Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Surface-modified solid lipid nanoparticles for oral delivery of docetaxel: enhanced intestinal absorption and lymphatic uptake
Authors Cho H, Park JW ![]() , Yoon I, Kim D
, Yoon I, Kim D ![]()
Received 26 October 2013
Accepted for publication 6 December 2013
Published 13 January 2014 Volume 2014:9(1) Pages 495—504
DOI https://doi.org/10.2147/IJN.S56648
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Hyun-Jong Cho1, Jin Woo Park2, In-Soo Yoon2, Dae-Duk Kim3
1College of Pharmacy, Kangwon National University, Chuncheon, 2College of Pharmacy and Natural Medicine Research Institute, Mokpo National University, Jeonnam, 3College of Pharmacy and Research Institute of Pharmaceutical Sciences, Seoul National University, Seoul, Republic of Korea
Abstract: Docetaxel is a potent anticancer drug, but development of an oral formulation has been hindered mainly due to its poor oral bioavailability. In this study, solid lipid nanoparticles (SLNs) surface-modified by Tween 80 or D-alpha-tocopheryl poly(ethylene glycol 1000) succinate (TPGS 1000) were prepared and evaluated in terms of their feasibility as oral delivery systems for docetaxel. Tween 80-emulsified and TPGS 1000-emulsified tristearin-based lipidic nanoparticles were prepared by a solvent-diffusion method, and their particle size distribution, zeta potential, drug loading, and particle morphology were characterized. An in vitro release study showed a sustained-release profile of docetaxel from the SLNs compared with an intravenous docetaxel formulation (Taxotere®). Tween 80-emulsified SLNs showed enhanced intestinal absorption, lymphatic uptake, and relative oral bioavailability of docetaxel compared with Taxotere in rats. These results may be attributable to the absorption-enhancing effects of the tristearin nanoparticle. Moreover, compared with Tween 80-emulsified SLNs, the intestinal absorption and relative oral bioavailability of docetaxel in rats were further improved in TPGS 1000-emulsified SLNs, probably due to better inhibition of drug efflux by TPGS 1000, along with intestinal lymphatic uptake. Taken together, it is worth noting that these surface-modified SLNs may serve as efficient oral delivery systems for docetaxel.
Keywords: solid lipid nanoparticles, vitamin E TPGS, docetaxel, lymphatic uptake, bioavailability, toxicity
Introduction
Docetaxel, a second-generation taxane, is widely used in the treatment of breast cancer, non-small cell lung cancer, prostate cancer, gastric adenocarcinoma, and head/neck cancers.1 It acts as a promoter of microtubule polymerization, leading to cell cycle arrest at G2/M, apoptosis, and cytotoxicity.2,3 An intravenous formulation of docetaxel is currently marketed (Taxotere®, Sanofi SA, Paris, France). However, it contains a high concentration of Tween 80, a nonionic surfactant that has been associated with severe hypersensitivity reactions.4 Moreover, intravenous administration has several drawbacks, including morbidity related to the intravenous access site, risk of catheter-related infection, potential thrombosis and extravasation, and the presence of particulate matter in infusion preparations.5
Oral chemotherapy would have advantages over the current intravenous chemotherapy regimen.6,7 Oral treatment of cancer is noninvasive and cost-saving in terms of time and labor, and is available to outpatients, resulting in better patient compliance and improved quality of life, particularly for patients with advanced or relapsed cancer and the elderly.8–10 Moreover, oral administration of anticancer drugs can provide a prolonged systemic exposure profile with less fluctuation, which may lead to lower toxicity and improved efficacy.11,12 Thus, oral chemotherapy for docetaxel may be a desirable alternative to the current intravenous infusion regimen.
Unfortunately, clinical application of docetaxel via the oral route is hindered due to its poor oral bioavailability.13 It is generally believed that P-glycoprotein (Pgp)-mediated efflux in the intestine and cytochrome P450 (CYP)3A-mediated first-pass metabolism in the intestine and/or liver, together with poor aqueous solubility (0.025 μg/mL), are primarily responsible for the low oral bioavailability of docetaxel.14,15 Several studies have shown that the oral bioavailability of docetaxel can be enhanced significantly by coadministration of Pgp and/or CYP3A inhibitors, such as cyclosporin A, ritonavir, interferon-alpha, and ontogen (ONT-093).14,16–18 However, the usefulness of these drugs in clinical practice is limited, especially for repeated administration, because of the risk of side effects, which include immunosuppression.19
Solid lipid nanoparticles (SLNs) are submicron (50–1,000 nm) colloidal particulate systems composed of physiologically tolerable lipid components, which remain in the solid state at room temperature.20 SLNs represent an alternative drug delivery system to emulsions and polymeric nanoparticles.21 They can overcome the membrane stability and drug-leaching problems associated with emulsions and the toxicity problems of polymeric nanoparticles.22 SLN systems can solubilize poorly water-soluble drugs and provide controlled release.20 The lipid core of SLNs has been reported to stimulate chylomicron formation and facilitate lymphatic uptake, which can bypass hepatic first-pass drug metabolism.23,24 Moreover, SLNs generally contain lipophilic or hydrophilic surfactants as stabilizers, some of which have been reported to inhibit Pgp-mediated efflux.5,25 Thus, SLNs have attracted much interest as an oral delivery system for lipophilic drugs with poor bioavailability. To date, SLNs have been used successfully as one of the oral drug delivery systems for enhancing the bioavailability of lipophilic drugs, such as cyclosporin A, nitrendipine, testosterone, halofantrine, paclitaxel, vinpocetine, quercetin, and lopinavir.5,20,23,26 These characteristics make SLNs an attractive oral delivery system for docetaxel.
