Back to Journals » OncoTargets and Therapy » Volume 10
Synaptotagmin-7 is overexpressed in hepatocellular carcinoma and regulates hepatocellular carcinoma cell proliferation via Chk1–p53 signaling
Authors Jin H, Xu G, Zhang Q, Pang Q ![]() , Fang M
, Fang M
Received 9 June 2017
Accepted for publication 14 July 2017
Published 29 August 2017 Volume 2017:10 Pages 4283—4293
DOI https://doi.org/10.2147/OTT.S143619
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Ingrid Espinoza
Hao Jin,1–3 Geliang Xu,2 Qiang Zhang,3 Qing Pang,3 Meifang Fang3
1School of Medicine, Shandong University, Jinan, 2Department of Hepatic Surgery, Anhui Provincial Hospital, Hefei, 3Department of Hepatobiliary Surgery, The First Affiliated Hospital, Bengbu Medical College, Bengbu, Anhui, People’s Republic of China
Background: Synaptotagmin-7 (Syt-7) is a member of the synaptotagmin (Syt) family, which plays an important role in many physiological and pathological processes. However, to the best of our knowledge, there is no study describing its function in tumors, particularly in hepatocellular carcinoma (HCC). Therefore, in this study, we examined the role of Syt-7 in HCC and attempted to elucidate its underlying mechanism.
Materials and methods: We examined the expression levels of Syt-7 in HCC cell lines and normal hepatocytes by real-time quantitative polymerase chain reaction analysis. The effects of Syt-7 knockdown on in vitro cell growth were assessed by Celigo image cytometry, MTT assay, colony formation assay, and cell cy cle analysis. In vivo tumorigenesis was evaluated using a nude mouse model. The underlying molecular mechanism was evaluated using a PathScan Stress Signaling Antibody Array.
Results: Syt-7 mRNA levels were highly expressed in Huh-7 and Hep3B cells; moderately expressed in SMMC-7721, HepG2, and BEL-7402 cells; and lowly expressed in normal hepatocytes L-O2. Functional experiments demonstrated that Syt-7 knockdown significantly suppressed cell proliferation and induced cell cycle arrest by increasing phosphorylation of Chk1 and p53. Furthermore, Syt-7 knockdown remarkably reduced the growth of xenograft tumors in mice.
Conclusion: The results of this study suggest that Syt-7 plays a vital role in tumorigenesis and in the development of HCC. Syt-7 can be used as a new diagnostic and therapeutic target in HCC.
Keywords: hepatocellular carcinoma, Syt-7, Chk1, p53, cell cycle
Introduction
Hepatocellular carcinoma (HCC) is a common malignant tumor of the hepatobiliary system and is the second most common cause of cancer-related deaths worldwide.1 It is commonly diagnosed in patients of East Asian and African descent. At present, the incidence of HCC is second highest in People’s Republic of China, and the number of incident cases and deaths is continuing to increase every year.2 The pivotal method of treatment of HCC is radical surgical resection; however, the recurrence rate and mortality remain very high owing to insensitivity to chemotherapy and rapid increase in drug resistance.3 Therefore, many researchers are currently focusing on the identification of specific molecular markers and the elucidation of underlying molecular mechanisms in HCC. Several genes are responsible in the development of HCC.4 Synaptotagmin-7 (Syt-7) is an important member of the synaptotagmin (Syt) family, which comprises 17 human isoforms.5,6 Syts have been identified as membrane trafficking proteins with an N-terminal single transmembrane domain and C-terminal tandem C2 domains. Syt-7 is an individually Ca2+-dependent protein, which interacts with SNARE proteins7 and has been found to be expressed in several systems, playing a crucial role in many physiological processes. For example, Syt-7 associates with specific SNAREs to trigger exocytosis in neuronal cells.8 Syt-7 functions as the principal Ca2+ sensor for glucagon secretion9 and exocytosis, triggering fusion of secretory granule and insulin secretion.10 Glavan et al reported that Syt-7 is involved in seizures. They found that kainate or pilocarpine induced differential patterns of overexpression of striatal Syt-7 mRNA in rats with pilocarpine-induced seizures; the muscarinic antagonist, scopolamine, modified the pattern of expression.11 Syt-7 is required in the calcium-dependent lysosomal fusion; thus, it promotes leukocyte migration via Syt-mediated vesicle fusion.12 Elucidation of this molecular mechanism was beneficial in the study of Syt-immune-mediated, autoimmune disorders, and inflammatory diseases, which raises the following question: could Syt-7 enhance tumor cell migration and have a role in tumorigenesis and tumor development? To the best of our knowledge, there are no studies reporting the role of Syt-7 in HCC. Therefore, in this study, we investigated the expression of Syt-7 in several human HCC cell lines. We adopted lentivirus-mediated specific siRNA targeting Syt-7 to identify the effect of Syt-7 silencing on the growth, proliferation, colony formation, and cell cycle progression of HCC cells and xenograft tumorigenesis in nude mice.
Materials and methods
Cell culture
Human HCC cell lines, namely, SMMC-7721, Huh-7, HepG2, BEL-7402, Hep3B, and L-O2 normal hepatocytes, were purchased from GeneChem Corporation (Shanghai, People’s Republic of China) and cultured in RPMI-1640 medium. All media were supplemented with 10% FBS, 100 U/mL penicillin G, and 100 μg/mL streptomycin. All cell cultures were maintained at 37°C in a humidified atmosphere with 5% CO2 in an incubator.
Real-time quantitative polymerase chain reaction (RT-qPCR) analysis
Total RNA was extracted using Trizol reagent (Pufei Biotech Co, Ltd, Shanghai, People’s Republic of China) according to the manufacturer’s instructions. Synthesis of cDNA was performed by reverse transcription using Reverse Transcription System reagents (Promega Corporation, Fitchburg, WI, USA). The expression of Syt-7 was quantified by RT-qPCR analysis using SYBR Premix Ex Taq II (Takara, Kyoto, Japan) with GAPDH as an internal reference. The following sequences of primers were used: 5′-TGACTTCAACAGCGACACCCA-3′ (upstream) and 5′-CACCCTGTTGCTGTAGCCAAA-3′ (downstream) for SYT-7 and 5′-ACTCCATCATCGTGAACATCATC-3′ (upstream) and 5′-TCGAAGGCGAAGGACTCATTG-3′ (downstream). Quantitative PCR was performed according to Takara SYBR Master Mix kit instructions: 95°C for 15 s, followed by 45 cycles of 95°C for 5 s and 60°C for 20 s. The relative gene expression levels were calculated and statistically compared using the 2−ΔΔCT analysis program.13
Construction of lentivirus for RNAi
We generated shRNAs targeting Syt-7 mRNA against the Syt-7 mRNA (NM_004200) sequence as follows: Syt-7 shRNA forward: 5′-CCGGGCTCACCGTGAAGATCATGAACTCGAGTTCATGATCTTCACGGTGAGCTTTTTG-3′, reverse: 5′-AATTCAAAAAGCTCACCGTGAAGATCATGAACTCGAGTTCATGATCTTCACGGTGAGC-3′ and negative shRNA sequences (scrambled control shRNA) forward: 5′-CCGTTTCTCCGAACGTGTCACGTTTCAAGAGAACGTGACACGTTCGGAGAATTTTTG-3′, reverse: 5′-AATTCAAAAATTCTCCGAACGTGTCACGTTCTCTTGAAACGTGACACGTTCGGAGAA-3′, used as a control. None of the sequences showed homology to other known human genes. Syt-7 shRNA and scrambled control shRNA were connected with GV115 linearized vectors (GeneChem Corporation) digested with AgeI and EcoRI, respectively, to construct the lentivirus interference plasmids, PSC39360 and PSC3741. The ligation products were transformed into TOP10 competent cells (Tiangen, Beijing, People’s Republic of China; Cat. #CB104-03), and positive colonies were selected for PCR authentication (GeneChem Corporation). The vectors pHelper1.0 and pHelper2.0 were used to pack the virus particles, which were constructed as previously described.14 SMMC-7721 cells (2×105 cells/well) were seeded in six-well plates; when cell growth reached 70%–80% confluency, appropriate volumes of lentivirus were added to the cells at a multiplicity of infection of 10. The rate of infected cells was determined 48–72 h after infection by observation under a fluorescence microscope (IX71, Olympus Corporation, Tokyo, Japan) to detect the expression of green fluorescent protein. The cells were used in subsequent experiments when the rate of infected cells reached 70%.
