Back to Journals » Cancer Management and Research » Volume 11
Synergistic interaction of gemcitabine and paclitaxel by modulating acetylation and polymerization of tubulin in non-small cell lung cancer cell lines
Authors Effendi WI, Nagano T ![]() , Tachihara M
, Tachihara M ![]() , Umezawa K, Kiriu T
, Umezawa K, Kiriu T ![]() , Dokuni R
, Dokuni R ![]() , Katsurada M
, Katsurada M ![]() , Yamamoto M
, Yamamoto M ![]() , Kobayashi K, Nishimura Y
, Kobayashi K, Nishimura Y
Received 7 November 2018
Accepted for publication 5 April 2019
Published 29 April 2019 Volume 2019:11 Pages 3669—3679
DOI https://doi.org/10.2147/CMAR.S193789
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Kenan Onel
Wiwin Is Effendi,1,2 Tatsuya Nagano,1 Motoko Tachihara,1 Kanoko Umezawa,1 Tatsunori Kiriu,1 Ryota Dokuni,1 Masahiro Katsurada,1 Masatsugu Yamamoto,1 Kazuyuki Kobayashi,1 Yoshihiro Nishimura1
1Division of Respiratory Medicine, Department of Internal Medicine, Kobe University Graduate School of Medicine, Kobe 650-0017, Japan; 2Department of Pulmonology and Respiratory Medicine, Airlangga University Medical Faculty, Surabaya 60131, Indonesia
Background: The combination of gemcitabine (GEM) and paclitaxel (PTX) was appealing for clinical exploration due to different mechanisms of action and partially non-overlapping toxicities.
Purpose: The aim of this study was to elucidate a potential effect of this combination on the proliferation of two non-small cell lung cancer (NSCLC) cell lines, A549 and H520.
Materials and methods: Cell lines were treated with GEM and PTX for 48 hours to evaluate the half maximal inhibitory concentration (IC50). To determine the combination index (CI), cell lines were exposed to GEM and PTX, in a constant ratio of IC50, by various combination treatments. GEM`s effect on tubulin was assessed by western blotting and immunofluorescent staining. GEM was combined with nanoparticle albumin-bound-paclitaxel (NP) in evaluating tumor growth inhibition.
Results: The IC50 of GEM and PTX in A549 and H520 were 6.6 nM and 46.1 nM, and 1.35 nM and 7.59 nM, respectively. Among the sequences explored (GEM→PTX, PTX→GEM, and GEM plus PTX simultaneously [GEM+PTX]), GEM→PTX produced a mean CI <1 in both cell lines. Western blotting and immunofluorescent staining revealed the intention expressions of acetylated tubulin protein and enhancement of tubulin polymerization within GEM→PTX group. A combination order GEM→NP also worked synergistically to suppress tumor growth.
Conclusion: The GEM→PTX sequence may represent a promising candidate regimen for the treatment of NSLCL.
Keywords: IC50, combination index, tubulin
Introduction
Globally, lung cancer is a highly lethal form of cancer; in 2018, approximately 19% of the 9.6 million cancer deaths recorded were associated with lung malignancy.1 Approximately 85–90% of lung cancer cases were diagnosed specifically as non-small cell lung cancer (NSCLC).2 An improved understanding of the mechanisms underlying lung cancer would lead to the discovery of novel molecular-targeted therapies and immunotherapies. However, anti-cancer cytotoxic drugs are still important, particularly because of recurrent disease and acquired drug resistance.3
Gemcitabine (GEM), a deoxycytidine analog, is used as single or combination chemotherapy for solid tumors including NSCLC and pancreatic cancer.4 Insertion of its active phosphorylated metabolite, difluorodeoxycytidine triphosphate (dFdCTP), into DNA synthesis will induce cell apoptosis.5 The combination of GEM and other anti-cancer drugs are recommended for systemic therapy in advanced NSCLC.6
Paclitaxel (PTX) is a microtubule-interfering drug that promotes the polymerization of tubulin.7 Suppression of the microtubules leads to dynamic changes in a cell including mitotic block and cell apoptosis.8 Targeting drugs to specific cellular pathways that drive cancer cells is a highly promising treatment modality; therefore, a PTX-based combination was essential in the treatment of advanced NSCLC.9 Nanoparticle albumin-bound-paclitaxel (NP) is a novel drug that delivers PTX in a manner which makes the most of its advantageous pharmacokinetic profile.10
The combination of GEM and PTX was appealing for clinical exploration because these drugs exhibit different mechanisms of action and partially non-overlapping toxicities.11 Previous studies showed that this combination had similar activity in comparison with a carboplatin-based agent but lower efficacy against cisplatin-containing doublets.12 Subsequent phase II studies, involving first-line and second-line chemotherapy, confirmed a high disease control rate but raised awareness of the safety profile.11,13
Interestingly, most previous studies which have investigated the interaction of GEM and PTX have been performed by focusing on the function of PTX as an agent which reinforces the action of GEM. Indeed, PTX was shown to enhance the anti-tumor activity of GEM by increasing levels of the GEM-metabolizing enzyme, deoxycytidine kinase (dCK), which eventually helps to concentrate GEM in cancer cells.14 In addition, a recent study showed that the administration of NP elevated the concentration of GEM by reducing the levels of cytidine deaminase (CDA).15 However, few studies have focused on the function of GEM as a reinforcing agent of PTX.
In this study, we evaluated the potential effect of GEM in combination with PTX (in vitro) and NP (in vivo). Our hypothesis was that GEM would enhance the anticancer activities of PTX and influence the synergism between the two drugs.
Materials and methods
Cell culture and reagents
The human NSCLC cell lines, A549 (adenocarcinoma) and H520 (squamous cell carcinoma), were obtained from the American Type Culture Collection (Manassas, VA, USA). The culture medium was RPMI 1640 (Sigma, St. Louis, MO, USA) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (Wako, Osaka, Japan) under 5% CO2 at 37 °C. GEM and PTX were purchased from Mochida Pharmaceuticals Co., Ltd. (Tokyo, Japan) while NP was obtained from Taiho Pharmaceuticals Co., Ltd. (Tokyo, Japan).