Herein, we report on surface-modified SLNs for oral delivery of docetaxel. The SLNs were prepared by a solvent-diffusion method using biodegradable and biocompatible materials, including tristearin, Tween 80, and D-α-tocopherol polyethylene glycol 1000 succinate (TPGS 1000). Tween 80 is currently used as a surfactant in the intravenous formulation of docetaxel (Taxotere). Moreover, TPGS 1000 is widely used as a surfactant in the pharmaceutical industry, and has been reported to enhance the intestinal absorption and bioavailability of paclitaxel, the structural analog of docetaxel, due to its potent Pgp-inhibiting activity.27 Thus, Tween 80 and TPGS 1000 were employed as surfactants for preparing docetaxel-loaded SLNs in this study. The SLNs were then characterized in terms of their particle size distribution, zeta potential, drug loading, and particle morphology. Drug release from the SLNs was assessed using a dialysis method. Moreover, in situ intestinal absorption, in vivo intestinal lymphatic uptake, and in vivo oral pharmacokinetic and toxicity studies of docetaxel-loaded SLNs and Taxotere were undertaken in rat models.
Materials and methods
Materials
Docetaxel was purchased from Taihua Co (Xi’an, People’s Republic of China). Butyl 4-hydroxybenzoate (internal standard for the high-performance liquid chromatographic [HPLC] analysis of docetaxel), paclitaxel (internal standard for the liquid chromatography/tandem mass spectrometry [LC-MS/MS] of docetaxel), Tween 80, and TPGS 1000 were purchased from Sigma-Aldrich (St Louis, MO, USA). Tristearin was purchased from Tokyo Chemical Industry Co (Tokyo, Japan). Other chemicals were of reagent or HPLC grade.
Preparation of docetaxel-loaded SLNs
Docetaxel-loaded SLNs were prepared by the solvent-diffusion method in an aqueous system, as reported elsewhere but with slight modifications.28–30 Briefly, 150 mg of tristearin and 10 mg of docetaxel were dissolved completely in a 10 mL mixture of acetone and ethanol (1:1, v/v) in a water bath at 70°C. The resulting organic solution was dispersed quickly into 100 mL of an aqueous phase containing 0.01% (w/v) of Tween 80 (F1) or 0.01% (w/v) of TPGS (F2) under continuous mechanical agitation at 400 rpm in a water bath at 70°C for 5 minutes. The resulting pre-emulsion (melted lipid droplets) was ultrasonicated at 30 W for 10 minutes and subsequently transferred into an ice bath to solidify the lipid droplets. The dispersion was then purified by dialysis against distilled water for 12 hours to remove water-soluble impurities (organic solvents and nonadsorbed surfactants) and subsequently centrifuged (1,600 × g, 5 minutes) to remove large lipid particles and precipitate free docetaxel. The supernatant was lyophilized, redispersed in distilled water, vortex-mixed, centrifuged, and syringe-filtered (Minisart RC15 0.2 μm; Sartorius Biotechnology, Göttingen, Germany) to obtain a docetaxel-loaded SLN dispersion. A drug-free SLN dispersion was prepared in the same manner, with the exception that the 10 mg of docetaxel was replaced with 10 mg of tristearin.
Drug entrapment efficiency and loading content
Drug entrapment efficiency (EE) and loading content (LC) were determined as reported previously.31 An 0.1 mL aliquot of docetaxel-loaded SLNs was added to 1 mL of acetone and ethanol (1:1, v/v) at 70°C, and the mixture was vortex-mixed for 24 hours. The mixture was then centrifuged to separate the undissolved components, and the supernatant containing the drug extracted from the SLNs was analyzed using an HPLC method. The equations for drug EE and LC are as follows:
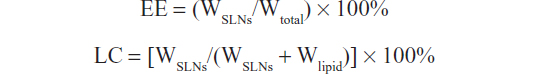
where WSLNs, Wtotal, and Wlipid represent the amount of drug in the SLNs, amount of drug added, and amount of excipients added, respectively.
Characterization of docetaxel-loaded SLNs
Particle size and zeta potential
The particle size, polydispersity index, and zeta potential of the drug-free (blank control) and docetaxel-loaded SLNs were measured using a dynamic light-scattering instrument (ELS-Z; Otsuka Electronics, Tokyo, Japan; dilution factor 1; refractive index 1.3328).
Transmission electron microscopy
The morphology of the docetaxel-loaded SLNs was examined using an energy-filtering transmission electron microscope (Libra120, Carl Zeiss, Göttingen, Germany) with an accelerating voltage of 80 kV. The SLNs were negatively stained by 2% sodium phosphotungstate (pH 7) and placed on carbon-coated, 400-mesh copper grids, followed by drying at room temperature before use.
In vitro drug release study
The in vitro release of docetaxel from SLNs, compared with Taxotere, a parenteral formulation of docetaxel, was evaluated using the dialysis method.32,33 Taxotere was prepared by dissolving docetaxel (10 mg/mL) in distilled water containing 25% (w/v) of Tween 80 and 9.75% (v/v) of ethanol. Aliquots of Taxotere and docetaxel-loaded SLNs (200 μL) were placed in mini dialysis kits (molecular weight cutoff 12–14 kDa; Gene Bio-Application Ltd., Kfar Hanagid, Israel) and immersed in 20 mL of release medium (0.5% of Tween 80 in phosphate-buffered saline, pH 7.4) in a shaking incubator at 100 rpm at 37°C. Aliquots of dissolution medium (0.5 mL) were then withdrawn, and the concentration of docetaxel was determined by HPLC after appropriate dilution with methanol. The cumulative percent release of docetaxel from the SLNs was calculated and plotted as a function of time.
Animals
Protocols for the animal studies were approved by the Institutional Animal Care and Use Committee of Seoul National University (Seoul, Republic of Korea). Male Sprague Dawley rats (aged 7–9 weeks, weighing 200–250 g) were purchased from Orient Bio Inc (Seongnam, South Korea). They were maintained in a clean room (Animal Center for Pharmaceutical Research, College of Pharmacy, Seoul National University) at a temperature of 20°C–23°C with 12/12-hour light (7 am to 7 pm)/dark (7 pm to 7 am) cycles, and a relative humidity of 50%±5%. The rats were housed in metabolic cages (Tecniplast, Varese, Italy) under filtered, pathogen-free air, with food (Agribrands Purina Korea, Pyeongtaek, South Korea) and water available ad libitum.
