Back to Journals » International Journal of Nanomedicine » Volume 12
Synergistic interactions between antimicrobial peptides derived from plectasin and lipid nanocapsules containing monolaurin as a cosurfactant against Staphylococcus aureus
Authors Umerska A, Cassisa V, Bastiat G, Matougui N, Nehme H, Manero F ![]() , Eveillard M, Saulnier P
, Eveillard M, Saulnier P
Received 13 April 2017
Accepted for publication 2 June 2017
Published 8 August 2017 Volume 2017:12 Pages 5687—5699
DOI https://doi.org/10.2147/IJN.S139625
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Anita Umerska,1 Viviane Cassisa,2 Guillaume Bastiat,1 Nada Matougui,1 Hassan Nehme,1 Florence Manero,3 Matthieu Eveillard,4 Patrick Saulnier1
1MINT, UNIV Angers, INSERM 1066, CNRS 6021, Université Bretagne Loire, Angers, Cedex, France; 2Laboratoire de bactériologie, CHU Angers, France; 3SCIAM (Service Commun d’Imagerie et d’Analyses Microscopiques), Angers, France; 4Equipe ATIP AVENIR, CRCINA, Inserm, Université de Nantes, Université d’Angers, Angers, France
Abstract: Development of effective antibacterial agents for the treatment of infections caused by Gram-positive bacteria resistant to existing antibiotics, such as methicillin-resistant Staphylococcus aureus (MRSA), is an area of intensive research. In this work, the antibacterial efficacy of two antimicrobial peptides derived from plectasin, AP114 and AP138, used alone and in combination with monolaurin-lipid nanocapsules (ML-LNCs) was evaluated. Several interesting findings emerged from the present study. First, ML-LNCs and both plectasin derivatives showed potent activity against all 14 tested strains of S. aureus, independent of their resistance phenotype. Both peptides displayed a considerable adsorption (33%–62%) onto ML-LNCs without having an important impact on the particle properties such as size. The combinations of peptide with ML-LNC displayed synergistic effect against S. aureus, as confirmed by two methods: checkerboard and time-kill assays. This synergistic interaction enables a dose reduction and consequently decreases the risk of toxicity and has the potential of minimizing the development of resistance. Together, these results suggest that ML-LNCs loaded with a plectasin derivative may be a very promising drug delivery system for further development as a novel antibacterial agent against S. aureus, including MRSA.
Keywords: nanoparticles, antimicrobial peptides, glycerol monolaurate, synergy, antibiotic resistance, MRSA, methicillin-resistant Staphylococcus aureus
Introduction
Staphylococcus aureus is a Gram-positive bacterium with spherical shape (cocci) that grows in clusters resembling grapes. This facultative anaerobe can frequently be found in the nose, respiratory tract, and on the skin. Although S. aureus is usually not pathogenic, it can cause skin infections such as abscesses or scalded skin syndrome, pyogenic infections (eg, endocarditis, septic arthritis), respiratory infections such as sinusitis or hospital-acquired pneumonia, toxic shock syndrome, and food poisoning.1 The emergence of S. aureus strains such as methicillin-resistant S. aureus (MRSA) that have developed multiresistance to beta-lactam antibiotics including the penicillins (methicillin, nafcillin, and so on) and the cephalosporins is a worldwide problem in clinical medicine.2 Strains unable to resist these antibiotics are referred to as methicillin-susceptible S. aureus (MSSA). Although MRSA strains are not necessarily more virulent than MSSA strains, some MRSA strains contain factors or genetic backgrounds that may enhance their virulence or may enable them to cause particular clinical syndromes.3 Moreover, the resistance makes the MRSA infections more difficult to treat with standard antibiotics and therefore more dangerous.4 MRSA infections are particularly problematic in places such as hospitals and nursing homes, where patients with open wounds, invasive devices, and weakened immune systems are at greater risk of infection than the general public.5 Hence, there is an urgent need to develop new antibacterial agents for the prevention and treatment of the infections caused by S. aureus, particularly by the MRSA strains.
Plectasin is a cationic antimicrobial peptide, which belongs to the class called defensins. It was obtained from the saprophytic ascomycete Pseudoplectania nigrella. It is composed of 40 amino acids and exhibits a potent activity against Gram-positive bacteria in vitro and in vivo.6–8 AP114 (formerly known as NZ2114) is a plectasin derivative with an improved activity against staphylococci and streptococci, including strains resistant to standard antibiotics.6,7,9–12 AP138 is the product of an extensive lead optimization program based on a set of in silico generated plectasin variants.13
In most cases, the single best antimicrobial agent should be selected for use because this minimizes side effects. However, there are several instances in which two or more drugs are commonly given, such as treatment of serious infections before the identity of the organism is known or achievement of a synergistic inhibitory effect against certain organisms. Using drug combinations also offers the advantage of prevention of the emergence of drug-resistant organisms – if bacteria become resistant to one drug, the second drug will kill them thereby preventing the emergence of resistant strains.1
Monolaurin, also known as glycerol monolaurate or 1-lauroyl-glycerol, is a monoester formed from lauric acid and glycerol. This compound exhibits a potent activity against Gram-positive cocci and Bacillus anthracis.14 Monolaurin has been proven to be effective against both susceptible and resistant strains of S. aureus.15 Glycerol monolaurate is generally recognized as safe for human use by Food and Drug Administration, and this compound is commonly used in the cosmetic and food industries.16,17 Unlike most antibiotics that have single targets for antibacterial activity, monolaurin appears to target nonspecifically many signal transduction systems present at bacterial surface through interactions with plasma membranes.14 The main disadvantage of monolaurin is poor solubility in water, which may cause problems with its administration via different routes. One of the strategies to resolve the problem of poor solubility in water is by using nanocarriers, such as lipid nanocapsules (LNCs).18
LNCs are biomimetic nanocarriers obtained by the phase inversion temperature method.19 LNCs have been widely studied as carriers for lipophilic compounds.20,21 Our recent study showed that LNCs are capable of effectively adsorbing peptides, including AP114.22 Recently, we proposed using fatty acids or monoglycerides as cosurfactants to obtain LNCs with antibacterial properties.18 Monolaurin proved to be the most promising cosurfactant among all tested fatty acids and monoglycerides. Monolaurin-LNCs (ML-LNCs) exhibited the lowest minimum inhibitory concentration (MIC) or minimum bactericidal concentration (MBC) against two strains of S. aureus: one MSSA and one MRSA. Moreover, they showed the lowest toxicity among the tested LNCs, with the bactericidal concentration being 32- or 64-fold higher than the hemolytic concentration.18
The aim of the present work was to study the antibacterial activity of plectasin derivatives, AP114 and AP138, against seven MSSA and seven MRSA strains and to further investigate the antibacterial activity of ML-LNCs. To date, no studies have been performed to analyze the interactions between the peptides and ML-LNCs. Therefore, another purpose of this paper was to evaluate the adsorption of plectasin derivatives onto these nanocarriers. Because ML-LNCs have antibacterial properties, it is very important to investigate their influence on the activity of the peptide. Hence, the last important aim of this work was to investigate in vitro interactions between ML-LNCs and plectasin derivatives using time-kill assay and checkerboard method.
Materials and methods
Materials
Antimicrobial peptide AP114 was synthesized and provided by Adenium Biotech (Denmark) and AP138 was synthesized and provided by PolyPeptide Laboratories (Limhamn, Sweden). Monolaurin was purchased from Sigma Aldrich (France). Labrafac® WL1349 (caprylic/capric acid triglycerides) was kindly provided by Gattefossé S.A. (Saint-Priest, France). Lipoid® S75–3 (soybean lecithin) was kindly provided by Lipoïd Gmbh (Ludwigshafen, Germany). Solutol® HS15 (macrogol 15 hydroxystearate, polyoxyl 15 hydroxystearate; CAS number: 70,142–34-6; molecular weight 963.24 g/mol; HLB 14–16) was kindly provided by BASF (Ludwigshafen, Germany). Brain–heart infusion (BHI) broth was obtained from bioMérieux (France). Plates with Columbia agar supplemented with sheep blood were purchased from Oxoïd (France). All other chemicals and solvents were of analytical grade.