For immunoblotting, mouse monoclonal anti-acetylated tubulin and β-actin antibodies were purchased from Sigma-Aldrich (Darmstadt, Germany) and Cell Signaling (Danvers, MA, USA), respectively. A rabbit anti-α-tubulin antibody was also used for immunofluorescent staining (Atlas Antibodies, Bromma, Sweden).
Cell viability assay and drug combination studies
To determine the half maximal inhibitory concentration (IC50), we carried out a cell proliferation assay using the Cell Counting Kit-8 (Dojindo, Kumamoto, Japan) based on the manufacturer’s instructions. Cell suspensions of A549 and H520, at a concentration of 1×104 cell/100 μl, were seeded in a 96-well plate. The next day, cells were treated with GEM or PTX in various concentrations for a total of 48 h. Absorbances were analyzed at a wavelength of 450 nm using a microplate reader (iMark, Biorad). The IC50 was then determined using the four-parameter logistic function y= D +(A-D)/1+10(x-logC)B, with parameter C representing the estimation of IC50.
Combination studies were completed by incubating 1×104 cell/100 μl in the 96-well plates for 24 h. Next, the cell lines were simultaneously exposed to the drugs in a different order: GEM and PTX sequentially (GEM→PTX), vice versa PTX and GEM sequentially (PTX→GEM) for 24 h per drug, or GEM and PTX simultaneously (GEM+PTX) for 48 h at a constant ratio. Medium containing the first drug was removed and the cells were washed with phosphate-buffered saline (PBS) before adding the second drug. Based on the principles of the median-effect equation by Chou,16 drug interactions were analyzed using specific software (Calcusyn, Biosoft, Oxford, UK) and results presented as a combination index (CI).
The mean CI for every fraction affected (Fa, % inhibition of cellular proliferation) at 0.5, 0.75, and 0.9 was described as the final CI. The range of CI <0.1, 0.1–0.3, 0.3–0.7, 0.7–0.85, 0,85–0.9, 0.9–1.1, 1.1–1.2, 1.2–1.45, 1.45–3.3, and >10 describes very strong synergism, strong synergism, synergism, moderate synergism, slight synergism, nearly additive, slight antagonism, moderate antagonism, antagonism, strong antagonism, and very strong antagonism, respectively.16
Western blot analysis
Measurement of acetylated tubulin protein was commenced by incubating cells in a 10 cm dish before treating with single drugs, or a combination of drugs, in a different sequence for a total of 48 h. On ice, we then applied cell lysis buffer (Cell Signaling Danvers, MA, USA) containing proteinase inhibitors (Roche Applied Science, Indianapolis, IN, USA) to detach cells, aided by manual scraping. An equal amount of extracted protein was then separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE; Wako, Osaka, Japan) and transferred onto nitrocellulose membranes. After blocking, membranes were incubated with a 1:100,000 dilution of primary antibody (anti-acetylated tubulin) overnight at 4 °C. Blotted protein samples were then detected with an enhanced chemiluminescence kit from Thermo Fisher Scientific (Waltham, MA, USA).
Immunofluorescence
Cell suspensions at a concentration 1 x 106/ml were seeded into 2 well chambers on Lab-Tek chamber slides (Thermo Fisher Scientific, Waltham, MA, USA): GEM (6.6 nM in A549 or 46.1 nM in H520) and PTX (1.35 nM in A549 or 7.59 nM in H520) sequentially (GEM→PTX); vice versa PTX and GEM sequentially (PTX→GEM) for 24 h per drug or GEM and PTX simultaneously (GEM+PTX) for 48 h at a constant ratio. After the administration of drugs for a total of 48 h, the cells were fixed with 20 °C methanol with EGTA (a calcium chelator) and permeabilized with 1% Triton. One hour after blocking, incubation overnight with primary antibody at a dilution of 1:250 and 1:500 for α-tubulin and β-tubulin, respectively. Nuclei were counterstained with 4’, 6-diamidino-2-phenylindole (DAPI) and the expression of tubulin was analyzed using a fluorescence microscope.
Apoptosis assay
The cells were incubated in 6-well plates and treated with their IC50 values of GEM, PTX, and concurrent GEM+PTX for 48 h. In sequence group, GEM or PTX was administrated for 24 h followed by combination GEM+PTX in next 24 h. Following drug treatment, cells were then collected and washed twice with cold PBS. After centrifuge, cells were resuspended within 100 μl solution binding buffer at a concentration of 1.0×106 cells/ml and added with 5 μl Fluorescein isothiocyanate (FITC) Annexin V and 5 μl Propidium Iodide (PI). Cells were incubated for 15 min at room temperature under dark conditions. 400 μl binding buffer was added and apoptotic cells were identified using the BD Pharmingen FITC Annexin V Apoptosis Detection kit (San Diego, CA, USA), according to the manufacturer’s instructions.
In vitro scratch assay
To assess the effect of combination drugs on cell migration, a wound-healing assay was performed. The cells were seeded on 6 well plates and incubated at 37 °C until cells reach 80–100% confluence. A scratch was made through confluent monolayer and each group of cells was exposed to single and combination for 48 h, GEM or PTX for 24 h followed combination for 24 h.
Scratch zones representative for each group were photographed at 0, 24, and 48 h by BZ-X700 microscope (Japan). Relative wound closure (RW) is the comparison of the distance of final wound (Wf) and initial wound (Wi); RW =(Wf/Wi), Wi: initial wound width (μm), Wf: final wound width (μm).
In vivo growth inhibition assay
Twenty-four female BALB/c nude mice were purchased from SLC (Shizuoka, Japan). The research was approved by the Institutional Animal Care and Use Committee of Kobe University (Permit Numbers: P171201) and carried out according to the Kobe University Animal Experimentation Regulations. Mice were inoculated subcutaneously with 5.0×106 cells/100 μl suspension of cell lines. When the tumor volume (TV) reached 100 mm3, mice were divided into six groups (n=4 per group). Each mouse was treated with sterile PBS intraperitoneally (i. p.) (day 1, 8, 15), GEM 50 mg/kg i. p. (day 1, 8, 15), NP 75 mg/kg intravenously (i. v.) (day 1, 8, 15), GEM i. p. (day 0, 7, 14) followed by NP i. v. (day 1,8,15) (GEM→NP), GEM combined NP (day 1, 8, 15) (GEM+NP) and an NP i. v. (day 0, 7, 14) followed by GEM i. p. (day 1, 8, 15) (NP→GEM).