In situ closed-loop study in rats
The absorption of docetaxel from Taxotere and from the docetaxel-loaded SLNs in various rat intestinal segments was evaluated using an in situ closed-loop study.10 After a minimal abdominal incision under light ether anesthesia and sufficient washing of the contents within the gastrointestinal tract, jejunum, ileum, and colon loops each 5 cm in length were closed by ligations made approximately 2 cm distal to the ends of each intestinal section. Special care was taken to avoid damaging blood vessels and to include as much of a complete mesenteric blood vessel arch as possible in each loop. After injection of the docetaxel formulations (1 mg docetaxel; ~0.5 mL) into each loop using a 1 mL syringe with a 31 gauge needle, the whole gastrointestinal tract was carefully replaced into the abdominal cavity, and the incision was closed using clamps and kept moist by covering with gauze pads presoaked in normal saline. The rat was warmed using a lamp. At 120 minutes after drug injection, each loop was removed, transferred into a beaker containing 50 mL of methanol, and the gastrointestinal tract was cut into small pieces using scissors to facilitate the extraction of unabsorbed docetaxel. After manual shaking and stirring with a glass rod for one minute, a 50 μL aliquot of the supernatant was collected from each beaker and stored at −80°C until assay of docetaxel by HPLC.
In vivo intestinal lymphatic uptake study in rats
The femoral vein was cannulated with a polyethylene tube (PE-50) under light ketamine anesthesia (50 mg/kg, intramuscularly). A small incision was made in the abdomen, the duodenum was located, and the docetaxel formulations were injected directly using a 1 mL syringe with a 31 gauge needle. The whole gastrointestinal tract was then carefully replaced in the abdominal cavity, and the incision was closed using clamps and kept moist by covering with gauze pads presoaked in normal saline. The rat was warmed using a lamp, and normal saline was then infused at a rate of 1.5 mL per hour during the experiment to rehydrate the rats. At 0.5 or 1.5 hours after administration of the docetaxel formulations (Taxotere, F1, or F2) at a dose of 10 mg/kg, the rats were sacrificed by cervical dislocation, and the mesenteric lymph node was isolated as reported previously.34,35 The mesenteric lymph nodes obtained were weighed and homogenized with four volumes of phosphate-buffered saline using a Polytron homogenizer (PT 3100D, Kinematica, Lucerne, Switzerland). After centrifugation, a 200 μL aliquot of homogenate was transferred to a new 2 mL tube, and 1.5 mL of methanol was added. The mixture was vortex-mixed for 3 hours, incubated for 30 minutes at 65°C, vortex-mixed, and centrifuged (16,000 × g, 10 minutes). The supernatant was then transferred to a new 1.5 mL tube and dried under a gentle stream of nitrogen gas at 40°C. Next, 100 μL of mobile phase was added to reconstitute the residue, and after centrifugation, 10 μL of supernatant was subjected to LC-MS/MS analysis.
In vivo pharmacokinetic study in rats
The femoral artery was cannulated with a polyethylene tube (PE-50; Clay Adams, Parsippany, NJ, USA) under light ketamine anesthesia (50 mg/kg, intramuscularly).36 Taxotere and docetaxel-loaded SLNs at a dose of 20 mg/kg were administered orally to rats using a feeding tube after overnight fasting with free access to water. Blood samples approximately 200 μL in volume were collected via the femoral artery at 0 (control), and 15, 30, 45, 60, 75, 90, 120, 180, and 240 minutes after oral administration of the docetaxel formulations. An approximately 300 μL aliquot of heparinized 0.9% NaCl injectable solution (20 U/mL) was used to flush the cannula immediately after each blood sampling to prevent blood clotting. After centrifugation of the blood sample, a 100 μL aliquot of plasma sample was obtained. Acetonitrile (150 μL) containing 25 ng/mL of paclitaxel (internal standard) and 100 μL of 0.1% acetic acid were added to 100 μL of plasma sample. After vortex-mixing and centrifugation, 10 μL of supernatant was subjected to LC-MS/MS.
In vivo toxicity study in rats
The jejunum (approximately 5 cm) was removed 8 hours after oral administration of distilled water, Taxotere, or docetaxel-loaded SLNs. The segment was then washed with phosphate-buffered saline and fixed in 4% paraformaldehyde for 24 hours. A vertical section was prepared, stained with hematoxylin and eosin, and observed under a light microscope (×200).
HPLC and LC-MS/MS analyses of docetaxel
Concentrations of docetaxel were determined using LC-MS/MS (plasma and lymph node homogenate samples in oral study) or HPLC (other samples) methods as reported previously with slight modifications.37 For LC-MS/MS analysis, 10 μL of the prepared samples were injected directly onto a C18 column (Symmetry BEH phenyl; 100 × 2.1 mm id; particle size, 1.7 mm; Waters, Milford, MA, USA). The mobile phase, 0.1% acetic acid:acetonitrile (50:50, v/v), was run at a flow rate of 0.3 mL per minute. An ABI/MDS Sciex model API 4000 triple quadrupole mass spectrometer (Framingham, MA, USA) was used. The source temperature was set at 150°C, the ion spray voltage was 5,000 V, the gases were set at 50 for the nebulizing and curtain gases, and at 20 and 50 for the auxiliary and CAD gases, respectively. The MS/MS transition of docetaxel measured was 808.4 → 527.4 and that of paclitaxel was 854.3 → 286.2. The detection limit of docetaxel in rat plasma was 1 ng/mL, based on a signal-to-noise ratio of 3. The interday and intraday coefficients of variation were each <12.7%.
For HPLC analysis, 200 μL of methanol containing 5 μg/mL butyl 4 hydroxybenzoate (internal standard) was added to 100 μL of sample. After vortex-mixing and centrifugation, the supernatant was collected and dried (Dry Thermobath; Eyela, Tokyo, Japan) under a gentle stream of nitrogen gas at 40°C. Next, 100 μL of mobile phase was added to reconstitute the residue, and after centrifugation, 50 μL of supernatant was injected directly onto a C18 column (Symmetry; 300 × 4.6 mm id; particle size, 5 mm; μ bondapak; Waters). The mobile phase, ie, distilled water:acetonitrile (55:45, v/v), was run at a flow rate of 1.0 mL per minute with an ultraviolet detector at 230 nm and room temperature. The detection limit of docetaxel was 50 ng/mL, based on a signal-to-noise ratio of 3. The interday and intraday coefficients of variation were each <10.8%.