Preparation of LNCs
ML-LNCs were prepared at a concentration of 200 mg/mL as described by Umerska et al.18 The components of the LNCs (polyoxyl 15 hydroxystearate 0.77 g, triglycerides 0.93 g, and monolaurin 0.3 g) and NaCl (0.089 g) were weighed, mixed with 3 mL of water, and heated to 60°C and then cooled to 20°C. The sample was treated with three heating–cooling cycles; during the last cooling cycle, at the phase inversion temperature (37°C), the system was diluted with water to a final volume of 10 mL (Figure 1). The LNC dispersions were diluted with an aqueous solution of the plectasin derivative or water (control) and incubated at 37°C for 2 hours (sufficient for peptide molecules to achieve a dynamic equilibrium between the LNCs and the surrounding medium).
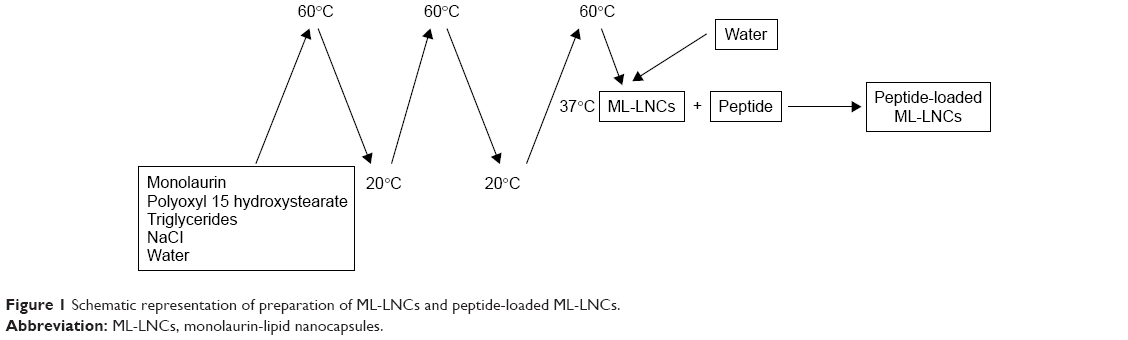 | Figure 1 Schematic representation of preparation of ML-LNCs and peptide-loaded ML-LNCs. |
Characterization of LNCs
The Z-average particle diameter (“cumulants mean”) and the polydispersity index of the LNCs were determined by dynamic light scattering (DLS) using 173 degree-angle backscatter detection. DLS measurements were conducted on a Zetasizer nano series Nano-ZS fitted with a 633 nm laser (Malvern Instruments, UK) as described previously.20
Rheological properties of ML-LNCs
Non-steady flow properties of the aliquots of ML-LNCs (concentration of 200 mg/mL) were studied using a Kinexus® rheometer (Malvern Instruments S.A., UK), with cone plate geometry (diameter 40 mm, angle: 2°) and with controlled shear rates ranging from 10 to 1,000 s−1 at 37°C (2 minute of equilibration time before the measurement). The profiles of shear viscosity versus shear rate were registered in triplicate. Viscosity results were expressed as a mean.
Peptide-loading studies
Separation of nonadsorbed peptide and extraction of adsorbed peptide
Nonadsorbed peptide was separated from the LNCs using a combined ultrafiltration–centrifugation technique as described previously.22
The adsorption efficiency (AE) and drug loading (DL) were calculated using the following equations:
|
(1) |
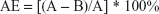
where A is the total amount (mass) of the peptide (AP114 or AP138), and B is the mass of the nonadsorbed peptide.
|
(2) |
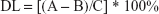
where C is the total weight of all components of the nanocarriers (the associated peptide and the mass of surfactants and oil used for the preparation of the LNCs).
Quantification of peptides by high-performance liquid chromatography
The AP114 and AP138 contents were analyzed using a high-performance liquid chromatography system described previously.22 Briefly, standard solutions of the APIs (0.1–1,000 μg/mL) were prepared in deionized water, and 20 μL of the standard or sample was injected into the SymmetryShield™ RP18 5 μm 4.6×250 mm column (Waters, USA). A flow rate of 1.0 mL/min was employed using mobile phase A composed of 0.1% trifluoroacetic acid (TFA) in water and mobile phase B composed of 0.1% TFA in acetonitrile. A linear gradient was run: 20% B for 25 minutes, 45% B for 0.1 minute, 100% B for 4.9 minutes, 20% B for 0.1 minute, and 20% B for 14.9 minutes. The API peaks had retention times of ~12 minutes for AP114 and 13 minutes for AP138, respectively. The UV detection was performed at 200 nm. Data collection and integration were accomplished using Empower®3 software.
The method showed good linearity for both molecules, AP114 and AP138 (R2≥0.9999). The quantification limits were 0.5 μg/mL for AP114 and 1 μg/mL for AP138, whereas the detection limits were 0.25 and 0.5 μg/mL for AP114 and AP138, respectively. More than 90% of peptides were recovered each time.
Microorganisms
The strains used in this study were seven methicillin-resistant clinical isolates of S. aureus, six methicillin-susceptible clinical isolates, and one reference strain of S. aureus (ATCC 25923).
Preparation of inoculum
One to two colonies were taken directly from the Columbia agar plate into 2 mL of 0.85% NaCl solution, and the density of the microorganism suspension was adjusted to 1.1 McFarland. The bacterial suspensions were then further diluted 100 times with BHI medium.
MIC determination
MIC was determined via a broth microdilution method, as described previously18,23 with slight modifications. Serial two-fold dilutions of the samples in BHI were prepared in order to obtain the desired concentration range. An amount of 50 μL of a bacterial suspension in BHI broth was added into each well of a sterile 96-well plate already containing 50 μL of the tested sample or control. The positive control wells contained BHI and the bacterial suspension without the tested sample, whereas the negative control wells contained BHI and the tested sample without the bacterial suspension. The samples were incubated for 24 hours at 37°C. The MIC was the lowest concentration that completely inhibited the growth of the bacteria as detected by the unaided eye.
Checkerboard titration
Combinations of ML-LNCs and plectasin derivatives were tested via a checkerboard titration method. Two-fold dilutions of ML-LNCs and peptide were prepared before mixing. The concentration range of each antimicrobial agent in combination ranged from 1/32× MIC to 4× MIC. The fractional inhibitory concentration (FIC) index was calculated using the following equation:
|
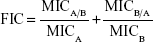
where MICA is the MIC of the compound A alone, MICA/B is the MIC of compound A in combination, and MICB and MICB/A are the MIC of the compound B alone and in combination, respectively.
Synergy was defined as an FIC index of ≤0.5, additivity/indifference as an FIC index of >5 but of ≤4. Antagonism was defined as an FIC index of >4.24
Time-kill assay
Samples were diluted with BHI medium to a volume of 1.98 mL, and 20 μL of bacterial suspension (see “Preparation of inoculum” section) was added into each tube. Samples were then incubated at 37°C. An amount of 100 μL was withdrawn from each tube after 0, 3, 6, 18, and 24 hours and serial 100-fold dilutions were prepared in distilled water. A total of 100 μL of the diluted and/or undiluted sample were delivered onto the surface of the agar, spread, and allowed to be absorbed into the agar. After overnight incubation of the agar plates at 37°C, the colonies were counted.
Transmission electron microscopy
Twenty microliters of inoculum was added to 0.98 mL of BHI broth and incubated for 10 hours at 37°C. One milliliter of sample dispersed in BHI or BHI on its own (control) was then added followed by incubation for 1 hour. The bacteria were collected by centrifugation (10,000× g, 5 minutes) and suspended in 0.1 M phosphate-buffered saline (PBS) (pH =7.4). After centrifugation, the bacteria were fixed with 2% paraformaldehyde and 2% glutaraldehyde in 0.1 M PBS (pH =7.4) and left overnight under vacuum. The bacteria were centrifuged, suspended in PBS, centrifuged, resuspended in water, centrifuged, resuspended in 1% osmium tetroxide in water, and incubated for 1 hour at room temperature. Samples were washed and dehydrated with graded ethanol solutions (twice with deionized water for 10 minutes, 50% ethanol for 20 minutes, 70% ethanol for 20 minutes, 95% ethanol for 20 minutes, and three times with 100% ethanol for 30 minutes). Samples were then embedded in Epon 812 resin, which was then left to polymerize for a few days. Thin slices (~60 nm) were cut from each sample using Leica UC7 ultramicrotome and deposited onto copper grids. The samples were stained with 3% uranyl acetate in 50% ethanol for 15 minutes and washed with deionized water. The samples were examined using JEOL JEM-1400 electron microscope.