Western blot
Total protein was extracted from cells by lysis in radio immunoprecipitation assay buffer (Ding Guo Biotechnology Corporation, Shanghai, People’s Republic of China). Whole-cell protein extracts were prepared and quantified using a BCA Protein Assay kit (Beyotime Biotechnology, Shanghai, People’s Republic of China). Protein lysates were subjected to 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and wet-transferred (Bio-Rad Laboratories Inc., Hercules, CA, USA) at 300 mA for 120 min onto PVDF membranes (Millipore, Billerica, MA, USA). The PVDF membrane was blocked using Tris-buffered saline/tween with 5% non-fat milk at room temperature for 2 h or at 4°C overnight and was incubated with primary antibody and subsequently with an appropriate secondary antibody. Protein bands formed were visualized using an ECL Western Blotting Substrate kit (Pierce, Thermo Fisher Scientific, Waltham, MA, USA). The primary Mouse Anti-Flag antibody was purchased from Sigma Biotechnology (Sigma-Aldrich Co., St Louis, MO, USA) and Mouse Anti-GAPDH was purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). The secondary antibody, Goat Anti-Mouse IgG, was purchased from Santa Cruz Biotechnology Inc.
Cell growth assay
As reported previously,15 we performed multiparametric high-content screening to measure cell growth. SMMC-7721 cells at the logarithmic phase, after infection with either PSC3741 or PSC39360, were seeded at a density of 1,000 cells/well into 96-well plates; the cells were then incubated at 37°C with 5% CO2 for 5 days. The cells were counted daily using the Celigo Imaging Cytometer (Nexcelom Bioscience, Lawrence, MA, USA), and each experiment was performed in triplicate.
MTT cell proliferation assay
Lentivirus-infected SMMC-7721 cells were seeded at a density of 2,000 cells/well into 96-well plates, and cell viability was assessed using MTT (Genview, Craigieburn, Australia). MTT (20 μL; 5 mg/mL) was added to each well and incubated for 4 h at 37°C. Formazan crystals formed were then dissolved by adding 100 μL dimethyl sulfoxide. The cells were shaken for 2–5 min, and the absorbance at 490 nm was measured using a microplate reader (Tecan Infinite, Tecan GmbH, Austria). Each experiment was performed in triplicate.
Colony formation assay
We performed colony formation assay to detect the growth of cells. Briefly, SMMC-7721 cells were infected with lentivirus; 3 days later, 1,000 cells/well were seeded into six-well plates and cultured for further 11 days at 37°C. The status of cells was observed, and the replacement of the medium was performed once every 2 or 3 days. At the end of the culture period, the cells were washed with phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde for 30–60 min. Then, they were washed with PBS, stained with Giemsa for 10–20 min, and washed several times with double-distilled water. The number of colonies formed was counted and photographed under a microscope. (Cai Kang Optical Instrument Co., Ltd, Shanghai, People’s Republic of China).
Cell cycle analysis
Cell cycle distribution was determined via flow cytometry analysis. SMMC-7721 cells infected with lentivirus were collected and washed once with pre-chilled D-Hanks solution (pH 7.2–7.4; Shanghai GeneChem Gene Chemical Technology Inc., Shanghai, People’s Republic of China). The cells were fixed in pre-chilled 75% ethanol for at least 1 h before washing once with D-Hanks solution. Samples were then stained with a solution containing propidium iodide (PI, 2 mg/mL; Sigma-Aldrich Co.), RNase concentrate (10 mg/mL), and D-Hanks solution at a ratio of 25:10:1,000, and the cells were analyzed on a flow cytometer (Guava easyCyte HT, Millipore). Each experiment was performed in triplicate.
Animal experiments
Twenty-four-week-old female BALB/c nude mice (Ling Chang Biological Technology Corporation, Shanghai, People’s Republic of China) were randomly divided into two groups – PSC3741 (NC) and LVpGCSIL-004PSC39360 (KD) – with 10 mice in each group. All nude mice were injected subcutaneously in the right front flank with 5×106 SMMC-7721 cells that were infected with PSC3741 (NC) or LVpGCSIL-004PSC39360 (KD). Tumor size was gauged first at 20 days after injection and then measured twice weekly. Tumor volume was calculated according to the formula: volume = length × width2 × 3.16/6. In addition, tumorigenesis was assessed in mice using live animal imaging technique. All the mice were injected intraperitoneally with D-luciferin (15 mg/mL) and pentobarbital sodium solution (70%) at 10 μL/g weight. After 5 min, anaesthetized mice were sent for in vivo imaging to inspect fluorescent expression throughout their bodies. All animal experiments were executed according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals, with the approval of the Animal Research Committee of the Medical School of Shandong University, Jinan, Shandong Province, People’s Republic of China.
PathScan intracellular signaling array
Infected SMMC-7721 cells were collected and lysed. A PathScan® Stress and Apoptosis Signaling Antibody Array Kit (Cell Signaling Technology, #7018) was then used, according to the manufacturer’s instructions,16 to detect variations in intracellular signaling pathways.
Statistical analysis
Student’s t-test and Kruskal–Wallis test were performed to analyze the raw data. All values in the text and figures are expressed as the mean ± standard deviation of these observations. Statistical analysis was performed using the SPSS 21.0 software package. Furthermore, P-values <0.05 were considered to be statistically significant.
Results
Syt-7 is overexpressed in HCC cells
We assessed the expression level of Syt-7 in cells by RT-qPCR. According to the results, Syt-7 mRNA was highly expressed in Huh-7 and Hep3B cells; moderately expressed in SMMC-7721, HepG2, and BEL-7402 cells; and lowly expressed in L-O2 cell (Table 1 and Figure 1A). SMMC-7721 is more stable than the other cell lines and was used for subsequent experiments.