Pharmacokinetic analysis
The total area under the plasma concentration-time curve from time zero to time infinity (AUC) was calculated using standard software (WinNonlin; Pharsight Corporation, Mountain View, CA, USA). The peak plasma concentration (Cmax) and time to reach Cmax (Tmax) were read directly from the experimental data. The extent of relative oral bioavailability was calculated by dividing the AUC after oral administration of SLNs (F1 and F2) by the AUC after oral administration of Taxotere.
Statistical analysis
A P-value<0.05 was considered to be statistically significant using a t-test between the two means for the unpaired data or a Duncan’s multiple range test of Statistical Package for the Social Sciences (version 18.0, SPSS Inc., Chicago, IL, USA) posteriori analysis of variance among the more than three means for unpaired data. All data are expressed as the mean ± standard deviation, except the median (range) for Tmax.
Results and discussion
Characterization of docetaxel-loaded SLNs
Table 1 lists the particle size, polydispersity, zeta potential, drug EE, and LC of the drug-free (F1-blank and F2-blank) and docetaxel-loaded SLNs (F1, F2). F1 and F2 were of submicron particle sizes ranging from 189 nm to 215 nm and had high EEs ranging from 80.7% to 83.1%. The polydispersity indices ranging from 0.191 to 0.214 for the SLNs indicate that they are polydispersed systems having neither a very narrow (polydispersity index <0.05) nor a very broad (polydispersity index >0.7) size distribution. In general, a colloidal dispersion having an absolute zeta potential value >30 mV could be stabilized by electric repulsion between particles.35 Thus, the negative zeta potentials for the SLNs, ranging from −35.0 to −27.7 mV, could improve their physical stability. The particle size, polydispersity index, and zeta potential of the drug-free SLNs were not significantly different from those of the docetaxel-loaded SLNs. The size distributions and transmission electron microscopic images of docetaxel-loaded SLNs are shown in Figure 1. Some spherical particles were observed, and their sizes seemed to be approximately 200–300 nm. However, no significant differences between F1 and F2 were observed regarding the above-mentioned characteristics.
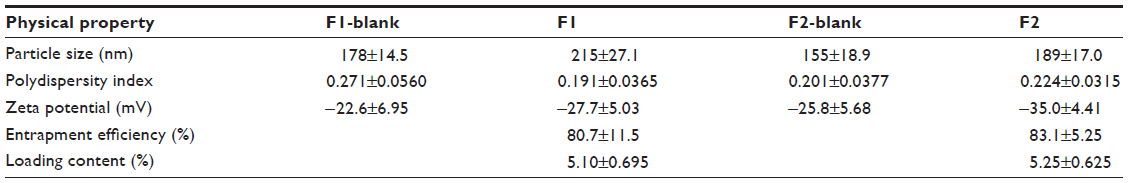 | Table 1 Physical properties of drug-free (F1-blank and F2-blank) and docetaxel-loaded SLNs (F1 and F2) |
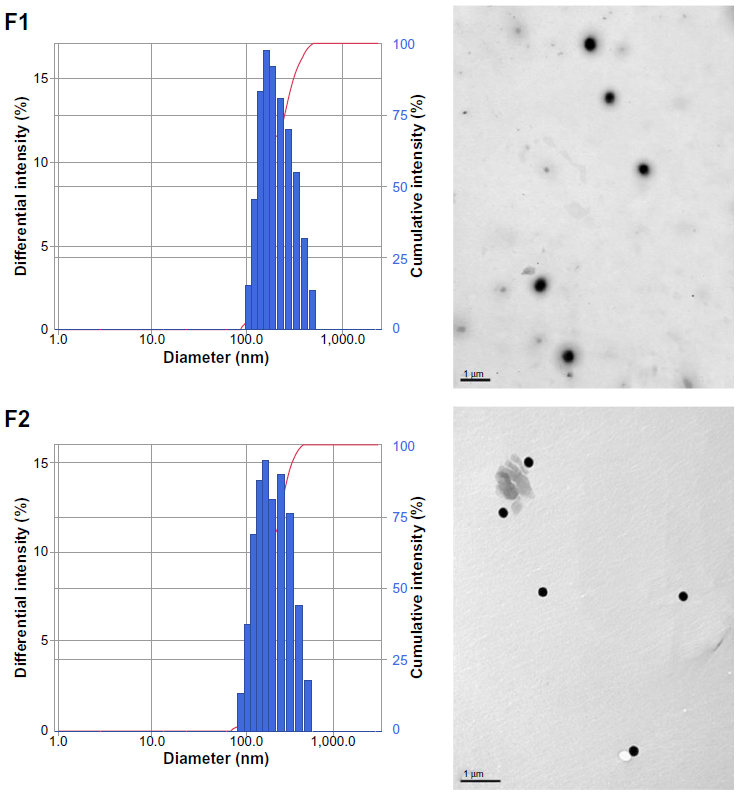 | Figure 1 Size distributions (left) and transmission electron microscopic images (right) of docetaxel-loaded SLNs (F1 and F2). |
In vitro drug release study
The release experiment was conducted under sink conditions, and a dialysis membrane was used to separate free drug from entrapped drug. Phosphate-buffered saline containing 0.5% (w/v) Tween 80 was used to dissolve the free docetaxel, a poorly water-soluble drug. The solubility of docetaxel in 0.5% (w/v) Tween 80 at 37°C was 52.3±7.16 μg/mL, which was sufficient to maintain sink conditions. Figure 2 shows the time profiles of in vitro release of docetaxel from Taxotere, F1, and F2 at 37°C in phosphate-buffered saline. Docetaxel release from SLNs was much slower than that of Taxotere. Drug release from orally administered SLNs occurs mainly via diffusion through the solid lipid matrix and/or lipid nanoparticle degradation in the gut.38 Thus, the diffusion process through the solid lipid matrix might have resulted in the slower release of docetaxel from SLNs compared with Taxotere. However, there was no significant difference in docetaxel release between F1 and F2.