Statistical analysis
The statistical significance of the differences between samples was determined using one-way analysis of variance. Differences were considered significant at P<0.05.
Results and discussion
Properties of ML-LNCs and peptide-loaded ML-LNCs
LNCs are composed of an oily core of capric/caprylic acid triglycerides that is surrounded by a shelf composed of a hydrophilic surfactant, for example, macrogol 15 hydroxystearate. Although it is possible to obtain LNCs using solely macrogol 15 hydroxystearate as a surfactant, usually a cosurfactant is used to increase the LNC stability and decrease their size. The most common cosurfactant is lecithin.18–20,25 The macrogol 15 hydroxystearate/triglycerides mass mixing ratio (MMR) of 0.82 was selected as this is the ratio present in the most commonly described LNCs.18,21 A previous study has shown that it is the monolaurin that makes the LNCs active against Gram-positive bacteria. It is important to use a high concentration of monolaurin to reduce the amount of the nanocarrier required to achieve a desired effect. LNCs composed of monolaurin (15.0%), macrogol 15 hydroxystearate (38.4%), and triglycerides (46.6%) were characterized by a hydrodynamic diameter of 36±0.3 nm and homogeneous size distribution (polydispersity index below 0.1). The zeta potential of ML-LNCs was close to neutral (−4.77±2.19).18 This is not surprising, as ML-LNCs do not contain ionizable molecules. The incorporation of monolaurin resulted in an important decrease in phase inversion temperature from 80 to 85°C (LNCs composed solely of macrogol 15 hydroxystearate and triglycerides or lecithin-LNCs) to 37°C (ML-LNCs). The concentration of surfactants and oil in ML-LNCs at 200 mg/mL was sufficiently high for the system to undergo phase inversion on heating. Because monolaurin is not soluble in water, the use of LNCs offers the advantage of obtaining pharmaceutically acceptable dosage form without using organic solvents or cosolvents.
The shape of the flow curves (Figure 2) indicates that ML-LNC dispersion at a concentration of 200 mg/mL exhibits a shear thickening flow. In this non-Newtonian flow phenomenon, shear viscosity decreases (about 10-fold) as the shear rate is increased (about 100-fold). The non-Newtonian behavior can be observed in concentrated particle systems because of asymmetrical microstructure caused by particle–particle flow field interactions.26
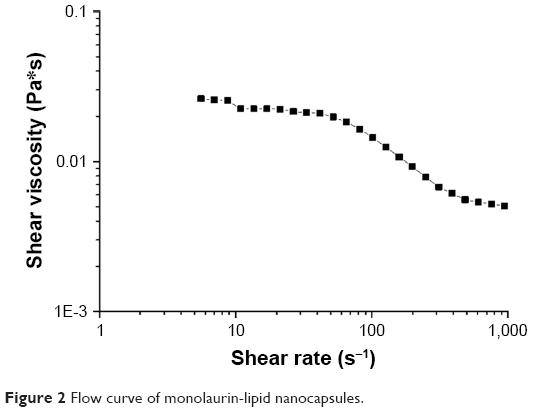 | Figure 2 Flow curve of monolaurin-lipid nanocapsules. |
In addition to covalent attachment and encapsulation within the nanocarrier, surface adsorption is one approach for incorporating drug molecules into drug delivery systems.27 Due to their small size and large surface area, nanocarriers may interact with biomolecules such as peptides. Such interaction is a multifactorial process that depends on the properties of both, the peptide and the nanocarrier, and also on experimental conditions such as concentration and the medium.22,28 Both plectasin derivatives, AP114 and AP138, are peptides composed of 40 amino acids with very similar amino acid sequence and molecular weight (Table 1). They differ only by 3 amino acids (positions 9, 17, and 26 from the amino end). The net charge at pH 7 is +3 and +4, and the theoretical isoelectric points are 8.62 and 8.87 for AP114 and AP138, respectively. The cationic property of the antimicrobial peptides is very important, because the electrostatic forces between the positively charged peptide and the negatively charged bacterial surface are critical determinants for their interactions, which is a key factor for antibacterial activity whether the membrane itself is targeted or when an intracellular target must be reached by means of translocation.29
 | Table 1 Structure and properties of antimicrobial peptides |
It is very unlikely that the antimicrobial peptides showing good solubility in water, for example, plectasin derivatives, would penetrate into the oily core of the LNCs. Instead, they are expected to be localized within or on the particle shell.22 The adsorption of the peptide did not cause an important change in the properties of the LNCs (Table 2). The size distribution remained unaffected, and the increase in the nanocapsule diameter, although statistically significant at the lowest LNC/peptide MMR, can be considered as negligible, because the difference in particle size was considerably small (<1 nm). It is in agreement with previously reported data that the adsorption of peptide onto LNCs without ionizable surfactants did not change the properties of the nanocarrier.22 The adsorption yield ranged from 34% to 62%. The percentage of adsorbed peptide increased corresponding to an increasing LNC/peptide MMR. AP114 and AP138 showed comparable adsorption yield, which can likely be attributed to their similar composition and physical properties. Although the interactions between the peptides and the nanocarriers are governed mainly by electrostatic forces,30,31 it is possible to achieve significant adsorption of peptide onto LNCs even without the presence of an ionizable surfactant because of the enormous surface area of the nanocarriers and of the peptide–carrier interactions (such as dipol–ion interactions between the cationic groups of peptides and PEG dipoles on macrogol 15 hydroxystearate molecules).22 A peptide loading of 1.5% or even 2.7% can be considered as high for lipidic nanocarriers – they are usually characterized by smaller loading of molecules than the nanocarriers made of hydrophilic polymers encapsulating the drug based on electrostatic interactions.30,31 The DL is a key parameter when developing an effective nanoparticulate delivery system, because low loading may limit the use of nanoparticulate systems because a substantial amount of the formulation must be administered to achieve the therapeutic effect.32
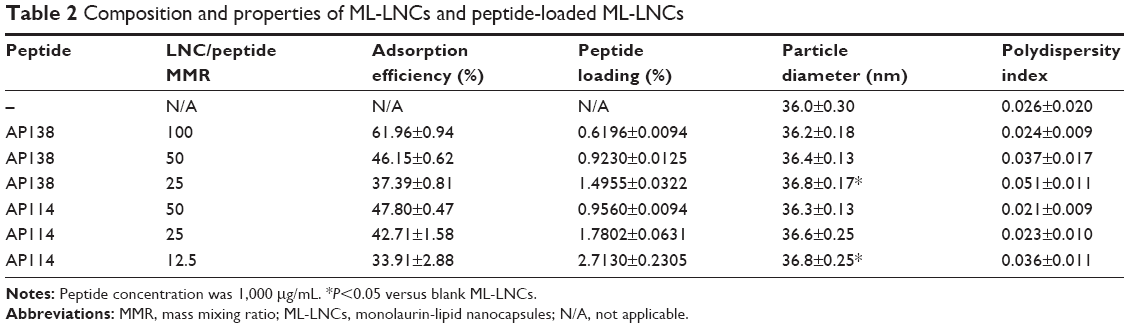 | Table 2 Composition and properties of ML-LNCs and peptide-loaded ML-LNCs |
Antibacterial properties of ML-LNCs
The MICs of ML-LNCs against different strains of S. aureus are shown in Table 3. The MICs of ML-LNCs were 100 or 200 or 400 μg/mL depending on the strain (corresponding to 15, 30, and 60 μg/mL of ML, respectively). MSSA strains seemed to be slightly more sensitive to ML-LNCs – five strains were inhibited by ML-LNCs at 100 μg/mL (15 μg/mL of ML), whereas among MRSA strains only one strain was inhibited by 100 μg/mL of ML-LNCs; most of the strains were inhibited by ML-LNCs at 200 μg/mL (30 μg/mL). The highest MIC (400 μg/mL of ML-LNCs, which contains 60 μg/mL of monolaurin) was observed for MSSA.