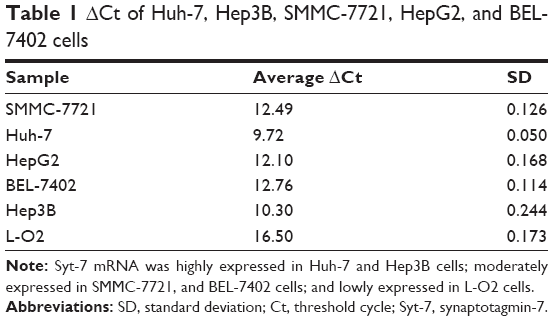 | Table 1 ΔCt of Huh-7, Hep3B, SMMC-7721, HepG2, and BEL-7402 cells |
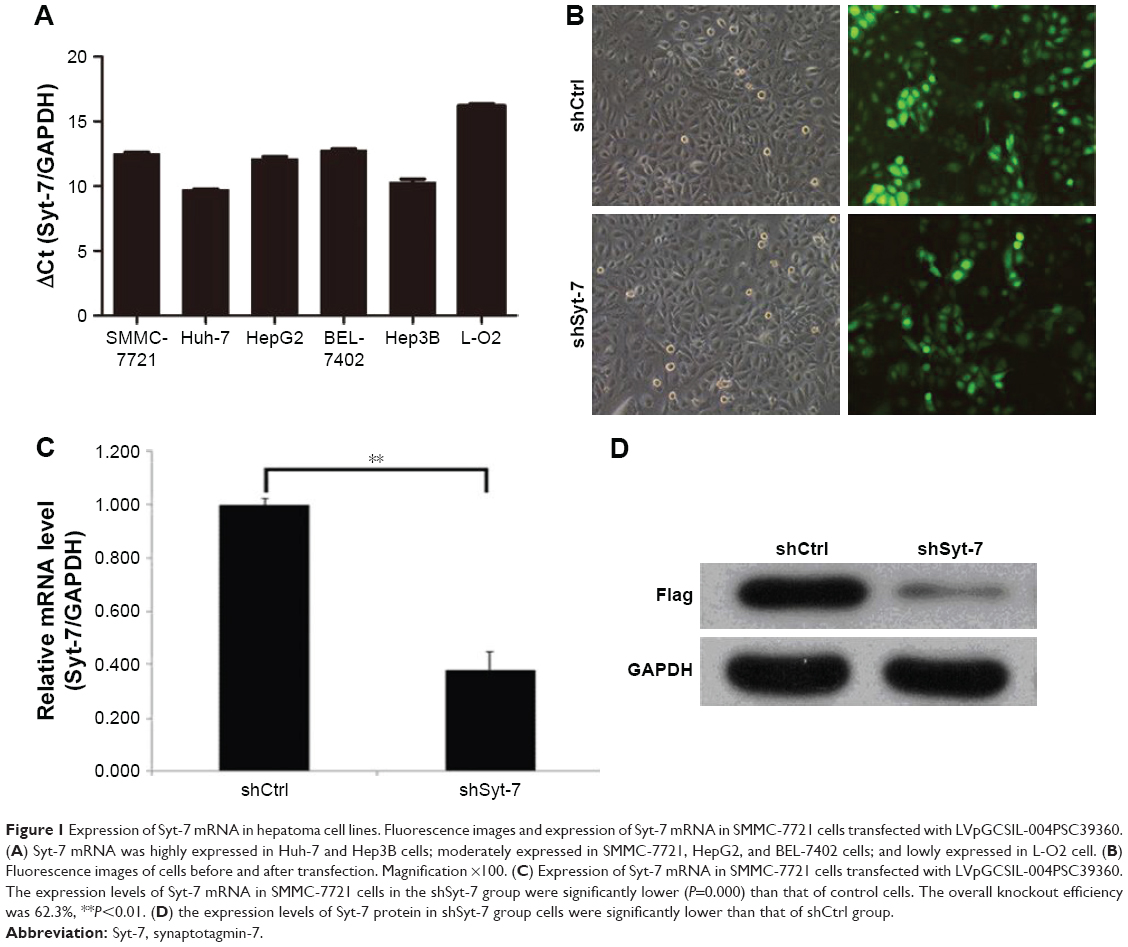 | Figure 1 Expression of Syt-7 mRNA in hepatoma cell lines. Fluorescence images and expression of Syt-7 mRNA in SMMC-7721 cells transfected with LVpGCSIL-004PSC39360. (A) Syt-7 mRNA was highly expressed in Huh-7 and Hep3B cells; moderately expressed in SMMC-7721, HepG2, and BEL-7402 cells; and lowly expressed in L-O2 cell. (B) Fluorescence images of cells before and after transfection. Magnification ×100. (C) Expression of Syt-7 mRNA in SMMC-7721 cells transfected with LVpGCSIL-004PSC39360. The expression levels of Syt-7 mRNA in SMMC-7721 cells in the shSyt-7 group were significantly lower (P=0.000) than that of control cells. The overall knockout efficiency was 62.3%, **P<0.01. (D) the expression levels of Syt-7 protein in shSyt-7 group cells were significantly lower than that of shCtrl group. |
shRNA-mediated Syt-7 knockdown efficiency in HCC cells
To evaluate the role of Syt-7, we knocked down Syt-7 in SMMC-7721 cells. As shown in Figure 1B, 72 h after transfection, the proportion of infected cells in both shCtrl and shSyt-7 groups had reached 70%. According to the results of RT-qPCR (Figure 1C) and Western blot analysis (Figure 1D), the expression levels of Syt-7 mRNA (P=0.000) and Syt-7 protein in shSyt-7 group of cells were significantly lower than those of the shCtrl group.
Knockdown of Syt-7 suppressed HCC cell growth, proliferation, and colony formation in vitro
To assess the effect of Syt-7 on cell growth, shCtrl and shSyt-7 SMMC-7721 cells were counted every day for 5 days. The rate of cell growth was defined as: cell count of Nth day/cell count of the first day, where N=2, 3, 4, and 5. According to the results, knockdown of Syt-7 significantly reduced the total number of cells and rate of cell growth (Table 2, Figure 2A and B).