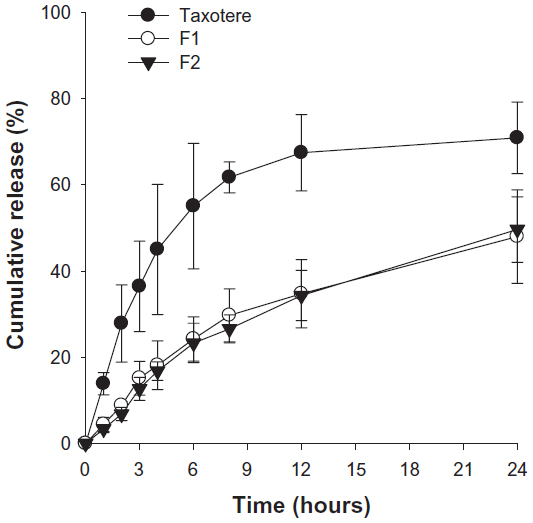 | Figure 2 Time profiles for in vitro release of docetaxel from Taxotere (●), F1 (○), and F2 (▾) at 37°C in phosphate-buffered saline. |
In situ closed-loop study in rats
The fractions of docetaxel remaining 2 hours after injection of Taxotere and docetaxel-loaded SLNs in the rat jejunal, ileal, and colonic loops are shown in Figure 3. The mean values of the remaining fractions of docetaxel after injection of Taxotere ranged from 72.8% to 79.0%, indicating limited intestinal absorption of docetaxel from Taxotere. It is known that intestinal Pgp-mediated efflux and poor water solubility are primarily responsible for the limited intestinal absorption of docetaxel.13,17 In the jejunal and colonic loops, the fractions of docetaxel remaining after F1 injection were significantly lower than that of Taxotere, indicating that F1 enhanced the intestinal absorption of docetaxel. Considering that Taxotere contains a high concentration of Tween 80 (54%, w/v), the enhanced intestinal absorption of docetaxel from F1, compared with Taxotere, seems to be attributable primarily to the lipid core (tristearin) of F1 rather than the emulsifier, Tween 80. It has been reported that oral SLNs can be absorbed as intact nanoparticles through Peyer’s patches and microfold cells, mainly in the ileum and colon, which is generally accepted as the main oral absorption mechanism for other nanoparticles.26 Moreover, oral SLNs may be digested in the intestinal fluid, and the lipid degradation products (fatty acids and monoglycerides or diglycerides) can promote intestinal drug absorption by formation of mixed micelles with bile acids and subsequent uptake into enterocytes.38 Thus, tristearin nanoparticles could enhance the intestinal absorption of docetaxel by bypassing Pgp-mediated efflux via these mechanisms.
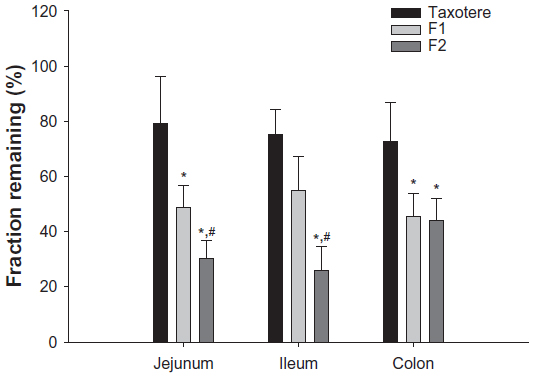 | Figure 3 Remaining fractions of docetaxel at 2 hours after injection of Taxotere® (Sanofi SA, Paris, France), F1, and F2 into rat jejunum, ileum, and colon loops. |
Interestingly, in the jejunal and ileal loops, the fractions of docetaxel remaining after injection of F2 were significantly lower than those of F1, indicating that F2 further enhanced the intestinal absorption of docetaxel. Because F1 and F2 contain the same tristearin lipid core, the further enhancement of intestinal absorption of docetaxel from F2 would seem to be attributable to the emulsifier, TPGS 1000. TPGS 1000 is a water-soluble derivative of natural vitamin E, consisting of a lipophilic head (D-α-tocopherol) and a hydrophilic tail (polyethylene glycol 1000).27 It has been reported to inhibit Pgp-mediated efflux by ATPase inhibition with an IC50 value of 4.81±2.98 μg/mL.39 Moreover, the absorptive transport of paclitaxel, a compound structurally similar to docetaxel, across rat ileum was markedly enhanced (by more than 4.7-fold) in the presence of 2–1,000 μg/mL TPGS 1000.27 In addition to TPGS 1000, various other nonionic surfactants, including Tween 80, have been reported to inhibit Pgp-mediated efflux activity.40,41 However, their Pgp inhibition characteristics may vary greatly with the type and concentration of nonionic surfactants and Pgp substrates. The IC50 values of TPGS 1000 and Tween 80 for the inhibition of Pgp-mediated efflux in Caco-2 cells have been reported to be 6 μg/mL and 200 μg/mL, respectively.40 Moreover, the enhancing effects of 500 μg/mL Tween 80 on the absorptive transport of paclitaxel were only moderate (1.47-fold) in CaCo-2 cells and negligible in MDR1-MDCK cells.42 Although the contents of Tween 80 and TPGS 1000 incorporated into docetaxel-loaded SLNs are unknown, further enhancement of intestinal absorption of docetaxel from F2 could have been due partly to greater Pgp inhibition by TPGS 1000 than by Tween 80. Taken together, the possible mechanisms for enhancement of intestinal docetaxel absorption by the SLNs include the cellular uptake of intact SLNs (into Peyer’s patches and microfold cells) and/or lipid degradation product-based mixed micelles (into enterocytes) to bypass Pgp-mediated efflux (Taxotere versus F1) and the higher Pgp-inhibiting activity of TPGS 1000 compared with that of Tween 80 (F1 versus F2).