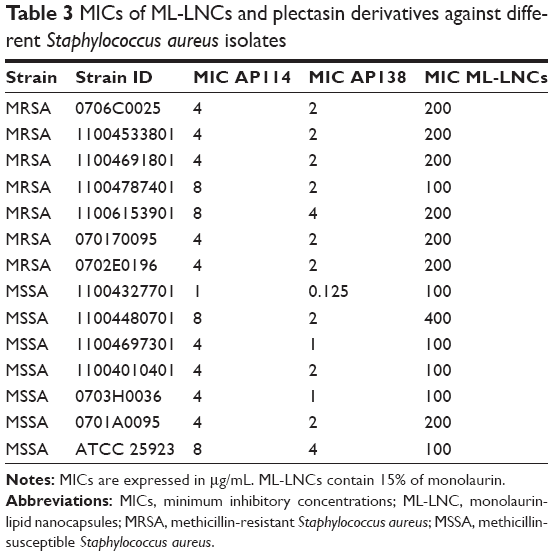 | Table 3 MICs of ML-LNCs and plectasin derivatives against different Staphylococcus aureus isolates |
Time-kill curves revealed that ML-LNCs were bactericidal (Figure 3). In the absence of ML-LNCs, bacterial counts reached their maximum 18–24 hours after inoculation. The killing was time dependent rather than concentration dependent. The killing occurred slowly – even at such a high concentration as 50 mg/mL (500× MIC) the reduction in the colony-forming unit (CFU) number after 3 hours was relatively small (~2 Log). This is in good agreement with the previously reported data.18
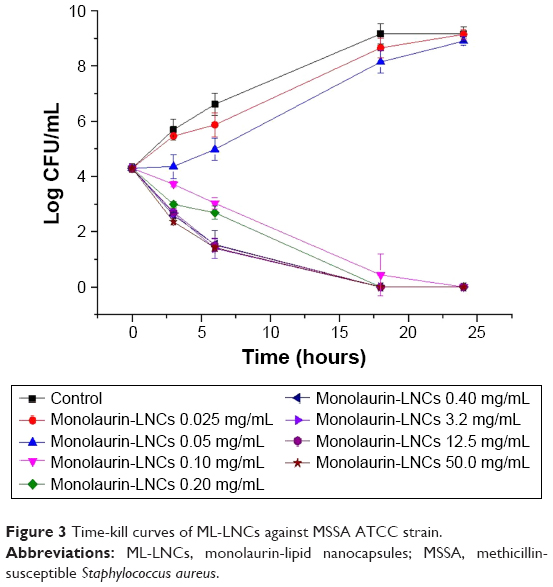 | Figure 3 Time-kill curves of ML-LNCs against MSSA ATCC strain. |
Because ML-LNCs were characterized by low killing rates even at very high concentrations (500× MIC), it can be concluded that they do not solubilize the bacterial membrane as do other detergents, including monoglycerides or fatty acids with shorter chains.18 It is very likely that monolaurin decreases the fluidity of the membrane, because it has a melting point of 63°C and, therefore, is solid at 37°C.18 This may modulate the activity of membrane proteins thereby playing an important role in cellular functions and homeostasis.33 Monolaurin has been shown to inhibit the production of toxic shock syndrome toxin 1 and staphylococcal enterotoxins. The production of exotoxins by S. aureus was inhibited at monolaurin concentration that did not inhibit bacterial growth.14,17,34,35 Monolaurin has 12 carbon molecules in its fatty acid backbone chain, which makes it span exactly one-half the width of a lipid bilayer.36 Interestingly, it has been reported that the optimum chain length of fatty acids and their corresponding monoglycerides to achieve the maximum antibacterial activity is 12 carbons.18,37,38 Because of its size and lipophilic nature, it has been postulated that glycerol monolaurate is inserted into cytoplasmic membrane and exerts its antibacterial effect by interfering with many transmembrane signal transduction systems.14,39
The influence of ML-LNCs on S. aureus morphology was studied by transmission electron microscopy (Figure 4). TE micrographs of control (untreated bacteria – Figure 4A) showed proliferating cells that were uniformly shaped with intact cell walls and well-defined cell membranes. In ML-LNC-treated cells, disappearance of plasma membrane was observed (Figure 4B). ML-LNCs also exerted the effects on the cell wall – in some cells, the disappearance of cell wall was observed. This is in agreement with the effects of monocaprin on group B streptococci observed by Bergsson et al.40 Interestingly, ML-LNCs affected the shape of bacteria – characteristic concave deformations were observed in numerous cells. Some lysed cells were observed with the leakage of cytoplasm contents. Proliferating cells were observed, therefore suggesting that ML-LNCs do not stop cell division, but numerous abnormalities described above occurred during cell division.
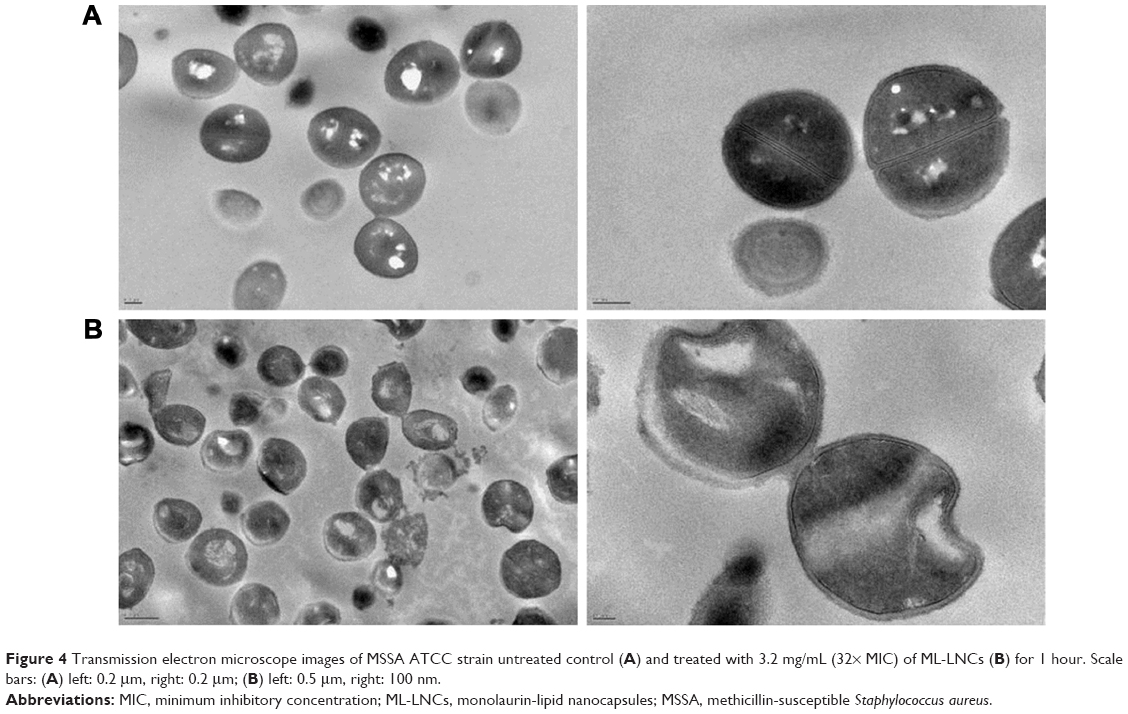 | Figure 4 Transmission electron microscope images of MSSA ATCC strain untreated control (A) and treated with 3.2 mg/mL (32× MIC) of ML-LNCs (B) for 1 hour. Scale bars: (A) left: 0.2 μm, right: 0.2 μm; (B) left: 0.5 μm, right: 100 nm. |
Antibacterial activity of plectasin derivatives
The MICs of plectasin derivatives against 14 studied strains are shown in Table 3. The MICs of AP114 varied eightfold (range 1–8 μg/mL), whereas those of AP138 varied 16-fold (range 0.125–4 μg/mL). One of the MSSA strains was very sensitive to plectasin derivatives (MICs of 0.125 and 1 μg/mL for AP138 and AP114, respectively). The existence of S. aureus mutants that are more sensitive to host defense peptides such as plectasin has been described previously.41 It can be concluded that AP138 was more effective than AP114. Both plectasin derivatives effectively inhibited all tested strains of S. aureus, independent of their resistance pattern. This is in good agreement with the previously reported data indicating that the antibacterial spectrum of NZ2114 or AP138 includes staphylococci such as MRSA.7,9,13,42