We then determined a range of parameters: longest diameter (a), widest diameter (b), TV =a x b2/2, TVn= the tumor volume on n day, TV0= the tumor volume on day 0, relative tumor volume (RTV)= TVn/TV0, body weight (BW), and relative body weight (RBW)= BWn/BW0. TV and BW were measured twice a week after injection of an anti-cancer drug.
Statistical analysis
Data are presented as mean ± standard error (SE). Independent two-sample t-test was used to determine the statistical difference between all combination drugs and control groups. A P-value <0.05 was considered to be statistically significant.
Results
The sensitivity of NSCLC cells to GEM and PTX
The sensitivities of A549 and H520 to anticancer drugs are summarized in Table 1. A549 cells were more sensitive than H520. As predicted, PTX inhibited cell proliferation more conclusively in both A549 and H520 than GEM. The IC50 value of PTX in A549 and H520 cells were 1.35 nM and 7.59 nM, respectively (Figure 1). The IC50 value of GEM in A549 cells (6.6 nM) was approximately 7-fold stronger compared to the IC50 value of GEM in H520 cells (46.1 nM) (Figure 1).
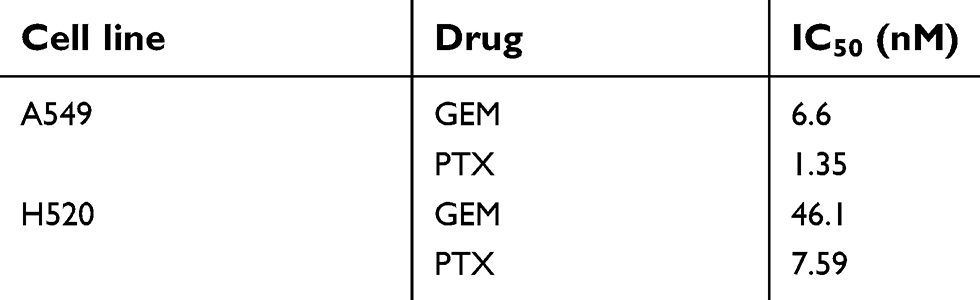 | Table 1 The sensitivity of cell lines to gemcitabine and paclitaxel |
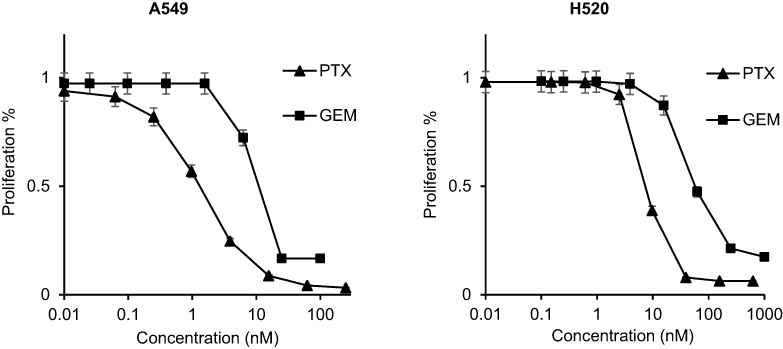 | Figure 1 Inhibitory effect of gemcitabine (GEM) or paclitaxel (PTX) on human non-small cell lung cancer (NSCLC) cell lines. After incubating cell lines with various concentrations of drugs for a total of 48 h, we measured the absorbance at a wavelength of 450 nm. The IC50 of GEM in A549 and H520 cells was 6.6 nM and 46.1 nM, respectively. The IC50 of PTX in A549 and H520 cells was 1.35 nM and 7.59 nM, respectively. |
The synergistic effects of GEM and PTX
Calculations were carried out at a constant molar ratio of PTX: GEM of 1:4.8 and 1:6.1 for A549 and H520 cells, respectively. Table 2 shows the mean CI for cell lines while Figure 2 illustrates the median-effect and the combination index plots.
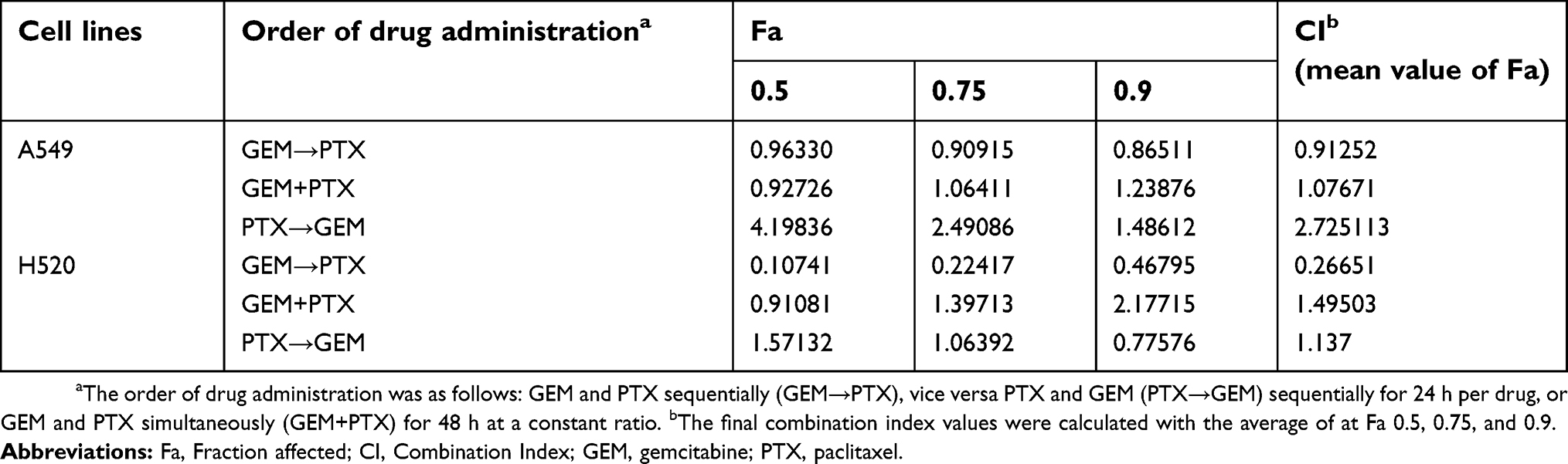 | Table 2 Drug-drug combination analysis |
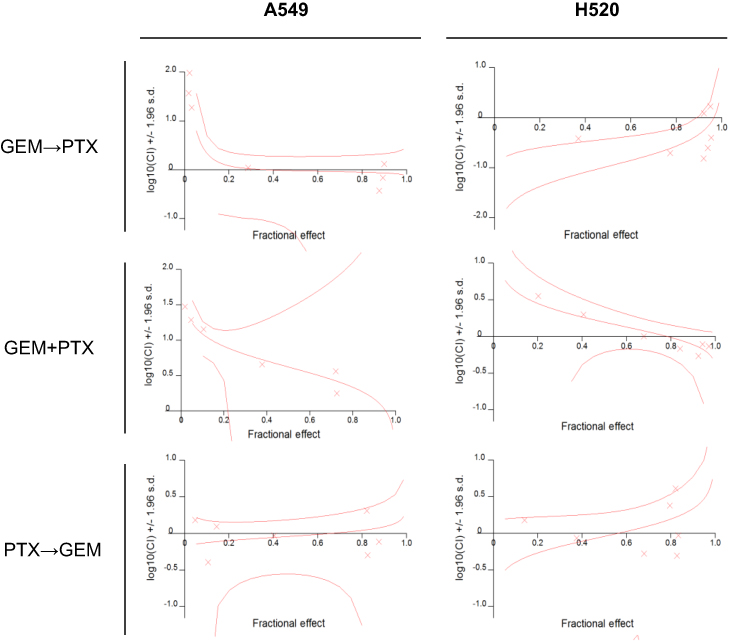 | Figure 2 Dose-dependent inhibition of cell growth after the administration of various sequences of gemcitabine (GEM) and/or paclitaxel (PTX). Using a constant ratio of IC50, drug-drug interaction was evaluated in various sequences as follows: GEM and PTX sequentially (GEM→PTX), vice versa PTX and GEM (PTX→GEM) sequentially for 24 h per drug, or GEM and PTX simultaneously (GEM+PTX) for 48 h. The sequence of GEM→PTX showed synergism in both cell lines. |