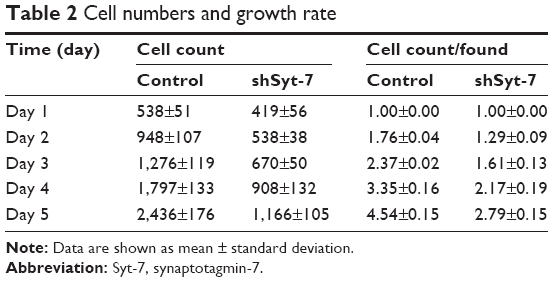 | Table 2 Cell numbers and growth rate |
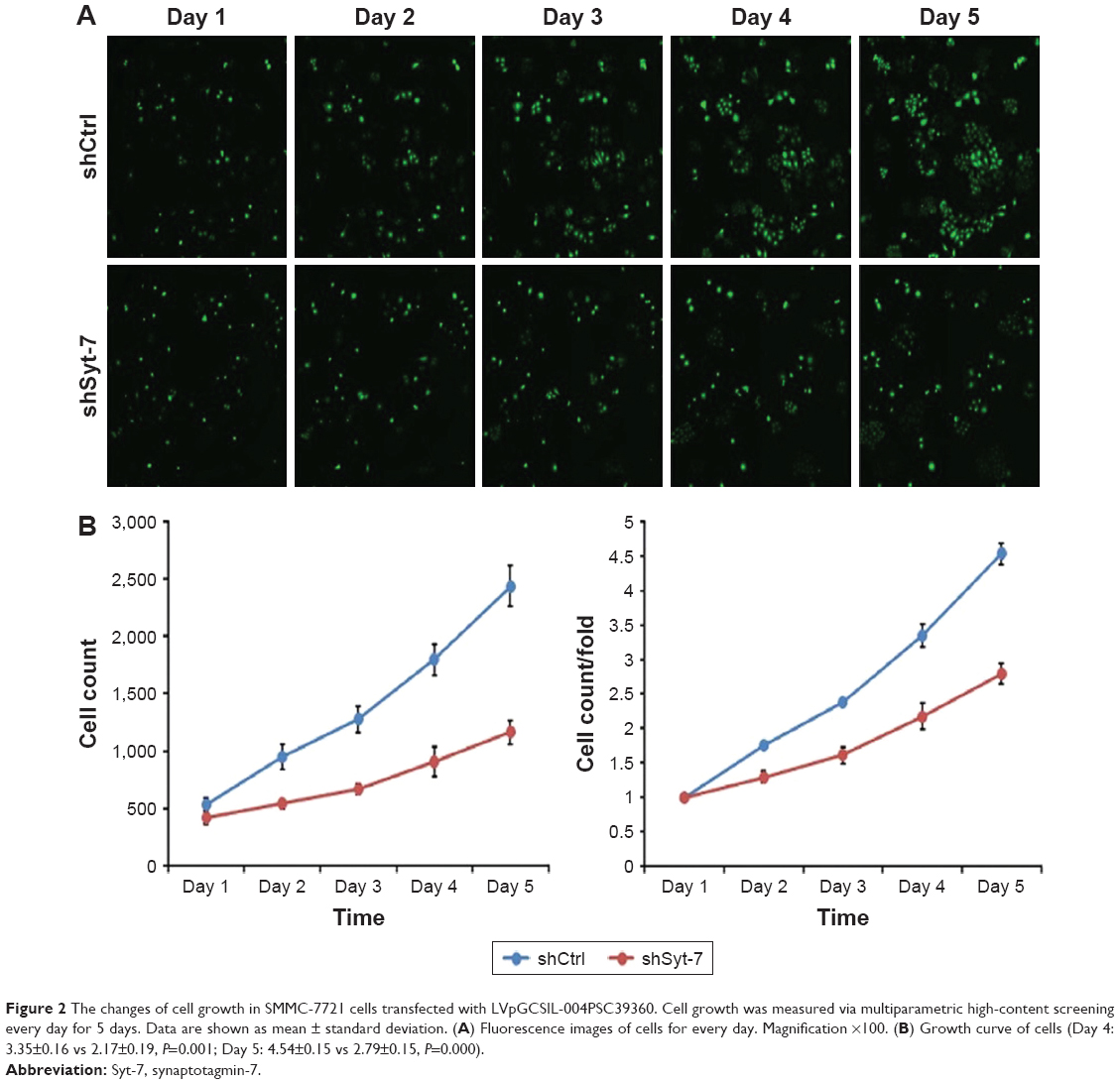 | Figure 2 The changes of cell growth in SMMC-7721 cells transfected with LVpGCSIL-004PSC39360. Cell growth was measured via multiparametric high-content screening every day for 5 days. Data are shown as mean ± standard deviation. (A) Fluorescence images of cells for every day. Magnification ×100. (B) Growth curve of cells (Day 4: 3.35±0.16 vs 2.17±0.19, P=0.001; Day 5: 4.54±0.15 vs 2.79±0.15, P=0.000). |
We evaluated the effect of Syt-7 on cell proliferation via MTT assay. The results showed that the number of viable cells in the shSyt-7 group was significantly higher than that of the control group on days 4 and 5 (P=0.000), that is, knockdown of Syt-7 inhibited cell proliferation (Figure 3).
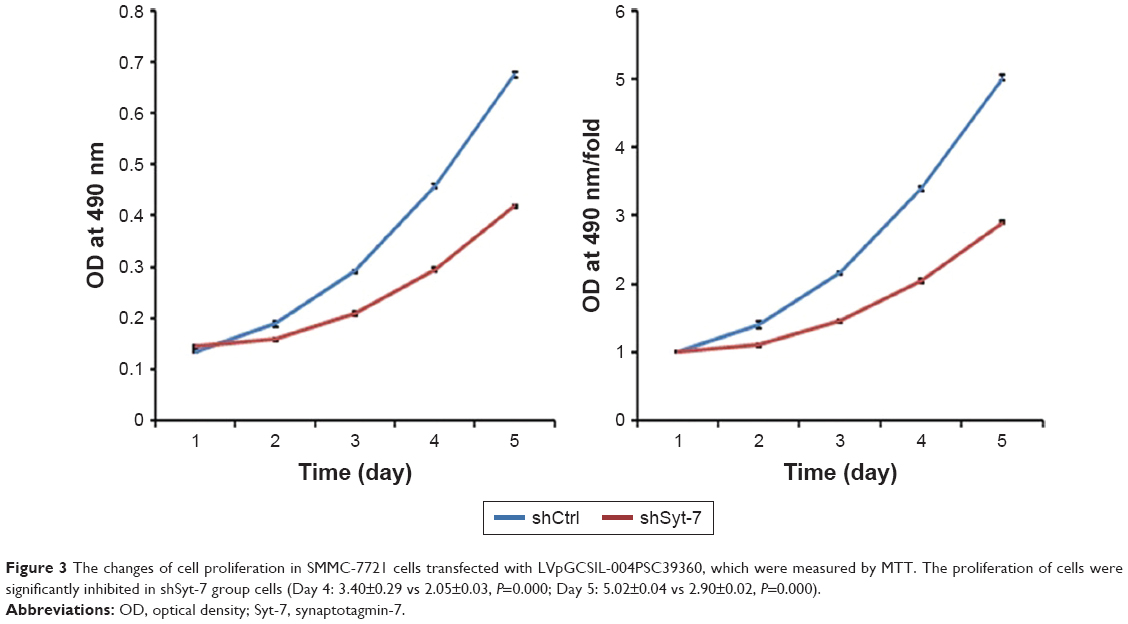 | Figure 3 The changes of cell proliferation in SMMC-7721 cells transfected with LVpGCSIL-004PSC39360, which were measured by MTT. The proliferation of cells were significantly inhibited in shSyt-7 group cells (Day 4: 3.40±0.29 vs 2.05±0.03, P=0.000; Day 5: 5.02±0.04 vs 2.90±0.02, P=0.000). |
Furthermore, we studied the function of Syt-7 knockdown on SMMC-7721 cell tumorigenesis in vitro by assaying colony formation. The results indicated that compared with cells in the control group, cells in the shSyt-7 group had significantly inhibited the ability of SMMC-7721 cells to form colonies (P<0.01). Thus, with this experiment, we have demonstrated that knockdown of Syt-7 restricted the tumorigenicity of hepatoma cells in vitro (Figure 4).