Time-kill assay of plectasin derivatives was performed to determine the effect of an increasing peptide concentration on the extent of bacteria killing. Time-kill curves of ATCC 25293 (MSSA) exposed to AP114 are shown in Figure 5A. In the presence of 1 or 2 μg/mL of AP114 (0.125× or 0.25× MIC, respectively), the bacterial counts grew more slowly than with the control. At 4 μg/mL (0.5× MIC), an evident growth inhibition was observed – the effect can be considered as bacteriostatic; however, after 18–24 hours of incubation, large standard deviations were observed and it was possible to observe visually slight bacterial growth. At 8 μg/mL of AP114 (1× MIC), a reduction by ~1.6 Log in the number of CFU was observed after 3 hours, 2.5 Log after 6 hours, and 4.3 Log after 18 hours. At 16 μg/mL of AP114 (2× MIC), a dramatic reduction of bacterial density by 3.3 Log was observed after 3 hours, and the bactericidal effect was maintained for at least 24 hours. Time-kill curves of ATCC 25293 (MSSA) exposed to AP138 are shown in Figure 5B. At 1 μg/mL of AP138 (0.25× MIC), the bacterial count grew more slowly than that of control. At 2 μg/mL (0.5× MIC), an inhibition of bacterial growth was observed; the effect can be considered as bacteriostatic with the increase in the number of CFU by 1 Log compared with the initial inoculum. At 4 μg/mL (1× MIC), a gradual decrease in the CFU number by 2.3 was observed within 24 hours – at this concentration, the action can be considered as bacteriostatic. At 8 and 16 μg/mL (2× and 4× MIC, respectively), a reduction by 1.5 Log was observed after 3 hours, by 2.3–2.7 Log after 6 hours, and the bactericidal effect was reached after 18 hours. Time-kill results confirmed that AP138 was more potent than AP114. However, AP114 killed bacteria more rapidly than AP138. At 16 μg/mL, the bactericidal effect, that is, a reduction of more than 3 Log in CFU number compared with the starting inoculum, was observed after 3 hours for AP114, whereas AP138 was not bactericidal even after 6 hours of incubation.
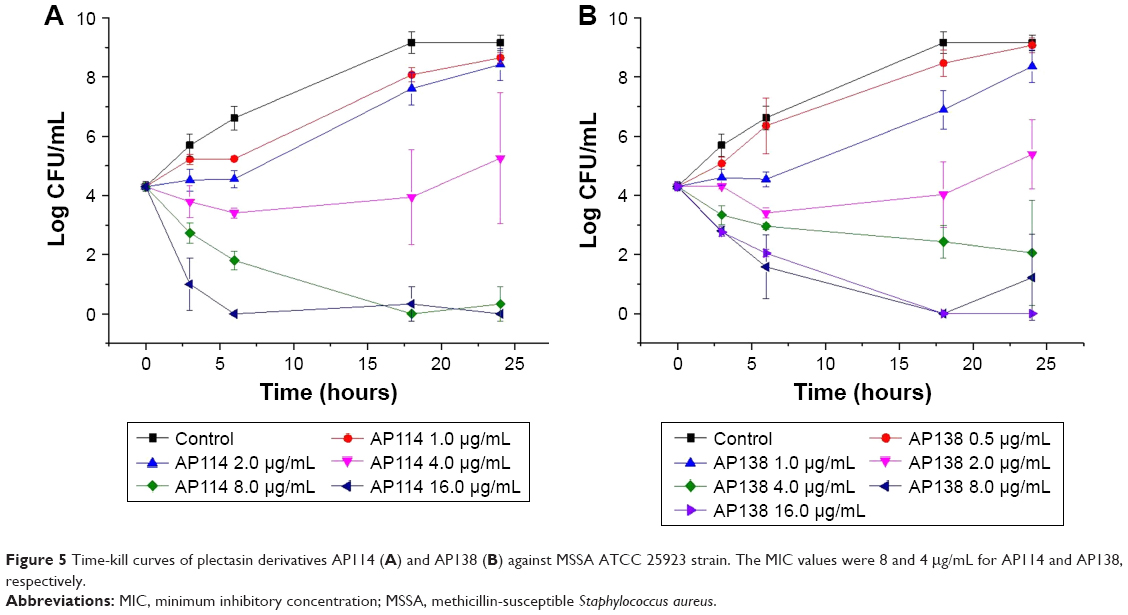 | Figure 5 Time-kill curves of plectasin derivatives AP114 (A) and AP138 (B) against MSSA ATCC 25923 strain. The MIC values were 8 and 4 μg/mL for AP114 and AP138, respectively. |
Although many antimicrobial peptides act on and disintegrate bacterial cytoplasmic membrane, this is not the case for plectasin. In spite of its amphipathic structure, this peptide does not compromise membrane integrity, thereby reducing the risk of unspecific toxicity.7 Indeed, AP114 (NZ2114) showed very low hemolytic activity (<0.1% of hemolysis at 128 μg/mL).42 Another study has confirmed that plectasin is neither hemolytic for human erythrocytes (half maximal effective concentration [EC50] >400 μg/mL) nor cytotoxic for human epidermal keratinocytes (EC50 >400 μg/mL) or for murine L929 fibroblasts (EC50 >400 μg/mL).6 Plectasin and its derivatives exhibit their antibacterial activity by interfering with cell wall biosynthesis. These peptides directly bind to bacterial cell wall precursor lipid II forming an equimolar stoichiometric complex.7 It has been demonstrated that AP114 shows bactericidal mode of action, with the MBC/MIC ratios ranging from 1 to 2 for either MSSA or MRSA strains.41,42 These results are in a good agreement with our time-kill data. Time-kill studies of plectasin performed by Schneider et al7 demonstrated that the time-kill behavior of plectasin was similar to that of agents interfering with cell wall synthesis (such as vancomycin or penicillin). Plectasin did not exhibit similar time-kill profile characteristic as the rapidly lytic membrane-active agents such as polymyxin B.7
Figure 6 shows influence of AP114 (Figure 6A) and AP138 (Figure 6B) on the morphology of S. aureus. Incubation with plectasin derivatives resulted in a destruction of cell walls. It was possible to observe cell walls that have separated from the cells, disappearance of fragments of cell membrane, and the leakage of cytoplasmic contents. Shape deformations and abnormalities in shape division were also observed. These observations are in agreement with the fact that plectasin derivatives exert their antibacterial effect by interfering with cell wall synthesis.