The drug interaction for the GEM→PTX sequence produced synergism with a mean CI of 0.91252 and 0.26651 for A549 and H520 cells, respectively. Various synergistic interactions were observed at a Fa of 0.5, 0.75, and 0.9 with CI values of 0.96330, 0.90915, 0.86511 for A459 cells, and 0.10741, 0.22417, 0.46795 for H520 cells, respectively.
The strongest synergism in H520 was obtained at low Fa. There were also synergisms in reverse sequence with a CI of 0.77576 in H520 cells, and concurrent sequence with a CIs of 0.91081 and 0.92726 for H520 and A549 cells. Overall, the mean CI of GEM+PTX and PTX→GEM sequences did not meet the criteria of synergism.
GEM-mediated activation of tubulin acetylation
To clarify the synergistic role of GEM in the GEM→PTX sequence, we performed Western blot analysis of acetylated tubulin protein, the main target of PTX. PTX alone, and the combination of drugs, increased the level of acetylated tubulin protein. Among the variant sequences, the highest expression of acetylated tubulin protein was observed in cells treated with GEM→PTX (Figure 3).
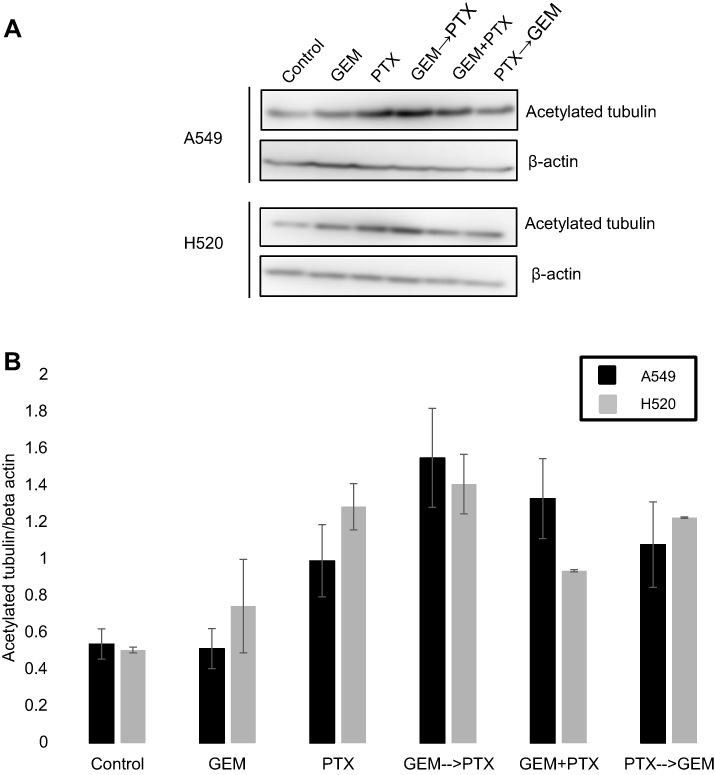 | Figure 3 The role of gemcitabine (GEM) in the expression of acetylated tubulin protein in non-small cell lung cancer cell lines. Representative Western blotting of cellular extracts from A549 and H520 cells after the administration of single or combined drug treatments in various sequence, as follows: GEM and paclitaxel (PTX) sequentially (GEM→PTX), vice versa PTX and GEM (PTX→GEM) sequentially for 24 h per drug, or GEM and PTX simultaneously (GEM+PTX) for 48 h. (A) PTX alone and all combinations of drugs enhanced the level of α-tubulin acetylated protein. Prominent bands were found in cells treated with GEM→PTX. (B) As quantified by densitometer analysis, treatment with GEM followed by PTX produced highest acetylated tubulin expression. |
The effect of GEM on microtubule polymerization
PTX-treated cell lines with increased levels of polymerized α- and β-tubulin protein were characterized by thicker and denser microtubule bundles (green and red) forming ring-like structures around the nucleus (blue) (Figure 4). As evidenced by immunofluorescence, the sequence of GEM→PTX in both of the cell lines appeared to enhance polymerization. Cells exposed to GEM, and controls, showed an extensive fine microtubule structure within the cytoplasm.