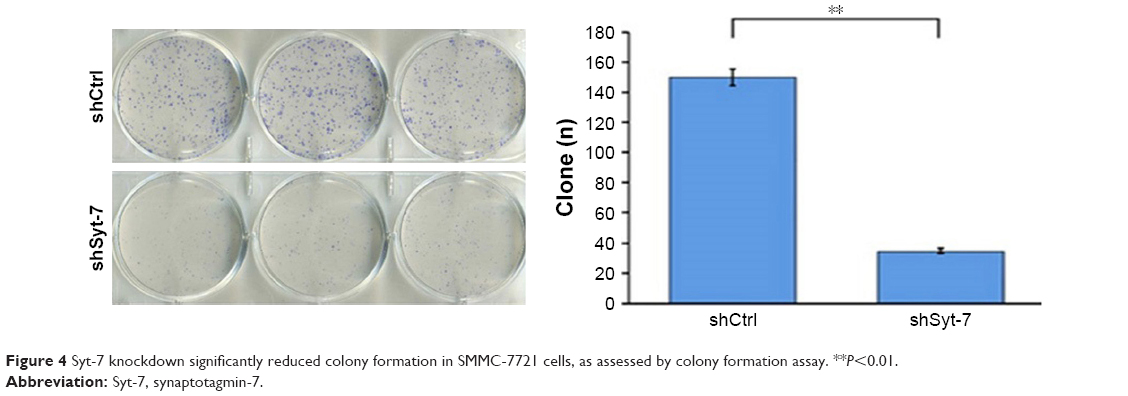 | Figure 4 Syt-7 knockdown significantly reduced colony formation in SMMC-7721 cells, as assessed by colony formation assay. **P<0.01. |
Knockdown of Syt-7 induced cell cycle arrest
To explore alterations in the cell cycle, we analyzed cells stained with PI using flow cytometry. The results show that compared with cells in the control group the proportion of SMMC-7721 cells in G1 phase in the shSyt-7 group significantly decreased (61.74±0.35 vs 65.50±0.68, P=0.0001), whereas the proportion of cells in S phase significantly increased (21.29±1.14 vs 16.59±0.03, P=0.0004). There was no significant change between the groups of cells in G2/M phase (16.97±0.79 vs 17.91±0.71, P>0.05) (Figure 5).
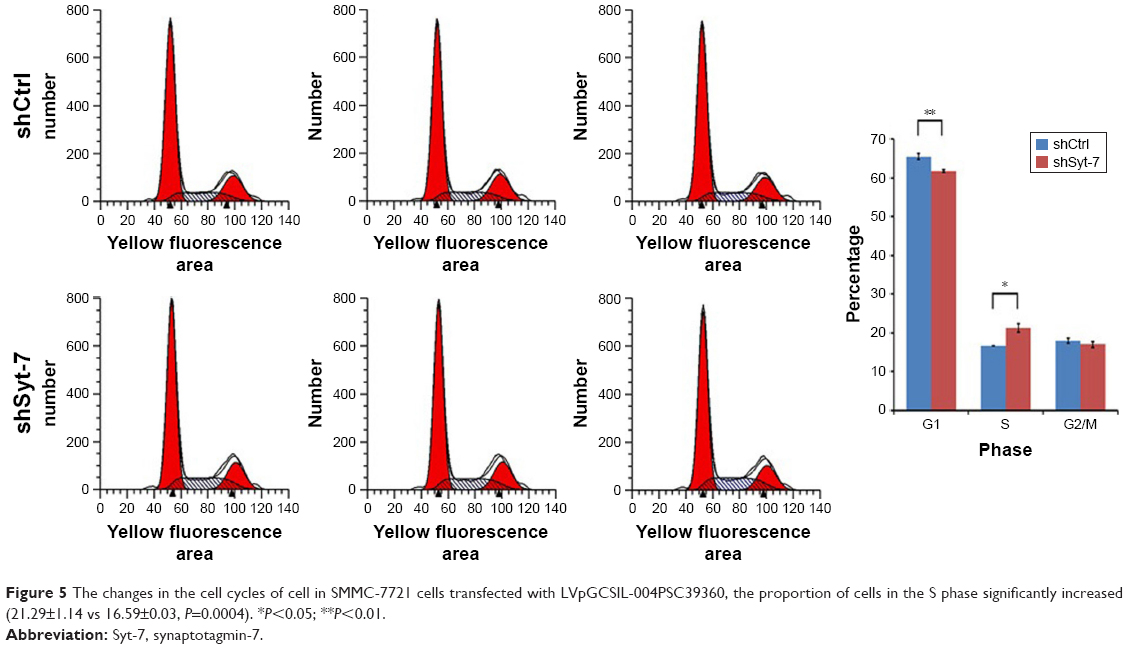 | Figure 5 The changes in the cell cycles of cell in SMMC-7721 cells transfected with LVpGCSIL-004PSC39360, the proportion of cells in the S phase significantly increased (21.29±1.14 vs 16.59±0.03, P=0.0004). *P<0.05; **P<0.01. |
Effect of Syt-7 knockdown on tumorigenesis in nude mice
We explored the effect of Syt-7 knockdown on tumor progression in vivo using BALB/c nude mice. We initially studied the levels of fluorescence expression (μW/cm2) in the bodies of anaesthetized mice. As shown in Figure 6A and B, the levels in the shSyt-7 group were significantly lower than those in the control group (2.83×1010±4.29×109 vs 5.55×1010±9.07×109, P<0.05). After sacrificing the animals, we further measured the size, volume, and weight of the tumors. Tumors in the shSyt-7 group were significantly smaller than those in the control group (Figure 6C), and on day 6, tumor volumes (mm3) were significantly reduced compared to those of the control group (2.61±7.71 vs 823.55±401.62, P<0.05) (Figure 6D). The weight (g) of tumors in the shSyt-7 group was also significantly lower than that of the control group (0.005±0.011 vs 0.830±0.396, P<0.05) (Figure 6E).