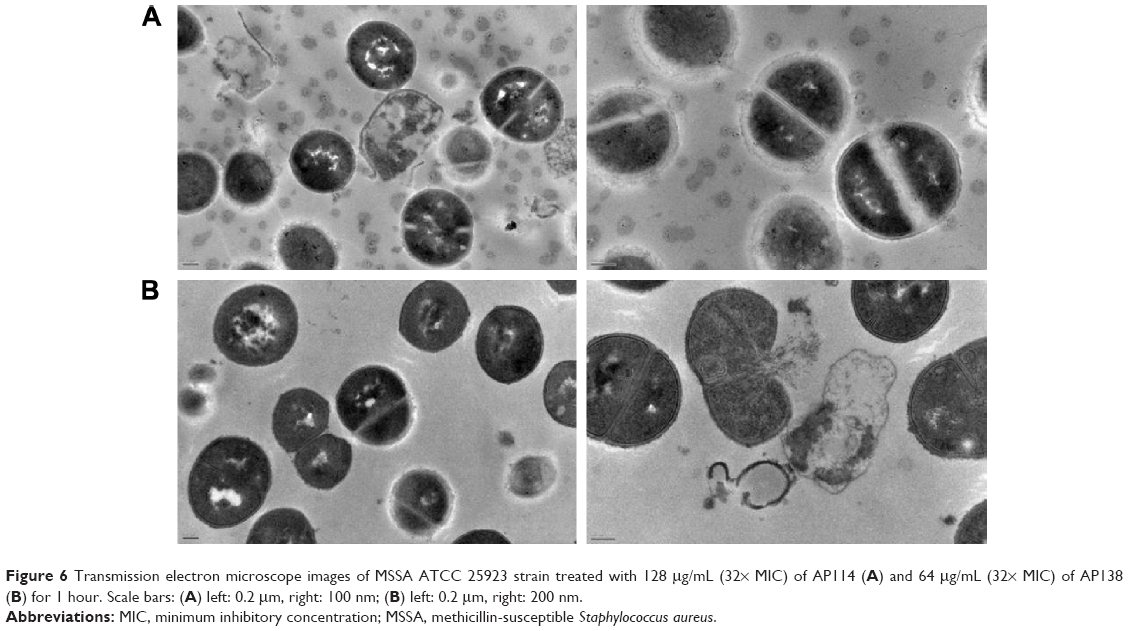 | Figure 6 Transmission electron microscope images of MSSA ATCC 25923 strain treated with 128 μg/mL (32× MIC) of AP114 (A) and 64 μg/mL (32× MIC) of AP138 (B) for 1 hour. Scale bars: (A) left: 0.2 μm, right: 100 nm; (B) left: 0.2 μm, right: 200 nm. |
Synergistic interactions between plectasin derivatives and ML-LNCs
Combinations containing a plectasin derivative and the ML-LNCs were tested on one MSSA reference strain and one MRSA strain to evaluate whether they can still enhance SA killing. The efficacies of these combinations were assessed using checkerboard method and time-kill assay. The antibacterial activity of peptide-loaded ML-LNCs was the same as the activity of combinations containing the same concentrations of ML-LNCs and a plectasin derivative that were prepared by mixing of the components in the 96-well plate just before the addition of bacteria (not shown). In the checkerboard method, the values of FIC index were equal to 0.375 or 0.5, indicating synergistic interactions between each plectasin derivative (AP114 or AP138) and ML-LNCs against both MSSA and MRSA strains (Table 4). By using the combinations, it was possible to reduce the concentrations of both, peptide and LNCs, fourfold.
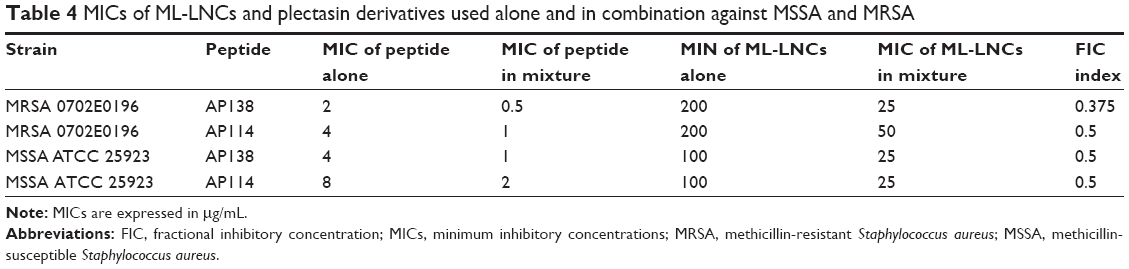 | Table 4 MICs of ML-LNCs and plectasin derivatives used alone and in combination against MSSA and MRSA |
Time-kill study showed that none of the ingredients used alone was capable of effectively inhibiting bacterial growth at tested concentrations (Figure 7). The combination of ML-LNCs at 25 μg/mL and AP114 at 1 μg/mL can be considered as synergistic (the CFU number was ~3 Log smaller than that of each of ingredients alone), but after 24 hours an increase in CFU number by 1.2 Log was observed compared with the initial inoculum (Figure 7A). An increase in the concentration of either AP114 or ML-LNCs or both of them yielded synergistic and bactericidal combinations. The reduction in CFU number was gradual (after 6 hours only the combination containing AP114 2 μg/mL and ML-LNCs 50 μg/mL proved to be bactericidal). The combinations showed at least an 8 Log reduction in the CFU number compared with the ingredients alone after 24 hours of incubation.
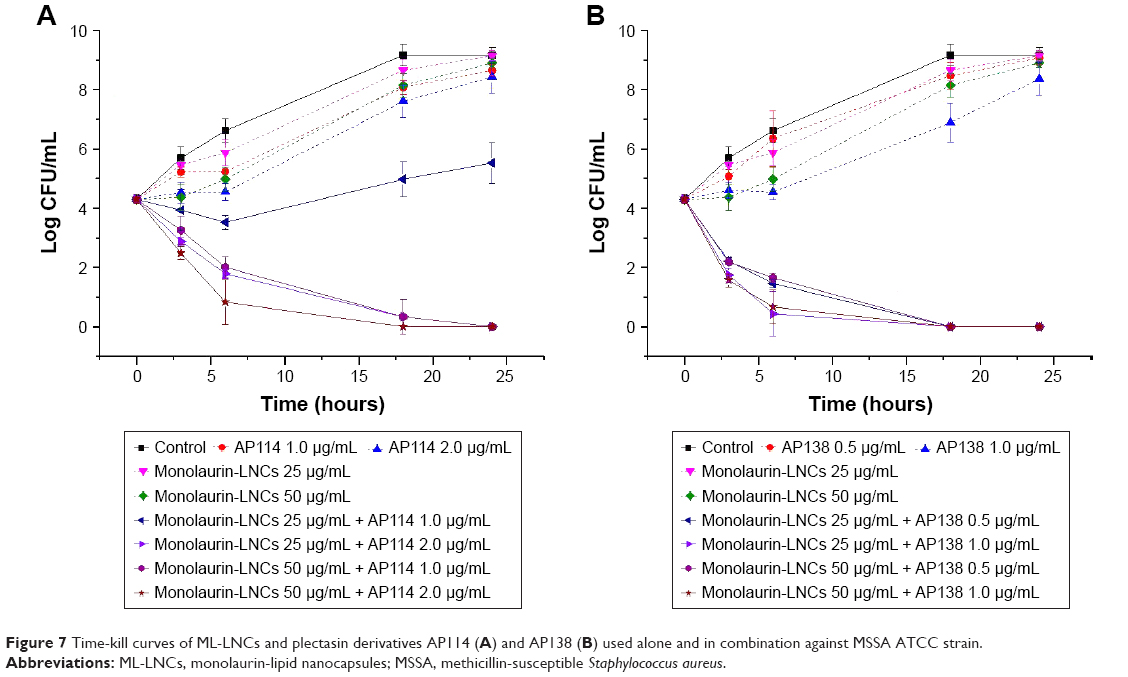 | Figure 7 Time-kill curves of ML-LNCs and plectasin derivatives AP114 (A) and AP138 (B) used alone and in combination against MSSA ATCC strain. |
All tested combinations containing AP138 and ML-LNCs showed synergistic effects (Figure 7B). Both samples containing 0.5 μg/mL AP138 reduced the CFU number by 2.1 Log after 3 hours and 2.6–2.8 Log after 6 hours. The samples containing more AP138 (1 μg/mL) showed larger reductions in CFU number: by 2.6–2.7 Log after 3 hours and after 6 hours they showed bactericidal effect reducing CFU number by 3.6–3.9 Log. All combinations showed bactericidal effect after 18 and 24 hours with at least an 8 Log reduction in the CFU number compared with the ingredients alone.
Figure 8 shows influence of synergistic combinations containing ML-LNCs and AP114 (Figure 8A) or AP138 (Figure 8B) on S. aureus morphology. The changes described for ML-LNCs and the plectasin derivatives were also observed in their combinations. These include deformations in shape of bacterial cells, disappearance of cell walls and cell membranes, fragments of cell walls that have separated from bacterial cells, and leakage of cytoplasmic contents from the cells.