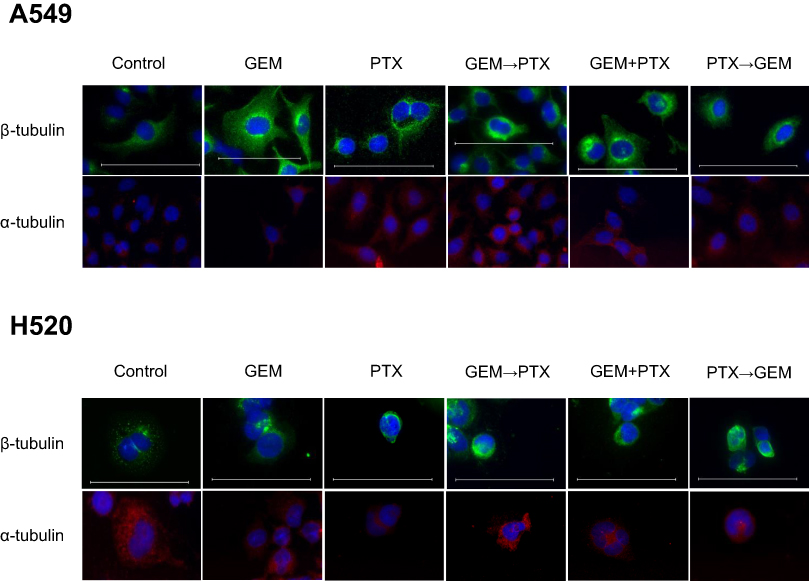 | Figure 4 The effect of gemcitabine (GEM) in polymerization microtubule. Cell lines were prepared for immunostaining β-tubulin (green) and α-tubulin (red) after stimulation with the indicated drugs for a total of 48 h. Nuclei were counter-stained with 4’, 6-diamidino-2-phenylindole (DAPI) (blue). Scale bar, 100 μm. Immunofluorescence staining revealed alterations of cellular microtubule structures characterized by the formation of a microtubule bundle. The GEM→PTX sequence appeared to enhance α- and β-tubulin polymerization. |
Apoptosis assay
To confirm treatment-induced apoptosis, flow cytometry using Annexin-V and Propidium Iodide was performed to allowed specific differentiation of the apoptotic and necrotic cell. The combination using GEM as a first drug followed by GEM+PTX resulted in a significance number of apoptotic cells, 8% and 9.4%, in A549 and H520 cell line respectively. Administration PTX→GEM+PTX and concurrent GEM+PTX produced a different result in each cell line (Figure 5A and B).
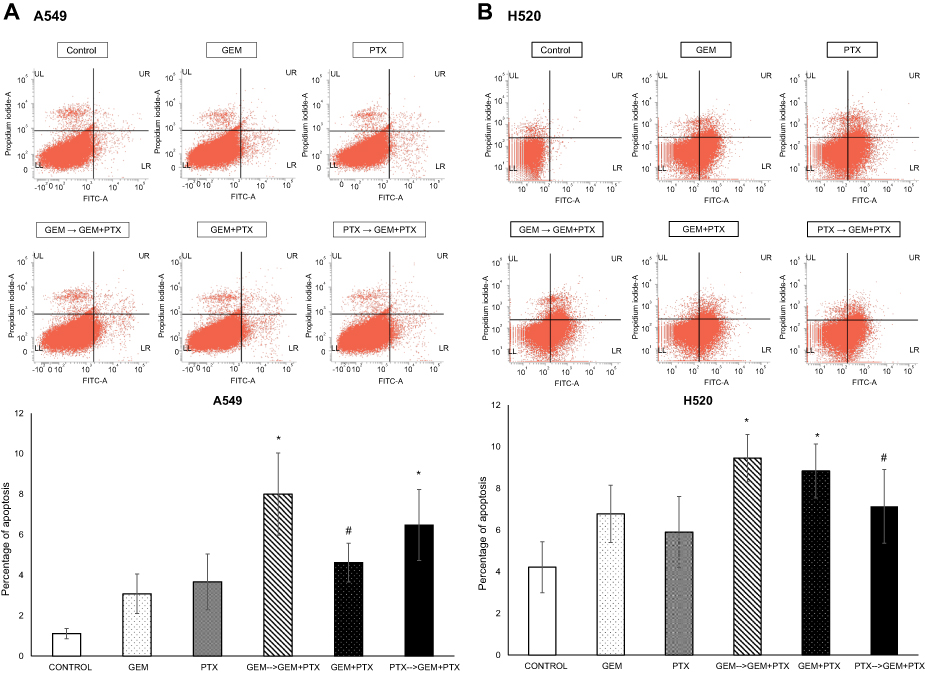 | Figure 5 Apoptosis assay. Flow cytometry analysis of both cell line that treated with sequence drug GEM→GEM+PTX resulted in significance meaning. (A) A percentage apoptotic of A549 cell within group GEM→GEM+PTX and vice versa were greater than concurrent GEM+PTX. (B) In the case of H520 cell line, GEM→GEM+PTX and concurrent GEM+PTX produced a similar percentage of apoptotic. *P<0.05 compared with control; #not significant. |
Scratch assay
The migration of cells in the scratch assay was followed over 48 hrs. Both cell lines displayed a different migratory profile during the wound healing process. A549 cells migrated to the scratched area and approached each other to close the wound faster than H520 cells. Although relative wound closure in A549 was almost similar in all treatment group (Figure 6A), administration of GEM for 24 hrs as the first drug continued by the next 24 hrs GEM+PTX in H520 cell line leads to a significant decrease of migration rate (0.82) compared with control (Figure 6B).