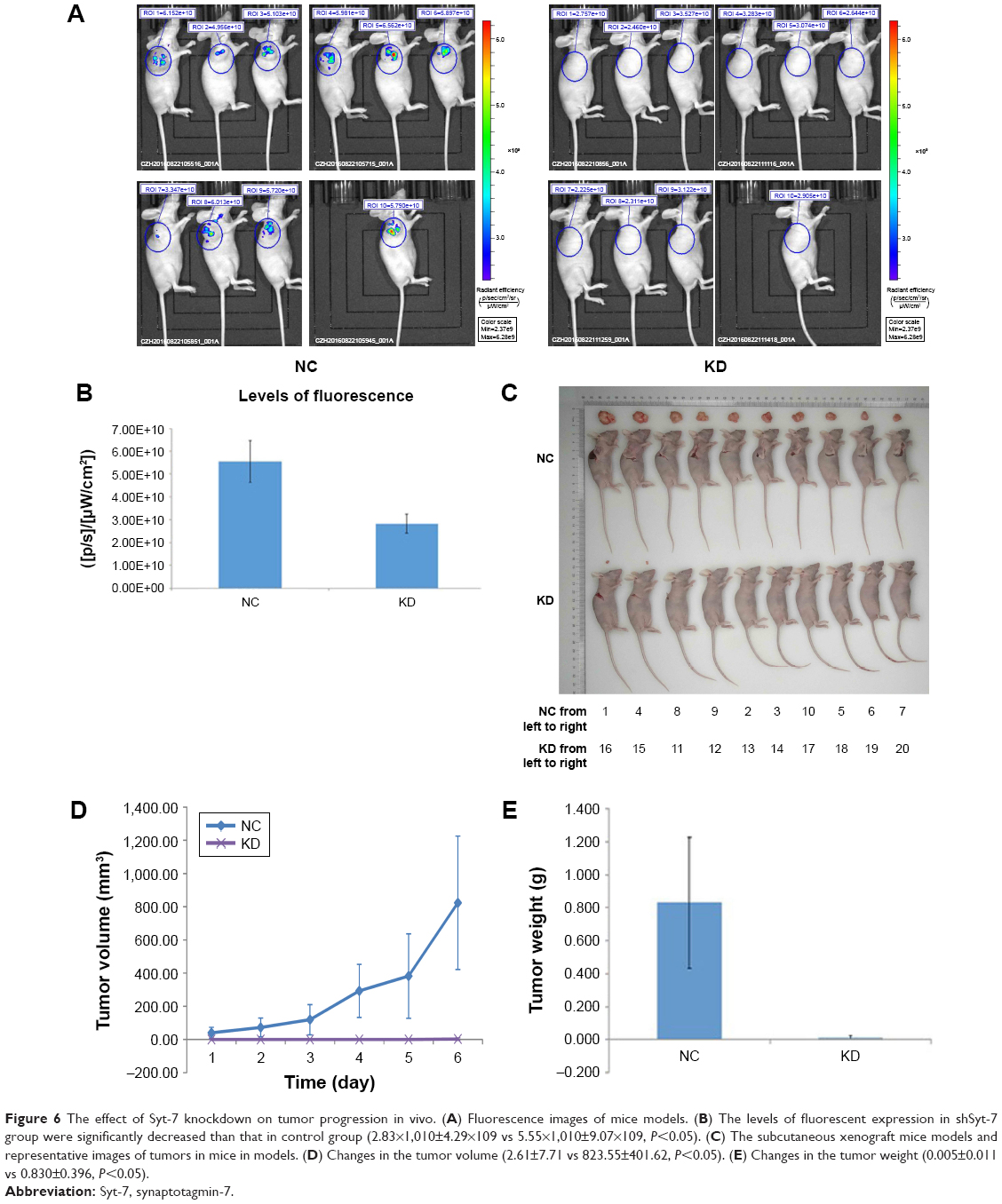 | Figure 6 The effect of Syt-7 knockdown on tumor progression in vivo. (A) Fluorescence images of mice models. (B) The levels of fluorescent expression in shSyt-7 group were significantly decreased than that in control group (2.83×1,010±4.29×109 vs 5.55×1,010±9.07×109, P<0.05). (C) The subcutaneous xenograft mice models and representative images of tumors in mice in models. (D) Changes in the tumor volume (2.61±7.71 vs 823.55±401.62, P<0.05). (E) Changes in the tumor weight (0.005±0.011 vs 0.830±0.396, P<0.05). |
Elucidation of mechanism of Syt-7 in HCC cells
According to the results of the previous experiments, Syt-7 knockdown in SMMC-7721 cells inhibited proliferation and induced S phase arrest. To investigate the involvement of Syt-7 in tumorigenesis in HCC, we analyzed signaling pathways in SMMC-7721 cells after Syt-7 knockdown. The results showed that Syt-7 knockdown significantly increased the phosphorylation of Chk1 at Ser345 and p53 at Ser15, indicating that Syt-7 knockdown inhibited proliferation of SMMC-7721 cells and induced S phase arrest via the Chk1–p53 signaling pathway (Figure 7).
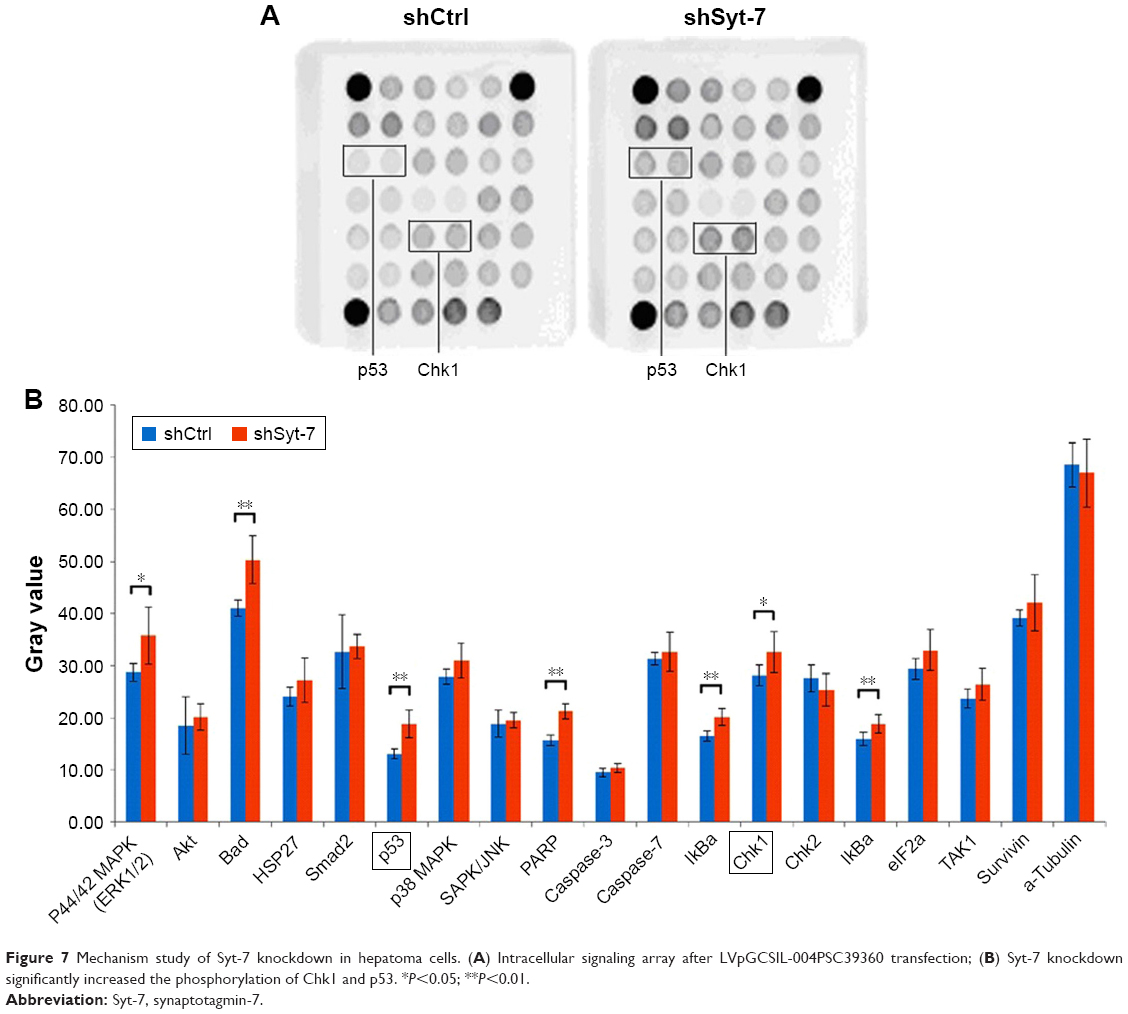 | Figure 7 Mechanism study of Syt-7 knockdown in hepatoma cells. (A) Intracellular signaling array after LVpGCSIL-004PSC39360 transfection; (B) Syt-7 knockdown significantly increased the phosphorylation of Chk1 and p53. *P<0.05; **P<0.01. |
Discussion
Previously, researchers have focused on identifying potential biomarkers that can be of value in the diagnosis and treatment of HCC. There is abundant information about HCC at genetic and molecular levels; however, owing to the restricted clinical value of past confirmed molecular markers, scientists have been encouraged to find new genes. Elucidating suitable targets and understanding the related signaling pathways are of crucial importance. Previous studies have demonstrated that many genes play important roles in the tumorigenesis and development of HCC; the results of experiments have shown that RNAi-mediated gene regulation could significantly influence the biological behavior of hepatoma cells.17–20
Syt-7 is a vesicle exocytosis protein that plays pivotal roles in many physiological and pathological processes. Shih et al21 reported that Syt-7 is important for the Ca2+ sensors for norepinephrine-containing vesicle exocytosis in cardiac sympathetic nerve terminals. Peng et al22 studied the function of PLXNC1 in macrophage migration and collagen accumulation and found that both of these functions could be ameliorated by genetic deletion of Syt-7. This finding demonstrated that Syt-7 has a function in mediating macrophage migration and propelling lung fibrosis. Research on Syts in tumors is scarce. One study reported that expression levels of Syt-13 were significantly lower in left-sided colon carcinoma than those in the right-sided colon carcinoma.23 Calmodulin inhibition of parathyroid hormone secretion is likely due to the absence of Syt-1 in parathyroid adenoma.24 Lung cancer research has also shown that CD74-ROS, a novel ROS receptor tyrosine kinase, can enhance invasive and metastatic abilities of cells through the activation of a novel pathway.25 The core of this pathway is phosphorylation of the extended Syt-like protein E-Syt1.
As far as we know, there are no reports regarding the role of Syt-7 in tumors, particularly HCC, and this is the first study to report the same. We initially investigated the expression of Syt-7 in human hepatoma cell lines and in normal hepatocytes. Our results showed that Huh-7 and Hep3B cells expressed high levels of Syt-7, whereas SMMC-7721, HepG2, and BEL-7402 cells expressed moderate levels. The reason for this difference is potentially complex and may be attributed to tumor heterogeneity. In subsequent experiments, we chose SMMC-7721 cells to probe the effect of Syt-7 knockdown both in vitro and in vivo to elucidate