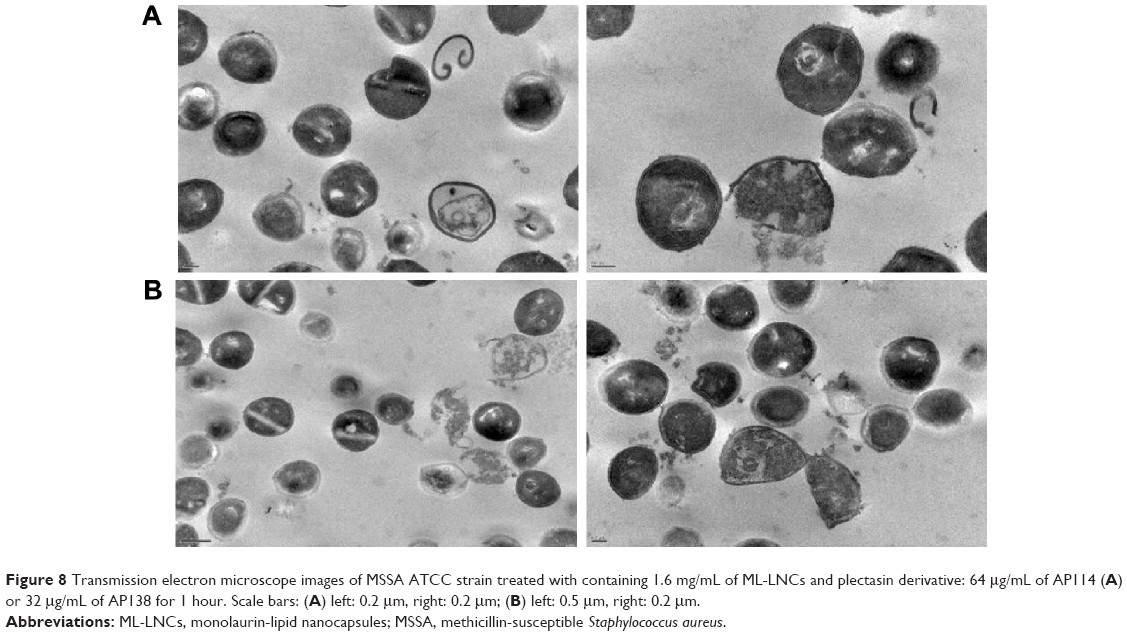 | Figure 8 Transmission electron microscope images of MSSA ATCC strain treated with containing 1.6 mg/mL of ML-LNCs and plectasin derivative: 64 μg/mL of AP114 (A) or 32 μg/mL of AP138 for 1 hour. Scale bars: (A) left: 0.2 μm, right: 0.2 μm; (B) left: 0.5 μm, right: 0.2 μm. |
Both plectasin derivatives have been shown to act in synergy with ML-LNCs against S. aureus. A synergistic effect can result from a variety of mechanisms. Synergistic antibacterial effect is often observed when between the compounds that attack different targets. A possible explanation of synergistic effect between ML-LNCs and plectasin derivatives is that the peptides damage cell wall sufficiently to enhance the entry of ML and/or ML-LNCs. Indeed, inhibitors of cell wall synthesis, for example, penicillin, show synergistic interactions with some antibiotics (eg, gentamicin) against enterococci by facilitating their entry as a result of cell wall damage.1
Antimicrobial peptides represent one of the most promising and innovative groups of anti-infective agents.6,29,41 The in vitro emergence of resistance to antimicrobial peptides is less common and less rapid than that observed for conventional antibiotics.43,44 More recently, however, a number of resistance mechanisms against antimicrobial peptides have been discovered and investigated in bacteria. The examples of such mechanisms include the release of glycosaminoglycans and other polyanionic molecules to scavenge positively charged peptides or membrane modification resulting in a decrease of negative surface potential of bacterial membranes.45–47 The latter mechanisms have been described for S. aureus.48 On the other hand, it has been demonstrated that the resistance to monolaurin does not develop even after a year of passaging of S. aureus on media containing a subinhibitory concentration of this compound.14,17 Moreover, it has been shown that glycerol monolaurate inhibits the induction of resistance to vancomycin in Enterococcus faecalis.49 In conclusion, combined administration of ML-LNCs and plectasin derivatives could markedly decrease the probability of development of resistance to the latter. Therefore, ML-LNCs can be considered as a very promising carrier for plectasin derivatives and possibly for other antimicrobial peptides.
Conclusion
Several interesting findings emerged from the present study. First, plectasin derivatives were adsorbed on the LNCs with the efficiency between 34% and 62% without having an important impact on the nanocarrier properties such as size. Second, when used alone, ML-LNCs and both plectasin derivatives were effective against all tested MSSA and MRSA strains with the MIC values of 100–400 μg/mL and 0.125–8 μg/mL for the nanocarriers and peptides, respectively. Both the peptides and the ML-LNCs showed bactericidal activity. Third, when used in combination, synergistic effect was observed between plectasin derivatives and ML-LNCs as confirmed by both, checkerboard and time-kill assay. This enables the dose reduction and consequently decreases the risk of toxic effects and minimizes the possibility of resistance development. Together, these results suggest that ML-LNCs loaded with a plectasin derivative may be a very promising drug delivery system for further development as a novel antibacterial agent against MRSA and MSSA.
Acknowledgments
The research leading to these results received funding from the European Union’s Seventh Framework Program (FP7/2007–2013) under grant agreement no 604182 (http://ec.europa.eu.research). It was carried out within the project FORMAMP-Innovative Nanoformulation of Antimicrobial Peptides to Treat Bacterial Infectious Diseases.
Disclosure
The authors report no conflicts of interest in this work.
References
Levinson WE. Review of Medical Microbiology and Immunology. 13th ed. Copenhagen, Denmark: The McGraw-Hill Companies; 2014. | ||
Fitzgerald JR, Sturdevant DE, Mackle SM, Gill SR, Musser JM. Evolutionary genomics of Staphylococcus aureus: insights into the origin of methicillin-resistant strains and the toxic shock syndrome epidemic. Proc Natl Acad Sci U S A. 2001;98(15):8821–8826. | ||
Gordon RJ, Lowy FD. Pathogenesis of methicillin-resistant Staphylococcus aureus infection. Clin Infect Dis. 2008;46(Suppl 5):S350–S359. | ||
Jensen SO, Lyon BR. Genetics of antimicrobial resistance in Staphylococcus aureus. Future Microbiol. 2009;4(5):565–582. | ||
Hardy KJ, Hawkey PM, Gao FO, Oppenheim BA. Methicillin resistant Staphylococcus aureus in the critically ill. Br J Anaesth. 2004;92(1):121–130. | ||
Mygind PH, Fischer RL, Schnorr KM, et al. Plectasin is a peptide antibiotic with therapeutic potential from a saprophytic fungus. Nature. 2005;473(7061):975–980. | ||
Schneider T, Kruse T, Wimmer R, et al. Plectasin, a fungal defensin, targets the bacterial cell wall precursor Lipid II. Science. 2010;328(5982):1168–1172. | ||
Xiong YQ, Hady WA, Deslandes A, et al. Efficacy of NZ2114, a novel plectasin-derived cationic antimicrobial peptide antibiotic, in experimental endocarditis due to methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2011;55(11):5325–5330. | ||
Andes D, Craig W, Nielsen LA, Kristensen HH. In vivo pharmacodynamic characterization of a novel plectasin antibiotic, NZ2114, in a murine infection model. Antimicrob Agents Chemother. 2009;53(7):3003–3009. | ||
Ostergaard C, Sandvang D, Frimodt-Møller N, Kristensen HH. High cerebrospinal fluid (CSF) penetration and potent bactericidal activity in CSF of NZ2114, a novel plectasin variant, during experimental pneumococcal meningitis. Antimicrob Agents Chemother. 2009;53(4):1581–1585. | ||
Brinch KS, Frimodt-Møller N, Høiby N, Kristensen HH. Influence of antidrug antibodies on plectasin efficacy and pharmacokinetics. Antimicrob Agents Chemother. 2009;53(11):4794–4800. | ||
Brinch KS, Tulkens PM, Van Bambeke F, Frimodt-Møller N, Høiby N, Kristensen HH. Intracellular activity of the peptide antibiotic NZ2114: studies with Staphylococcus aureus and human THP-1 monocytes, and comparison with daptomycin and vancomycin. J Antimicrob Chemother. 2010;65(8):1720–1724. | ||
Lociuro S, Neve S, Kjærulf S, Nordkild P. AP138, a second generation plectasin, shows good bactericidal properties and long post-antibiotic effect (PAE) in ATCC29213. 25th ECCMID. Eur Cong Clin Microbiol Infect Dis. 2015. | ||
Schlievert PM, Peterson ML. Glycerol monolaurate antibacterial activity in broth and biofilm cultures. PLoS One. 2012;7:e40350. | ||