In vivo lymphatic uptake study in rats
To evaluate the intestinal lymphatic uptake of docetaxel from Taxotere and SLNs, the docetaxel content recovered from the mesenteric lymph node at 0.5 and 1.5 hours after intraduodenal administration of docetaxel formulations to rats was measured. At both time points, the docetaxel content of F1 and F2 was significantly higher than that of Taxotere, while there was no significant difference between F1 and F2 with regard to the docetaxel content recovered from mesenteric lymph nodes (Figure 4). These results suggest that tristearin nanoparticles may enhance the lymphatic uptake of docetaxel. It is well established that oral SLNs facilitate the lymphatic uptake of drugs (eg, lopinavir, tobramycin, and methotrexate), mainly via chylomicron formation.23,24,43 In particular, tristearin nanoparticles have been reported to be effective in enhancing the lymphatic uptake of oral methotrexate.24
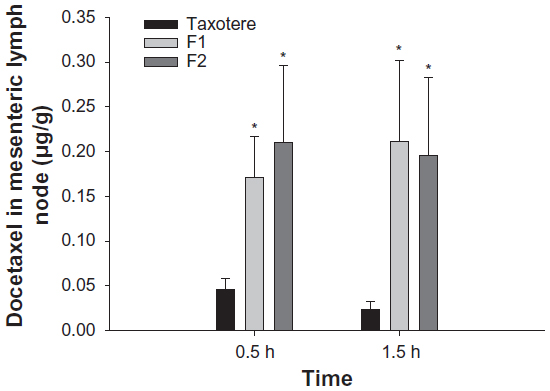 | Figure 4 Amount of docetaxel recovered from the mesenteric lymph nodes at 0.5 and 1.5 hours (h) after intraduodenal administration of Taxotere® (Sanofi SA, Paris, France), F1, and F2 at a dose of 10 mg/kg. |
In vivo pharmacokinetic study in rats
Figure 5 shows the time profiles for arterial plasma concentrations of docetaxel after oral administration of Taxotere, F1, and F2 at a dose of 20 mg/kg in rats. The relevant pharmacokinetic parameters of docetaxel are listed in Table 2. Oral docetaxel doses of 10–30 mg/kg were used in previous studies of the oral delivery of docetaxel.13,15,37 Moreover, docetaxel has been reported to exhibit linear pharmacokinetics at intravenous doses of 2–20 mg/kg and oral doses of 20–100 mg/kg.37 Thus, 20 mg/kg oral doses were used.
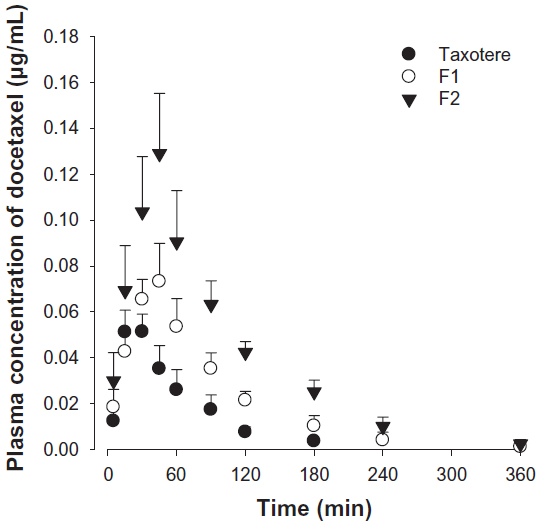 | Figure 5 Time profiles of arterial plasma docetaxel concentrations after oral administration of Taxotere® (Sanofi SA, Paris, France) (●), F1 (○), and F2 (▾) to rats at a dose of 20 mg/kg. |
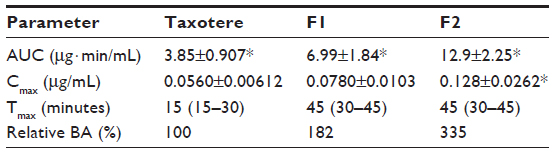 | Table 2 Pharmacokinetic parameters of docetaxel after oral administration of Taxotere®, F1, and F2 at a dose of 20 mg/kg to rats |
After oral administration, the AUC, Cmax, and F values of the three docetaxel formulations were significantly different, in the following order: F2 > F1 > Taxotere. Moreover, the Tmax values of F1 and F2 were relatively delayed compared with Taxotere, indicating prolonged systemic exposure. The results in Figures 3 and 4 suggest that the SLN formulations enhance the oral bioavailability of docetaxel by facilitating intestinal absorption and lymphatic uptake. In particular, the enhanced lymphatic uptake of docetaxel can reduce CYP3A-mediated hepatic first-pass metabolism and improve oral bioavailability, because intestinal lymph vessels drain directly into the thoracic duct, and then into the venous blood, bypassing the portal circulation.44
A good correlation between the fraction of the oral dose absorbed in rats and humans has been reported.45 Moreover, there is a similar level of Pgp expression and overlapping substrate specificity with quantitatively similar affinities for many Pgp substrates in rat and human MDR1.46 Sequential homologies of CYPs (including CYP3A) between rats and humans are also high (more than 70%); there are generally conserved regions (for cytochrome P450 reductase, heme, and signal peptide) that increase this similarity.47 Thus, these in situ and in vivo rat data may predict the intestinal absorption and oral bioavailability of docetaxel in humans.
In vivo toxicity study in rats
The toxicity of docetaxel-loaded SLNs in rat intestinal mucosa was evaluated by histological staining. As shown in Figure 6, there was no evidence of damage to the intestinal wall, such as villi fusion, occasional epithelial cell shedding, and congestion of mucosal capillary with blood and focal trauma, in the parts of the jejunum examined. There was no discernible difference between the control (phosphate-buffered saline), Taxotere, and SLN groups (Figure 6A–D).