its role in HCC. In further experiments, lentivirus-mediated Syt-7 knockdown significantly inhibited hepatoma cell proliferation and induced cell cycle arrest. The results of cell growth and MTT analysis showed that the cell number and cell growth rate in the shSyt-7 group were significantly reduced compared with those of the control group. Colony formation numbers in the shSyt-7 group were substantially reduced compared with those of the control group. Syt-7 knockdown also induced cell cycle arrest at S phase. Mice with SMMC-7721 cell-derived xenografts were inoculated with chemically altered Syt-7 siRNA for in vivo tumorigenesis assay. The results showed that levels of fluorescence expression, and the size, volume, and weight of tumors in the shSyt-7 group were significantly reduced compared with those of the control group, proving that Syt-7 knockdown retards tumor growth in vivo. Syt-7 knockdown effectively downregulates HCC progression both in vitro and in vivo and plays a vital role in the tumorigenesis and progression of HCC, implying an oncogenic role for Syt-7 in HCC.
Intracellular signaling pathway analysis showed that Syt-7 knockdown increased phosphorylation of Chk1 and p53. Chk1 is a cell cycle checkpoint protein kinase, activated by DNA damage and is involved in DNA damage repair and cell cycle arrest. Excessive activation of Chk1 is harmful to cell survival in the absence of DNA damage.26 Chemoresistance, an important reason for the poor prognosis of HCC, is dependent on the expression of DNA repair enzymes. Wu et al reported that the chemosensitivity of HCC cells could be improved by Mus81 knockdown, and the mechanism is the induction of S phase arrest via the Chk1 pathway.27 Furthermore, p53 is a well-known tumor suppressor gene; its function is to guard against malignant transformation. An abundance of cellular events, such as DNA damage and stress, can trigger p53 activation, resulting in regulation of many downstream genes related with DNA repair, cell cycle arrest, and so on.28 According to a previous publication, eriocitrin may be a natural chemopreventive agent for HCC, because it can restrict the proliferation of hepatoma cell lines by inducing cell cycle arrest in S phase via the activation of p53.29 Researchers have found that the relationship between Chk1 and p53 in DNA repair and cell cycle arrest is complicated; Chk1 can phosphorylate p53, and on the contrary, p53 can also activate Chk1.30 Lee et al studied the chemopreventive activity of pterostilbene against lung cancer and found that low doses of pterostilbene could inhibit proliferation and induce S phase arrest in human A549 lung adenocarcinoma cells. The underlying mechanism was found to be pterostilbene-induced activation of the ATR/ATM–Chk1–p53 axis, which induces Chk1 and p53 phosphorylation.31 We found that Syt-7 knockdown induced phosphorylation of Chk1 and p53 in SMMC-7721 cells. Our findings indicated that Syt-7 silencing suppresses proliferation and arrests cell cycle progression at S phase via activation of the Chk1–p53 signaling pathway in wild-type p53 HCC. We deduce that overexpression of Syt-7 is responsible for the genomic instability thereby causing DNA damage. Thus, DNA damage may lead to cell cycle arrest via activation of Chk1 and p53; however, in HCC, mechanisms such as gene mutation may bypass this surveillance, in turn resulting in malignant transformation. In our experiments, Syt-7 silencing restored this surveillance, and DNA damage resulted in the phosphorylation of Chk1 and p53, thus suppressing proliferation and arresting the cell cycle in order to repair DNA damage.
Conclusion
Our preliminarily demonstrates the role of Syt-7 in HCC. However, there are several problems to be resolved, including the expression level of Syt-7 in specimens and its clinical significance. Our research provides a basis for utilizing Syt-7 as a new diagnostic and therapeutic target in HCC.