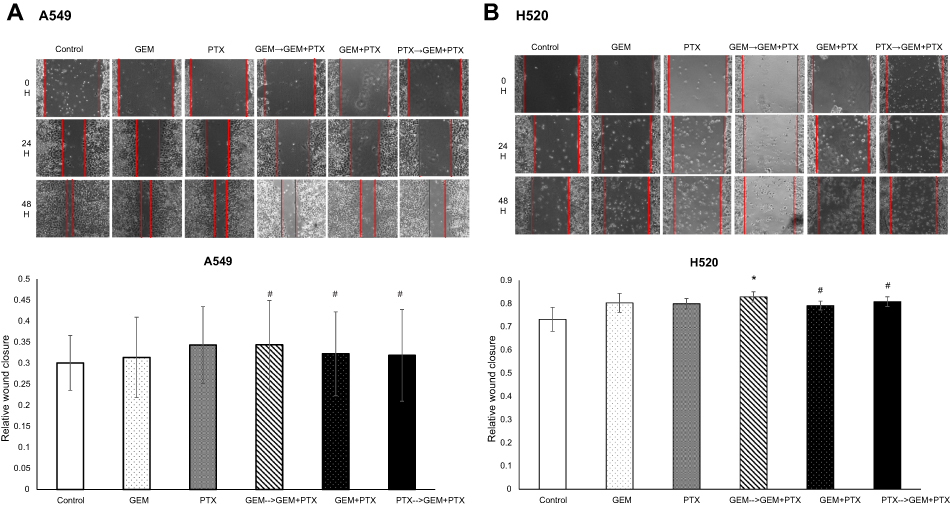 | Figure 6 Wound healing assay. (A) Relative wound closure of A549 cell line in all group combination drugs were similar and showed no significant results. (B) However, scratch assay showed the most significant deceleration of cell migration was in drug combination GEM→GEM+PTX-treated H520 cell. *P<0.05 compared with control; #not significant. |
Tumor growth inhibition assay
We evaluated the efficacy of drug combinations to inhibit tumor growth by RTV. The GEM→NP sequence grouped showed a significant tumor suppression in both of cell lines (Figure 7A and B). It was important for us to consider the progression of tumor size. Tumor growth observed in control and GEM treatments of A549 cells was terminated on day 7 and 18, respectively, and mice in control and GEM of H520 were only followed to day 14. In both types of tumor-bearing mice, there were tendencies for tumor regrowth after stopping the treatment injections. The smallest body weight might be influenced by the lowest growth of tumor size in group GEM→NP.
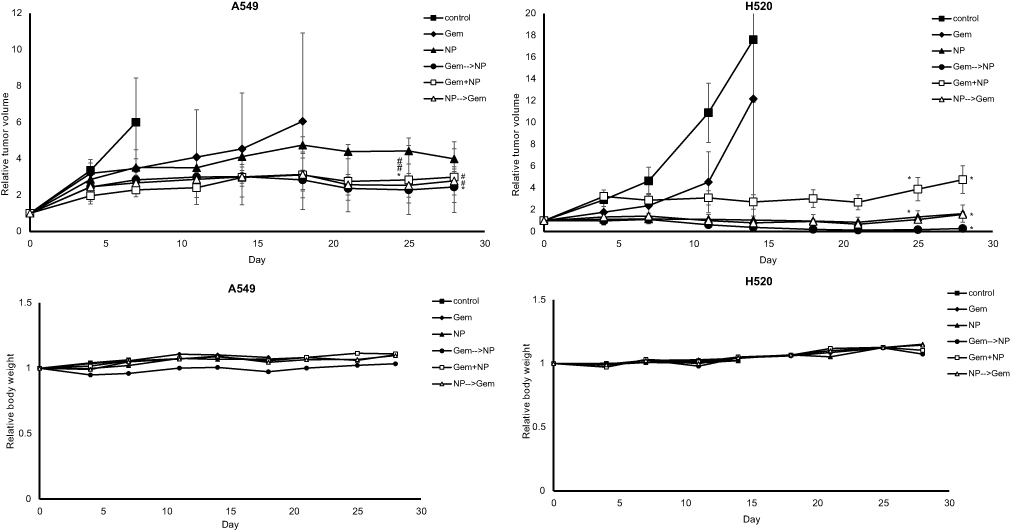 | Figure 7 Growth inhibition assays. The GEM→NP sequence had the significant inhibitory effect upon tumor growth in both groups of A549- and H520-bearing mice. (A) In the case of A549, the RTV of drug combinations were suppressed more strongly than that of single agents. (B) In the case of H520, NP singe and NP→GEM gave similar inhibition and suppressed tumor growth more strongly than GEM+NP and GEM. There was no evaluation of RBW which reflects toxicity effect in all groups both of tumor-bearing mice. *P<0.05 compared with control; #not significant. |
Discussion
This study utilized two major types of lung cancer cell line, the adenocarcinoma cell line A549 and the squamous cell carcinoma cell line H520. A549 was most sensitive to anti-cancer drugs (Table 1). The result was consistent with the results of previously published reports.17,18 On the other hand, our experiments on the H520 cell line showed that it was more sensitive to anti-cancer drugs than described in a previous report.19 Because of GEM and PTX have different action modes, the combination of these drugs could potentiate the action of these as single agents.20 Intriguingly, synergism was observed in the GEM→PTX sequence; this observation was apparent for both cell lines (Table 2) . The strongest synergism, with a CI of 0.10741 at 0.5 Fa, was apparent in H520 cells. This finding is of considerable clinical importance even though synergism at a high-level Fa for cancer treatment is more critical.16
This leaves us with the question of whether synergism and antagonism could be predicted? An earlier pharmacodynamic study showed that a low dose of GEM (1 ng/ml) induced cell cycle arrest in phase S of the cell cycle, and assisted the efficacy of the subsequent drug.21 A low dose of PTX will stabilize microtubule during mitosis at phase G2-M and lead to apoptosis rather than necrosis.22 Meanwhile, other studies described that low PTX may arrest of the cell only at phase G123 and also both of G2-M and/or G1.24 Furthermore, low PTX may induce aberrant mitosis resulting in aneuploidy and lead to apoptosis.25 We assume that administration of low GEM as the first drug in combination produce a transient accumulation cell in phase S that will facilitate efficacy of the next drug, low PTX. In reverse sequence, exposure to low PTX probably caused cells to arrest in phase G1, thereby the number of cells in phase S was reduced leading to insufficiency of cell cycle-specific cytotoxicity of GEM.