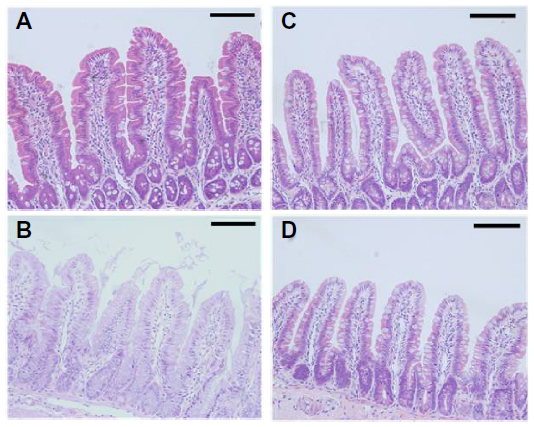 | Figure 6 Representative histological sections of jejunal segments at 8 hours after oral administration of phosphate-buffered saline (A), Taxotere® (Sanofi SA, Paris, France) (B), F1 (C), and F2 (D) to rats. |
Conclusion
SLNs surface-modified by Tween 80 or TPGS 1000 were prepared and evaluated in terms of their feasibility as oral delivery systems for docetaxel. Tween 80-emulsified and TPGS 1000-emulsified tristearin-based lipidic nanoparticles were prepared by a solvent-diffusion method, and their particle size distribution, zeta potential, drug loading, and particle morphology were characterized. The in vitro release study showed a sustained-release profile of docetaxel from the SLNs, compared with an intravenous docetaxel formulation (Taxotere). The Tween 80-emulsified SLNs showed enhanced intestinal absorption, lymphatic uptake, and relative oral bioavailability of docetaxel in rats compared with Taxotere. These results may be attributable to the absorption-enhancing effects of the tristearin nanoparticle system. Moreover, compared with Tween 80-emulsified SLNs, the intestinal absorption and relative oral bioavailability of docetaxel in rats were further improved in TPGS 1000-emulsified SLNs, probably due to higher inhibition of drug efflux by TPGS 1000 along with intestinal lymphatic uptake. Taken together, it is worth noting that the surface-modified SLNs developed in this study may serve as efficient oral delivery systems for docetaxel.
Acknowledgment
This work was supported by the National Research Foundation of Korea grant funded by the Korean government (2009-0083533, 2011-0016040).
Disclosure
The author reports no conflicts of interest in this work.
References
van Waterschoot RA, Lagas JS, Wagenaar E, et al. Absence of both cytochrome P450 3A and P-glycoprotein dramatically increases docetaxel oral bioavailability and risk of intestinal toxicity. Cancer Res. 2009;69(23):8996–9002. | |
Clarke SJ, Rivory LP. Clinical pharmacokinetics of docetaxel. Clin Pharmacokinet. 1999;36(2):99–114. | |
Yoon I, Han S, Choi YH, et al. Saturable sinusoidal uptake is rate-determining process in hepatic elimination of docetaxel in rats. Xenobiotica. 2012;42(11):1110–1119. | |
Gelderblom H, Verweij J, Nooter K, Cremophor EL. the drawbacks and advantages of vehicle selection for drug formulation. Eur J Cancer. 2001;37(13):1590–1598. | |
Peltier S, Oger JM, Lagarce F, Couet W, Benoit JP. Enhanced oral paclitaxel bioavailability after administration of paclitaxel-loaded lipid nanocapsules. Pharm Res. 2006;23(6):1243–1250. | |
DeMario MD, Ratain MJ. Oral chemotherapy: rationale and future directions. J Clin Oncol. 1998;16(7):2557–2567. | |
Le Lay K, Myon E, Hill S, et al. Comparative cost-minimisation of oral and intravenous chemotherapy for first-line treatment of non-small cell lung cancer in the UK NHS system. Eur J Health Econ. 2007;8(2):145–151. | |
Dong Y, Feng SS. Poly(d,l-lactide-co-glycolide)/montmorillonite nanoparticles for oral delivery of anticancer drugs. Biomaterials. 2005;26(30):6068–6076. | |
Bromberg L. Polymeric micelles in oral chemotherapy. J Control Release. 2008;128(2):99–112. | |
Kim JE, Cho HJ, Kim JS, et al. The limited intestinal absorption via paracellular pathway is responsible for the low oral bioavailability of doxorubicin. Xenobiotica. 2013;43(7):579–591. | |
Kalaria DR, Sharma G, Beniwal V, Ravi Kumar MN. Design of biodegradable nanoparticles for oral delivery of doxorubicin: in vivo pharmacokinetics and toxicity studies in rats. Pharm Res. 2009;26(3):492–501. | |
Zhang Z, Feng SS. Nanoparticles of poly(lactide)/vitamin E TPGS copolymer for cancer chemotherapy: synthesis, formulation, characterization and in vitro drug release. Biomaterials. 2006;27(2):262–270. | |
Yan YD, Kim DH, Sung JH, Yong CS, Choi HG. Enhanced oral bioavailability of docetaxel in rats by four consecutive days of pre-treatment with curcumin. Int J Pharm. 2010;399(1–2):116–120. | |
Malingre MM, Richel DJ, Beijnen JH, et al. Coadministration of cyclosporine strongly enhances the oral bioavailability of docetaxel. J Clin Oncol. 2001;19(4):1160–1166. | |
Yin YM, Cui FD, Mu CF, et al. Docetaxel microemulsion for enhanced oral bioavailability: preparation and in vitro and in vivo evaluation. J Control Release. 2009;140(2):86–94. | |
Ben Reguiga M, Bonhomme-Faivre L, Farinotti R. Bioavailability and tissular distribution of docetaxel, a P-glycoprotein substrate, are modified by interferon-alpha in rats. J Pharm Pharmacol. 2007;59(3):401–408. | |
Kuppens IE, Bosch TM, van Maanen MJ, et al. Oral bioavailability of docetaxel in combination with OC144-093 (ONT-093). Cancer Chemother Pharmacol. 2005;55(1):72–78. | |
Oostendorp RL, Huitema A, Rosing H, et al. Coadministration of ritonavir strongly enhances the apparent oral bioavailability of docetaxel in patients with solid tumors. Clin Cancer Res. 2009;15(12):4228–4233. | |
Woo JS, Lee CH, Shim CK, Hwang SJ. Enhanced oral bioavailability of paclitaxel by coadministration of the P-glycoprotein inhibitor KR30031. Pharm Res. 2003;20(1):24–30. | |
Hu L, Tang X, Cui F. Solid lipid nanoparticles (SLNs) to improve oral bioavailability of poorly soluble drugs. J Pharm Pharmacol. 2004;56(12):1527–1535. | |