Disclosure
The authors report no conflicts of interest in this work.
References
El-Serag HB, Kanwal F. Epidemiology of hepatocellular carcinoma in the United States: where are we? Where do we go? Hepatology. 2014;60(5):1767–1775. | ||
Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. | ||
Llovet JM, Ricci S, Mazzaferro V, et al; SHARP Investigators Study Group. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–390. | ||
Wang Z, Qu L, Deng B, et al. STYK1 promotes epithelial-mesenchymal transition and tumor metastasis in human hepatocellular carcinoma through MEK/ERK and PI3K/AKT signaling. Sci Rep. 2016;6:33205. | ||
Chapman ER. How does synaptotagmin trigger neurotransmitter release? Annu Rev Biochem. 2008;77:615–641. | ||
Gustavsson N, Han W. Calcium-sensing beyond neurotransmitters: functions of synaptotagmins in neuroendocrine and endocrine secretion. Biosci Rep. 2009;29(4):245–259. | ||
Fukuda M, Kanno E, Satoh M, Saegusa C, Yamamoto A. Synaptotagmin VII is targeted to dense-core vesicles and regulates their Ca2+ −dependent exocytosis in PC12 cells. J Biol Chem. 2004;279(50):52677–52684. | ||
Weber JP, Toft-Bertelsen TL, Mohrmann R, Delgado-Martinez I, Sorensen JB. Synaptotagmin-7 is an asynchronous calcium sensor for synaptic transmission in neurons expressing SNAP-23. PLoS One. 2014;9(11):e114033. | ||
Gustavsson N, Wei SH, Hoang DN, et al. Synaptotagmin-7 is a principal Ca2+ sensor for Ca2+ −induced glucagon exocytosis in pancreas. J Physiol. 2009;587(Pt 6):1169–1178. | ||
Wu B, Wei S, Petersen N, et al. Synaptotagmin-7 phosphorylation mediates GLP-1-dependent potentiation of insulin secretion from beta-cells. Proc Natl Acad Sci U S A. 2015;112(32):9996–10001. | ||
Glavan G, See RE, Zivin M. Differential patterns of synaptotagmin7 mRNA expression in rats with kainate- and pilocarpine-induced seizures. PLoS One. 2012;7(5):e36114. | ||
Colvin RA, Means TK, Diefenbach TJ, et al. Synaptotagmin-mediated vesicle fusion regulates cell migration. Nat Immunol. 2010;11(6):495–502. | ||
Svec D, Tichopad A, Novosadova V, Pfaffl MW, Kubista M. How good is a PCR efficiency estimate: recommendations for precise and robust qPCR efficiency assessments. Biomol Detect Quantif. 2015;3:9–16. | ||
Sun W, Yao L, Jiang B, Guo L, Wang Q. Spindle and kinetochore-associated protein 1 is overexpressed in gastric cancer and modulates cell growth. Mol Cell Biochem. 2014;391(1–2):167–174. | ||
Nabzdyk CS, Chun M, Pradhan Nabzdyk L, Yoshida S, LoGerfo FW. Differential susceptibility of human primary aortic and coronary artery vascular cells to RNA interference. Biochem Biophys Res Commun. 2012;425(2):261–265. | ||
Wang J, Yu S, Cui L, et al. Role of SMC1A overexpression as a predictor of poor prognosis in late stage colorectal cancer. BMC Cancer. 2015;15:90. | ||
Zhu Q, Gong L, Wang J, et al. miR-10b exerts oncogenic activity in human hepatocellular carcinoma cells by targeting expression of CUB and sushi multiple domains 1 (CSMD1). BMC Cancer. 2016;16(1):806. | ||
Figarola JL, Singhal J, Tompkins JD, et al. SR4 uncouples mitochondrial oxidative phosphorylation, modulates AMP-dependent kinase (AMPK)-mammalian target of rapamycin (mTOR) signaling, and inhibits proliferation of HepG2 hepatocarcinoma cells. J Biol Chem. 2015;290(51):30321–30341. | ||
Jiang P, Cao J, Bai WH. Lentivirus-mediated siRNA targeting ER-alpha inhibits tumorigenesis and induces apoptosis in hepatocarcinoma cells. BioMed Res Int. 2015;2015:490681. | ||
Qi J, Li T, Bian H, et al. SNAI1 promotes the development of HCC through the enhancement of proliferation and inhibition of apoptosis. FEBS Open Bio. 2016;6(4):326–337. | ||
Shih AM, Varghese L, Bittar A, Park SH, Chung JM, Shin OH. Dysregulation of norepinephrine release in the absence of functional synaptotagmin 7. J Cell Biochem. 2016;117(6):1446–1453. | ||
Peng X, Moore M, Mathur A, et al. Plexin C1 deficiency permits synaptotagmin 7-mediated macrophage migration and enhances mammalian lung fibrosis. FASEB J. 2016;30(12):4056–4070. | ||
Zhu H, Wu TC, Chen WQ, et al. Screening for differentially expressed genes between left- and right-sided colon carcinoma by microarray analysis. Oncol Lett. 2013;6(2):353–358. | ||
Lu M, Berglund E, Larsson C, Höög A, Farnebo LO, Bränström R. Calmodulin and calmodulin-dependent protein kinase II inhibit hormone secretion in human parathyroid adenoma. J Endocrinol. 2011;208(1):31–39. | ||
Jun HJ, Johnson H, Bronson RT, de Feraudy S, White F, Charest A. The oncogenic lung cancer fusion kinase CD74-ROS activates a novel invasiveness pathway through E-Syt1 phosphorylation. Cancer Res. 2012;72(15):3764–3774. | ||
Zhang Y, Hunter T. Roles of Chk1 in cell biology and cancer therapy. Int J Cancer. 2014;134(5):1013–1023. | ||
Wu F, Chen WJ, Yan L, et al. Mus81 knockdown improves chemosensitivity of hepatocellular carcinoma cells by inducing S-phase arrest and promoting apoptosis through CHK1 pathway. Cancer Med. 2016;5(2):370–385. | ||
Hientz K, Mohr A, Bhakta-Guha D, Efferth T. The role of p53 in cancer drug resistance and targeted chemotherapy. Oncotarget. 2017;8(5):8921–8946. | ||
Wang Z, Zhang H, Zhou J, et al. Eriocitrin from lemon suppresses the proliferation of human hepatocellular carcinoma cells through inducing apoptosis and arresting cell cycle. Cancer Chemother Pharmacol. 2016;78(6):1143–1150. | ||
Lezina L, Purmessur N, Antonov AV, et al. miR-16 and miR-26a target checkpoint kinases Wee1 and Chk1 in response to p53 activation by genotoxic stress. Cell Death Dis. 2013;4(12):e953. | ||
Lee H, Kim Y, Jeong JH, Ryu JH, Kim WY. ATM/CHK/p53 pathway dependent chemopreventive and therapeutic activity on lung cancer by pterostilbene. PLoS One. 2016;11(9):e0162335. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.