Synergism in combination using low GEM as a first drug were also represented by apoptosis and migration assay. Apoptosis was more pronounced in the sequence combination in which GEM preceded GEM+PTX. The migration rate of cells was also decreased after a scratch made and followed for 48 hrs. The increases in apoptotic cell and deceleration of migration assay may be related in the scheduling of drug combination.
Growth inhibition in GEM→NP seemed more favorable. The maximum tolerated dose (MTD) of NP was between 120–240 mg/kg and the lethal dose was 240 mg/kg.26 These might be explained that the more cell arrested by GEM in phase S will facilitate the low dose of NP (50 mg/kg) lead to apoptosis of the cell and suppress the tumor growth. GEM works effectively in phase S so that the use of NP as the first drug or concurrent combination will decrease the activities of GEM. Hence, the single use of NP might be inhibited the tumor growth similar to group NP→GEM and concurrent.
Microtubule stabilization, resulting in cell apoptosis, is a cornerstone that underlies drug-drug interaction. Microtubules, which consist of α and β tubulin, are highly dynamic structures and represent the target of tubulin-binding anti-cancer drugs.27 PTX interacts with an amino-terminal region of tubulins against depolymerization.28 Even at low concentrations, PTX can still exert the ability to suppress the dynamics of a microtubule.29 Tubulin acetylation is an indicator of microtubule stability.30 In contrast, histone deacetylase 6 (HDAC6), a class IIb deacetylase, is known to deacetylate substrates such as tubulin.31 Indeed, in a previous study, a combination of trichostatin A, a histone deacetylase inhibitor, and PTX resulted in a significant increase in tubulin acetylation and microtubule stabilization.32 On the other hand, GEM can influence microtubules indirectly. For example, GEM can play a vital role in apoptosis driven by caspase-3.33 The cleavage of the C-terminal ubiquitin-binding zinc finger of HDAC6 will promote apoptosis.34 Therefore, GEM can enhance the acetylation of tubulin and polymerization of microtubules, thus reinforcing the anti-cancer effect of PTX.
In conclusion, the present study provides evidence that the synergistic drug combination of low GEM and low PTX/NP can inhibit the growth of NSCLC cell lines. Furthermore, the administration of low GEM as a first drug in the combination was proven to enhance the anti-tumor activity of low PTX by modulating tubulin acetylation and microtubule polymerization.
Abbreviation list
NSCLC, non-small cell lung cancer; GEM, gemcitabine; dFdCTP, difluorodeoxycytidine triphosphate; PTX, paclitaxel; NP, nanoparticle albumin-bound-paclitaxel; dCK, deoxycytidine kinase; CDA, cytidine deaminase; FBS, fetal bovine serum; IC50, half maximal inhibitory concentration; PBS, phosphate-buffered saline; CI, combination index; Fa, fraction affected; TV, tumor volume; i. p, intraperitoneally; i. v, intravenously; RTV, relative tumor volume; BW, body weight; RBW, relative body weight; HDAC6, histone deacetylase 6; RW, relative wound closure; Wf, final wound; Wi, initial wound.
Acknowledgments
We would like to express our gratitude to members of the Division of Respiratory Medicine of Kobe University Graduate School of Medicine for their helpful discussion and the Indonesian Endowment Fund for Education (LPDP) Scholarship under Beasiswa Unggulan Dosen Indonesia-Luar Negeri (BUDI-LN) batch I 2016 (Number: PRJ-3712/LPDP.3/2016). This work was supported by Research Grants from Eli Lilly Japan K.K. [grant number LGO-1600002] to Motoko Tachihara.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Novello S, Barlesi F, Califano R, et al. Metastatic non-small-cell lung cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2016;27(March):V1–V27. doi:10.1093/annonc/mdw326
3. Hatakeyama Y, Kobayashi K, Nagano T, et al. Synergistic effects of pemetrexed and amrubicin in non-small cell lung cancer cell lines: potential for combination therapy. Cancer Lett. 2014;343(1):74–79. doi:10.1016/j.canlet.2013.09.019
4. van Moorsel CJ, Peters GJ, Pinedo HM. Gemcitabine: future prospects of single-agent and combination studies. Oncologist. 1997;2(3):127–134.
5. Mini E, Nobili S, Caciagli B, Landini I, Mazzei T. Cellular pharmacology of gemcitabine. Ann Oncol. 2006;17(SUPPL. 5):7–12. doi:10.1093/annonc/mdj941
6. Postmus PE, Kerr KM, Oudkerk M, et al. Early and locally advanced non-small-cell lung cancer (NSCLC): ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2017;28(March):iv1–iv21. doi:10.1093/annonc/mdx222
7. Rowinsky EK, Donehower RC. Paclitaxel (TAXOL). N Engl J Med. 1995;332(15):1004–1013. doi:10.1056/NEJM199504133321507
8. Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4(4):253–265. doi:10.1038/nrc1317
9. Ramalingam S, Belani CP. Paclitaxel for non-small cell lung cancer. Expert Opin Pharmacother. 2004;5(8):1771–1780. doi:10.1517/14656566.5.8.1771
10. Yardley DA. Nab-paclitaxel mechanisms of action and delivery. J Control Release. 2013;170(3):365–372. doi:10.1016/j.jconrel.2013.05.041
11. Douillard JY, Lerouge D, Monnier A, et al. Combined paclitaxel and gemcitabine as first-line treatment in metastatic non-small cell lung cancer: a multicentre phase II study. Br J Cancer. 2001;84(9):1179–1184. doi:10.1054/bjoc.2001.1784
12. Toschi L, Cappuzzo F. Gemcitabine for the treatment of advanced nonsmall cell lung cancer. OncoTargets Ther. 2009;2:209–217.