Tsai MJ, Huang YB, Wu PC, et al. Oral apomorphine delivery from solid lipid nanoparticles with different monostearate emulsifiers: pharmacokinetic and behavioral evaluations. J Pharm Sci. 2011;100(2):547–557. | |
Muller RH, Runge SA, Ravelli V, Thunemann AF, Mehnert W, Souto EB. Cyclosporine-loaded solid lipid nanoparticles (SLN): drug-lipid physicochemical interactions and characterization of drug incorporation. Eur J Pharm Biopharm. 2008;68(3):535–544. | |
Aji Alex MR, Chacko AJ, Jose S, Souto EB. Lopinavir loaded solid lipid nanoparticles (SLN) for intestinal lymphatic targeting. Eur J Pharm Sci. 2011;42(1–2):11–18. | |
Paliwal R, Rai S, Vaidya B, et al. Effect of lipid core material on characteristics of solid lipid nanoparticles designed for oral lymphatic delivery. Nanomedicine. 2009;5(2):184–191. | |
Dong X, Mattingly CA, Tseng MT, et al. Doxorubicin and paclitaxel-loaded lipid-based nanoparticles overcome multidrug resistance by inhibiting P-glycoprotein and depleting ATP. Cancer Res. 2009;69(9):3918–3926. | |
Li H, Zhao X, Ma Y, Zhai G, Li L, Lou H. Enhancement of gastrointestinal absorption of quercetin by solid lipid nanoparticles. J Control Release. 2009;133(3):238–244. | |
Varma MV, Panchagnula R. Enhanced oral paclitaxel absorption with vitamin E-TPGS: effect on solubility and permeability in vitro, in situ and in vivo. Eur J Pharm Sci. 2005;25(4–5):445–453. | |
Hu FQ, Jiang SP, Du YZ, Yuan H, Ye YQ, Zeng S. Preparation and characterization of stearic acid nanostructured lipid carriers by solvent diffusion method in an aqueous system. Colloids Surf B Biointerfaces. 2005;45(3–4):167–173. | |
Hu FQ, Yuan H, Zhang HH, Fang M. Preparation of solid lipid nanoparticles with clobetasol propionate by a novel solvent diffusion method in aqueous system and physicochemical characterization. Int J Pharm. 2002;239(1–2):121–128. | |
Trotta M, Debernardi F, Caputo O. Preparation of solid lipid nanoparticles by a solvent emulsification-diffusion technique. Int J Pharm. 2003;257(1–2):153–160. | |
Subedi RK, Kang KW, Choi HK. Preparation and characterization of solid lipid nanoparticles loaded with doxorubicin. Eur J Pharm Sci. 2009;37(3–4):508–513. | |
Liu D, Liu Z, Wang L, Zhang C, Zhang N. Nanostructured lipid carriers as novel carrier for parenteral delivery of docetaxel. Colloids Surf B Biointerfaces. 2011;85(2):262–269. | |
Luo Y, Chen D, Ren L, Zhao X, Qin J. Solid lipid nanoparticles for enhancing vinpocetine’s oral bioavailability. J Control Release. 2006;114(1):53–59. | |
Kobayashi H, Miura S, Nagata H, et al. In situ demonstration of dendritic cell migration from rat intestine to mesenteric lymph nodes: relationships to maturation and role of chemokines. J Leukoc Biol. 2004;75(3):434–442. | |
Selga E, Perez-Cano FJ, Franch A, et al. Gene expression profiles in rat mesenteric lymph nodes upon supplementation with conjugated linoleic acid during gestation and suckling. BMC Genomics. 2011;12:182. | |
Yoon IS, Choi MK, Kim JS, Shim CK, Chung SJ, Kim DD. Pharmacokinetics and first-pass elimination of metoprolol in rats: contribution of intestinal first-pass extraction to low bioavailability of metoprolol. Xenobiotica. 2011;41(3):243–251. | |
Choi YH, Suh JH, Lee JH, Cho IH, Lee MG. Effects of tesmilifene, a substrate of CYP3A and an inhibitor of P-glycoprotein, on the pharmacokinetics of intravenous and oral docetaxel in rats. J Pharm Pharmacol. 2010;62(8):1084–1088. | |
Muchow M, Maincent P, Muller RH. Lipid nanoparticles with a solid matrix (SLN, NLC, LDC) for oral drug delivery. Drug Dev Ind Pharm. 2008;34(12):1394–1405. | |
Collnot EM, Baldes C, Wempe MF, et al. Mechanism of inhibition of P-glycoprotein mediated efflux by vitamin E TPGS: influence on ATPase activity and membrane fluidity. Mol Pharm. 2007;4(3):465–474. | |
Bogman K, Erne-Brand F, Alsenz J, Drewe J. The role of surfactants in the reversal of active transport mediated by multidrug resistance proteins. J Pharm Sci. 2003;92(6):1250–1261. | |
Rege BD, Kao JP, Polli JE. Effects of nonionic surfactants on membrane transporters in Caco-2 cell monolayers. Eur J Pharm Sci. 2002;16(4–5):237–246. | |
Hugger ED, Novak BL, Burton PS, Audus KL, Borchardt RT. A comparison of commonly used polyethoxylated pharmaceutical excipients on their ability to inhibit P-glycoprotein activity in vitro. J Pharm Sci. 2002;91(9):1991–2002. | |
Cavalli R, Bargoni A, Podio V, Muntoni E, Zara GP, Gasco MR. Duodenal administration of solid lipid nanoparticles loaded with different percentages of tobramycin. J Pharm Sci. 2003;92(5):1085–1094. | |
Porter CJ, Charman WN. Intestinal lymphatic drug transport: an update. Adv Drug Deliv Rev. 2001;50(1–2):61–80. | |
Chiou WL, Barve A. Linear correlation of the fraction of oral dose absorbed of 64 drugs between humans and rats. Pharm Res. 1998;15(11):1792–1795. | |
Stephens RH, O’Neill CA, Warhurst A, Carlson GL, Rowland M, Warhurst G. Kinetic profiling of P-glycoprotein-mediated drug efflux in rat and human intestinal epithelia. J Pharmacol Exp Ther. 2001;296(2):584–591. | |
Soucek P, Gut I. Cytochromes P-450 in rats: structures, functions, properties and relevant human forms. Xenobiotica. 1992;22(1):83–103. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.