. Our work demonstrated that Apt modified T-DNT is not only a promising strategy for targeting delivery but also an efficient solution for the synergistic treatment of cancer.
Acknowledgments
We thank the financial support by National Natural Science Foundation of China (NSFC No. 81573011 and NSFC No. 81861138004). We thank Prof. Dr. B. Högberg (Karolinska Institutet, Sweden) for the donation of M13mp18. We appreciate the help from Instrumental Analysis Centre of Zhengzhou University. We thank Ziyi Wang for the UV-vis analysis and DLS measurements.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Mura S, Nicolas J, Couvreur P. Stimuli-responsive nanocarriers for drug delivery. Nat Mater. 2013;12(11):991–1003. doi:10.1038/nmat3776
2. Pan LM, He QJ, Liu JN, et al. Nuclear-targeted drug delivery of TAT peptide-conjugated monodisperse mesoporous silica nanoparticles. J Am Chem Soc. 2012;134(13):5722–5725. doi:10.1021/ja211035w
3. Merino S, Martín C, Kostarelos K, Prato M, Vázquez E. Nanocomposite hydrogels: 3D polymer–nanoparticle synergies for on-demand drug delivery. ACS Nano. 2015;9(5):4686–4697. doi:10.1021/acsnano.5b01433
4. Seeman NC. Nucleic acid junctions and lattices. J Theor Biol. 1982;99(2):237–247. doi:10.1016/0022-5193(82)90002-9
5. Zhang M, Wang WT, Zhou NL, et al. Near-infrared light triggered photo-therapy, in combination with chemotherapy using magnetofluorescent carbon quantum dots for effective cancer treating. Carbon. 2017;118:752–764. doi:10.1016/j.carbon.2017.03.085
6. Wu F, Zhang M, Lu HW, et al. Triple stimuli-responsive magnetic hollow porous carbon-based nanodrug delivery system for magnetic resonance imaging-guided synergistic photothermal/chemotherapy of cancer. Acs Appl Mater Inter. 2018;10(26):21939–21949. doi:10.1021/acsami.8b07213
7. Zhang M, Wang WT, Cui YJ, et al. Magnetofluorescent Fe3O4/carbon quantum dots coated single-walled carbon nanotubes as dual-modal targeted imaging and chemo/photodynamic/photothermal triple-modal therapeutic agents. Chem Eng J. 2018;338:526–538. doi:10.1016/j.cej.2018.01.081
8. Agudelo D, Bourassa P, Berube G, Tajmir-Riahi HA. Intercalation of antitumor drug doxorubicin and its analogue by DNA duplex: structural features and biological implications. Int J Biol Macromol. 2014;66:144–150. doi:10.1016/j.ijbiomac.2014.02.028
9. Zhang Q, Jiang Q, Li N, et al. DNA origami as an in vivo drug delivery vehicle for cancer therapy. ACS Nano. 2014;8(7):6633–6643. doi:10.1021/nn502058j
10. Li LL, Xie MY, Wang J, et al. A vitamin-responsive mesoporous nanocarrier with DNA aptamer-mediated cell targeting. Chem Commun. 2013;49(52):5823–5825. doi:10.1039/c3cc41072b
11. Kim D, Jeong YY, Jon S. A drug-loaded aptamer-gold nanoparticle bioconjugate for combined CT imaging and therapy of prostate cancer. ACS Nano. 2010;4(7):3689–3696. doi:10.1021/nn901877h
12. Wu JH, Song CC, Jiang CX, Shen X, Qiao Q, Hu YQ. Nucleolin targeting AS1411 modified protein nanoparticle for antitumor drugs delivery. Mol Pharmaceut. 2013;10(10):3555–3563. doi:10.1021/mp300686g
13. Lale SV, R GA, Aravind A, Kumar DS, Koul V. AS1411 aptamer and folic acid functionalized pH-responsive ATRP fabricated pPEGMA-PCL-pPEGMA polymeric nanoparticles for targeted drug delivery in cancer therapy. Biomacromolecules. 2014;15(5):1737–1752. doi:10.1021/bm5001263
14. Zhou J, Rossi J. Aptamers as targeted therapeutics: current potential and challenges. Nat Rev Drug Discovery. 2017;16(3):181–202. doi:10.1038/nrd.2016.199
15. Sun PC, Zhang N, Tang YF, Yang YN, Chu X, Zhao YX. SL2B aptamer and folic acid dual-targeting DNA nanostructures for synergic biological effect with chemotherapy to combat colorectal cancer. Int J Nanomed. 2017;12:2657–2672. doi:10.2147/IJN
16. Charoenphol P, Bermudez H. Aptamer-targeted DNA nanostructures for therapeutic delivery. Mol Pharm. 2014;11(5):1721–1725. doi:10.1021/mp500047b
17. Parekh P, Kamble S, Zhao NX, Portier BP, Zu YL. Immunotherapy of CD30-expressing lymphoma using a highly stable ssDNA aptamer. Biomaterials. 2013;34(35):8909–8917. doi:10.1016/j.biomaterials.2013.07.099
18. Zeng ZH, Parekh P, Li Z, Shi ZZ, Tung CH, Zu YL. Specific and sensitive tumor imaging using biostable oligonucleotide aptamer probes. Theranostics. 2014;4(9):945–952. doi:10.7150/thno.9246
19. Sun PC, Zhang N, Tang YF, Yang YA, Zhou J, Zhao YX. Site-specific anchoring aptamer C2NP on DNA origami nanostructures for cancer treatment. RSC Adv. 2018;8(46):26300–26308. doi:10.1039/C8RA04589E
20. Lehnert D, Wehrle-Haller B, David C, et al. Cell behaviour on micropatterned substrata: limits of extracellular matrix geometry for spreading and adhesion. J Cell Sci. 2004;117(1):41–52. doi:10.1242/jcs.00836
21. Shaw A, Lundin V, Petrova E, et al. Spatial control of membrane receptor function using ligand nanocalipers. Nat Methods. 2014;11(8):841–846. doi:10.1038/nmeth.3025
22. Ye H, Park YC, Kreishman M, Kieff E, Wu H. The structural basis for the recognition of diverse receptor sequences by TRAF2. Mol Cell. 1999;4(3):321–330. doi:10.1016/S1097-2765(00)80334-2
23. Rothemund PW. Folding DNA to create nanoscale shapes and patterns. Nature. 2006;440(7082):297–302. doi:10.1038/nature04586
24. Wang R, Li L, Zhang S, et al. A novel enediyne-integrated antibody-drug conjugate shows promising antitumor efficacy against CD30(+) lymphomas. Mol Oncol. 2018;12(3):339–355. doi:10.1002/1878-0261.12166
25. Zhang Y, Tu J, Wang D, et al. Programmable and multifunctional DNA-based materials for biomedical applications. Advan Mater. 2018;30(24):e1703658.
26. Zhao YX, Shaw A, Zeng XH, Benson E, Nystrom AM, Hogberg B. DNA origami delivery system for cancer therapy with tunable release properties. ACS Nano. 2012;6(10):8684–8691. doi:10.1021/nn3022662
27. Kennedy MK, Willis CR, Armitage RJ. Deciphering CD30 ligand biology and its role in humoral immunity. Immunology. 2006;118(2):143–152. doi:10.1111/imm.2006.118.issue-2
28. Pelicano H, Carney D, Huang P. ROS stress in cancer cells and therapeutic implications. Drug Resist Updates. 2004;7(2):97–110. doi:10.1016/j.drup.2004.01.004
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.