13. Dazzi C, Cariello A, Casanova C, et al. Gemcitabine and paclitaxel combination as second-line chemotherapy in patients with small-cell lung cancer: a phase II study. Clin Lung Cancer. 2013;14(1):28–33. doi:10.1016/j.cllc.2012.03.003
14. Kroep JR, Giaccone G, Tolis C, et al. Sequence dependent effect of paclitaxel on gemcitabine metabolism in relation to cell cycle and cytotoxicity in non-small-cell lung cancer cell lines. Br J Cancer. 2000;83(8):1069–1076. doi:10.1054/bjoc.2000.1399
15. Frese KK, Neesse A, Cook N, et al. Nab-paclitaxel potentiates gemcitabine activity by reducing cytidine deaminase levels in a mouse model of pancreatic cancer. Cancer Discov. 2012;2(3):260–269. doi:10.1158/2159-8290.CD-11-0242
16. Chou T. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58(3):623–642. doi:10.1124/pr.58.3.10
17. Li J, Pan YY, Zhang Y. Synergistic interaction between sorafenib and gemcitabine in EGFR-TKI-sensitive and EGFR-TKI-resistant human lung cancer cell lines. Oncol Lett. 2013;5(2):440–446. doi:10.3892/ol.2012.1017
18. Liebmann JE, Cook JA, Lipschultz C, Teague D, Fisher J, Mitchell JB. Cytotoxic studies of paclitaxel (Taxol) in human tumour cell lines. Br J Cancer. 1993;68(6):1104–1109.
19. Shord SS, Patel SR. Paclitaxel alters the expression and specific activity of deoxycytidine kinase and cytidine deaminase in non-small cell lung cancer cell lines. J Exp Clin Cancer Res. 2009;28(1):76. doi:10.1186/1756-9966-28-121
20. Kroep BJR, Giaccone G, Voorn DA, et al. Gemcitabine and paclitaxel : pharmacokinetic with non – small-cell lung cancer. J Clin Oncol. 1999;17(7):2190–2197. doi:10.1200/JCO.1999.17.7.2190
21. Hamed SS, Straubinger RM, Jusko WJ. Pharmacodynamic modeling of cell cycle and apoptotic effects of gemcitabine on pancreatic adenocarcinoma cells. Cancer Chemother Pharmacol. 2014;72(3):553–563.
22. Yeung TK, Germond C, Chen X, Wang Z. The mode of action of Taxol: apoptosis at low concentration and necrosis at high concentration. Biochem Biophys Res Commun. 1999;263(2):398–404. doi:10.1006/bbrc.1999.1375
23. Demidenko ZN, Kalurupalle S, Hanko C, Lim CU, Broude E, Blagosklonny MV. Mechanism of G1-like arrest by low concentrations of paclitaxel: next cell cycle p53-dependent arrest with sub G1 DNA content mediated by prolonged mitosis. Oncogene. 2008;27(32):4402–4410. doi:10.1038/onc.2008.82
24. Giannakakou P, Robey R, Fojo T, Blagosklonny MV. Low concentrations of paclitaxel induce cell type-dependent p53, p21 and G1/G2 arrest instead of mitotic arrest: molecular determinants of paclitaxel-induced cytotoxicity. Oncogene. 2001;20(29):3806–3813. doi:10.1038/sj.onc.1204487
25. Ikui AE, Chia-Ping HY, Matsumoto T, Horwitz SB. Low concentrations of taxol cause mitotic delay followed by premature dissociation of p55CDC from Mad2 and BubR1 and abrogation of the spindle checkpoint, leading to aneuploidy. Cell Cycle. 2005;4(10):1385–1388. doi:10.4161/cc.4.10.2061
26. Desai N, Trieu V, Soon-Shiong P, Hawkins M. Abraxane (ABI-007) vs taxotere: a preclinical comparison of toxicity and efficacy. Cancer Res. 2005;65(9Supplement):336 LP–337.
27. Parker AL, Kavallaris M, McCarroll JA. Microtubules and their role in cellular stress in cancer. Front Oncol. 2014;4(June):1–19. doi:10.3389/fonc.2014.00001
28. Arnal I, Wade RH. How does taxol stabilize microtubules? Curr Biol. 1995;5(8):900–908.
29. Jordan MA, Wendell K, Gardiner S, Derry WB, Copp H, Wilson L. Mitotic block induced in hela cells by low concentrations of paclitaxel (Taxol) results in abnormal mitotic exit and apoptotic cell death mitotic block induced in hela cells by low concentrations of paclitaxel (Taxol) results in abnormal mitotic exit. Cancer Res. 1996;56:816–825.
30. Shafer A, Zhou C, Gehrig PA, Boggess JF, Bae-Jump VL. Rapamycin potentiates the effects of paclitaxel in endometrial cancer cells through inhibition of cell proliferation and induction of apoptosis. Int J Cancer. 2010;126(5):1144–1154. doi:10.1002/ijc.24837
31. Li Y, Shin D, Kwon SH. Histone deacetylase 6 plays a role as a distinct regulator of diverse cellular processes. FEBS J. 2013;280(3):775–793. doi:10.1111/febs.12079
32. Dowdy SC, Jiang S, Zhou XC, et al. Histone deacetylase inhibitors and paclitaxel cause synergistic effects on apoptosis and microtubule stabilization in papillary serous endometrial cancer cells. Mol Cancer Ther. 2006;5(11):2767–2776. doi:10.1158/1535-7163.MCT-06-0209
33. Chandler NM, Canete JJ, Callery MP. Caspase-3 drives apoptosis in pancreatic cancer cells after treatment with gemcitabine. J Gastrointest Surg. 2004;8(8):1072–1078. doi:10.1016/j.gassur.2004.09.054
34. Husain M, Harrod KS. Influenza A virus-induced caspase-3 cleaves the histone deacetylase 6 in infected epithelial cells. FEBS Lett. 2009;583(15):2517–2520. doi:10.1016/j.febslet.2009.07.005
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.