Preuss HG, Echard B, Dadgar A, et al. Effects of essential oils and monolaurin on Staphylococcus aureus: in vitro and in vivo studies. Toxicol Mech Method. 2005;15(4):279–285. | ||
Hess DJ, Henry-Stanley MJ, Wells CL. Antibacterial synergy of glycerol monolaurate and aminoglycosides in Staphylococcus aureus biofilms. Antimicrob Agents Chemother. 2014;58(11):6970–6973. | ||
Mueller EA, Schlievert PM. Non-aqueous glycerol monolaurate gel exhibits antibacterial and anti-biofilm activity against Gram-positive and Gram-negative pathogens. PLoS One. 2015;10:e0120280. | ||
Umerska A, Cassisa V, Matougui N, Joly-Guillou ML, Eveillard M, Saulnier P. Antibacterial action of lipid nanocapsules containing fatty acids or monoglycerides as co-surfactants. Eur J Pharm Biopharm. 2016;108:100–110. | ||
Matougui N, Boge L, Groo AC, et al. Lipid-based nanoformulations for peptide delivery. Int J Pharm. 2016;502(1–2):80–97. | ||
Umerska A, Mouzouvi CR, Bigot A, Saulnier P. Formulation and nebulization of fluticasone propionate-loaded lipid nanocarriers. Int J Pharm. 2015;493(1–2):224–232. | ||
Valcourt C, Saulnier P, Umerska A, et al. Synergistic interactions between doxycycline and terpenic components of essential oils encapsulated within lipid nanocapsules against gram negative bacteria. Int J Pharm. 2016;498(1–2):23–31. | ||
Umerska A, Matougui N, Groo AC, Saulnier P. Understanding the adsorption of salmon calcitonin, antimicrobial peptide AP114 and polymyxin B onto lipid nanocpasules. Int J Pharm. 2016;506(1–2):191–200. | ||
Boge L, Bysell H, Ringstad L, et al. Lipid-based liquid crystals as carriers for antimicrobial peptides: phase behavior and antimicrobial effect. Langmuir. 2016;32(17):4217–4228. | ||
White RL, Burgess DS, Manduru M, Bosso JA. Comparison of three different in vitro methods of detecting synergy: time-kill, checkerboard, and E test. Antimicrob Agents Chemother. 1996;40(8):1914–1918. | ||
Minkov I, Ivanova Tz, Panaiotov I, Proust J, Saulnier P. Reorganization of lipid nanocapsules at air–water interface. I. Kinetics of surface film formation. Colloids Surf B Biointerfaces. 2005;45(1):14–23. | ||
Rampall I, Smart JR, Leighton DT. The influence of surface roughness on the particle-pair distribution function of dilute suspensions of non-colloidal spheres in simple shear flow. J Fluid Mech. 1997;339:1–24. | ||
Hartig SM, Greene RR, DasGupta J, et al. Multifunctional nanoparticulate polyelectrolyte complexes. Pharm Res. 2007;24(12):2353–2369. | ||
Saptarshi SR, Duschi A, Lopata AL. Interactions of nanoparticles with proteins: relation to bio-reactivity of the nanoparticle. J Nanobiotechnol. 2013;11:26–38. | ||
Mahlapuu M, Håkansson J, Ringstad L, Björn C. Antimicrobial peptides: an emerging category of therapeutic agents. Front Cell Infect Microbiol. 2016;6:194. | ||
Umerska A, Paluch KJ, Santos-Martinez MJ, Medina C, Corrigan OI, Tajber L. Chondroitin-based nanoplexes as peptide delivery systems – investigations into the self-assembly process, solid-state and extended release characteristics. Eur J Pharm Biopharm. 2015;93:242–253. | ||
Umerska A, Corrigan OI, Tajber L. Design of chondroitin sulfate-based polyelectrolyte nanoplexes: Formation of nanocarriers with chitosan and a case study of salmon calcitonin. Carbohydr Polym. 2017;156:276–284. | ||
Umerska A, Corrigan OI, Tajber L. Intermolecular interactions between salmon calcitonin, hyaluronate, and chitosan and their impact on the process of formation and properties of peptide-loaded nanoparticles. Int J Pharm. 2014;477(1–2):102–112. | ||
Sikkema J, de Bont JA, Poolman B. Mechanisms of membrane toxicity of hydrocarbons. Microbiol Rev. 1995;59(2):201–222. | ||
Schlievert PM, Deringer JR, Kim MH, Projan SJ, Novick RP. Effect of glycerol monolaurate on bacterial growth and toxin production. Antimicrob Agents Chemother. 1992;36(3):626–631. | ||
Lin YC, Schlievert PM, Anderson MJ, et al. Glycerol monolaurate and dodecylglycerol effects on Staphylococcus aureus and toxic shock syndrome toxin-1 in vitro and in vivo. PLoS One. 2009;4(10):e7499. | ||
Vetter SM, Schlievert PM. Glycerol monolaurate inhibits virulence factor production in Bacillus anthracis. Antimicrob Agents Chemother. 2005;49(4):1302–1305. | ||
Kabara JJ, Vrable R. Antimicrobial lipids: natural and synthetic acids and monoglycerides. Lipids. 1977;12(9):753–759. | ||
Batovska DI, Todorova IT, Tsvetkova IV, Najdenski HM. Antibacterial study of the medium chain fatty acids and their 1-monoglycerides: individual effects and synergistic relationships. Pol J Microbiol. 2009;58(1):43–47. | ||
Projan SJ, Brown-Skrobot S, Schlievert PM, Vandenesch F, Novick RP. Glycerol monolaurate inhibits the production of β-lactamase, toxic schock syndrome toxin-1, and other staphylococcal exoproteins by interfering with signal transduction. J Bacteriol. 1994;176(14):4204–4209. | ||
Bergsson G, Arfinnsson J, Steingrímsson Ó, Thormar H. Killing of Gram-positive cocci by fatty acids and monoglycerides. APMIS. 2001;109(10):670–678. | ||
Gottlieb CT, Thomsen LE, Ingmer H, Mygind PH, Kristensen HH, Gram L. Antimicrobial peptides effectively kill a broad spectrum of Listeria monocytogenes and Staphylococcus aureus strains independently of origin, sub-type, or virulence factor expression. BMC Microbiol. 2008;8:205. | ||
Zhang Y, Teng D, Mao R, et al. High expression of a plectasin-derived peptide NZ2114 in Pichia pastoris and its pharmacodynamics, postantibiotic and synergy against Staphylococcus aureus. Appl Microbiol Biotechnol. 2014;98(2):681–694. | ||
Zasloff M. Antimicrobial peptides of multicellular organisms. Nature. 2002;415(6870):389–395. | ||
Hancock RE, Sahl HG. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat Biotechnol. 2006;24(12):1551–1557. | ||
Nizet V. Understanding how leading bacterial pathogens subvert innate immunity to reveal novel therapeutic targets. J Allergy Clin Immunol. 2007;120(1):13–22. | ||
Pasupuleti M, Schmidtchen A, Malmsten M. Antimicrobial peptides: key components of the innate immune system. Crit Rev Biotechnol. 2012;32(2):143–171. | ||
Andersson DI, Hughes D, Kubicek-Sutherland JZ. Mechanisms and consequences of bacterial resistance to antimicrobial peptides. Drug Resists Updat. 2016;26:43–57. | ||
Peschel A, Jack RW, Otto M, et al. Staphylococcus aureus resistance to human defensins and evasion of neutrophil killing via the novel virulence factor MprF is based on modification of membrane lipids with l-lysine. J Exp Med. 2001;193(9):1067–1076. | ||
Ruzin A, Novick RP. Glycerol monolaurate inhibits induction of vancomycin resistance in Enterococcus faecalis. J Bacteriol. 1998;180(1):182–185. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.