Back to Journals » OncoTargets and Therapy » Volume 10
SUN1 silencing inhibits cell growth through G0/G1 phase arrest in lung adenocarcinoma
Authors Huang W, Huang H, Wang L, Hu J, Song W
Received 22 December 2014
Accepted for publication 6 May 2015
Published 2 June 2017 Volume 2017:10 Pages 2825—2833
DOI https://doi.org/10.2147/OTT.S79727
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Faris Farassati
Weiyi Huang,* Haihua Huang,* Lei Wang, Jiong Hu, Weifeng Song
Department of Oncology, The First People’s Hospital Affiliated to Shanghai Jiaotong University, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Purpose: Cytoskeleton is critical for carcinoma cell proliferation, migration, and invasion. Sad-1 and UNC-84 domain containing 1 (SUN1) is one of the core linkers of nucleoskeleton and cytoskeleton. However, the functions of SUN1 in lung adenocarcinoma are largely unknown.
Methods: In this study, we first transduced the lentivirus delivering the short hairpin RNA (shRNA) against SUN1 to lung adenocarcinoma cells (A549 and 95D cells) with high efficiency. After lentivirus infection, quantitative real-time polymerase chain reaction and Western blotting were used to detect the expressions of SUN1 mRNA and protein. The cell proliferation and colony formation were detected by MTT assay and colony formation assay, respectively. The cell distribution in the cell cycle was analyzed by flow cytometry.
Results: Both mRNA and protein levels of SUN1 were significantly decreased in A549 and 95D cells after lentivirus infection, as indicated by quantitative real-time polymerase chain reaction and Western blot. Next, we found that cell proliferation and colony formation were markedly reduced in SUN1 silenced cells. Moreover, suppression of SUN1 led to cell cycle arrest at G0/G1 phase. Furthermore, Cyclin D1, CDK6, and CDK2 expressions were obviously reduced in A549 cells after SUN1 silencing.
Conclusion: These results suggest that SUN1 plays an essential role in proliferation of lung adenocarcinoma cells in vitro and may be used as a potential therapeutic target for the treatment of lung adenocarcinoma in the future.
Keywords: SU
Introduction
The Sad-1 and UNC-84 (SUN) domain family, composed of SUN1 and SUN2, is an important component of nuclear envelope.1,2 Through interaction with Klarsicht, ANC-1, and Syne/Nesprin homology (KASH) domain proteins, they form the SUN–KASH protein complexes bridge across the inner nuclear membrane and outer nuclear membrane.3 The SUN–KASH protein complexes together with emerin and lamins form a mechanical link between nucleoskeleton and cytoskeleton, linker of the nucleoskeleton and the cytoskeleton complex.4,5 Recently, the structure of this nuclear envelope complex was widely studied.2,4,6 SUN proteins are localized to the inner nuclear membrane where they interact with lamins in the nucleoplasm and recruit KASH proteins to the outer nuclear membrane.1,7 The cytoplasmic domains of KASH proteins are associated with actin and tubulin cytoskeleton networks.1,8
SUN–KASH protein complexes play important roles in various cellular and developmental processes, including gametogenesis, neurogenesis, myogenesis, retinogenesis, and ciliogeneisis.1 SUN was first characterized in Caenorhabditis elegans through molecular analysis of mutant unc-84, which showed defects in cell nuclear migration.9,10 Defects in SUN–KASH proteins can result in misposition of nuclei, for they link nuclear to actin, microtubule, and intermediate filaments. SUN or KASH knockout mice present disrupted neurological and muscular development. Mutations of SUN and KASH proteins contribute to human diseases, such as laminopathies,11–13 ataxia, lissencephaly, and cancer.1
Recent studies show the relationship of SUN–KASH complexes with cancer. Expression of KASH protein Syne/Nesprin-1 is decreased 20–180 folds in early tumors.14 An increased risk of invasive ovarian cancer is found to be potentially associated with a polymorphism of Syne/Nesprin-1.15 Furthermore, mutations in Syne/Nesprin-1 and -2 are found in colorectal and breast cancers.16 Knockdown of Nucleoporin 153 impairs migration of human breast carcinoma cells. Interestingly, the localization of SUN1 changes along with rearrangement of the cytoskeleton in Nucleoporin 153 knockdown cells.17 All these studies imply potential roles of SUN–KASH protein complexes in cancer progression.
Lung adenocarcinoma, along with cancer of the trachea and bronchus, is among the top ten leading causes of death worldwide. According to the World Health Organization (WHO) report, more than 1.5 million deaths are related to lung adenocarcinoma in 2011. Common treatments, such as surgery, chemotherapy, and radiotherapy, are widely used for the treatment of lung adenocarcinoma. However, only 15% of patients diagnosed with lung adenocarcinoma survive 5 years after the diagnosis in the US.18 With the development of biomedical research, gene therapy is promising therapeutic methods in the future. There is a need to discover specific gene therapy targets.
Here, we studied the functions of SUN1 in human lung adenocarcinoma. Using recombinant lentivirus taking the short hairpin RNA (shRNA) against SUN1, we established that SUN1 silenced lung adenocarcinoma cell lines and studied the effects on cell proliferation, colony formation, and cell cycle progression.
Materials and methods
Cell culture
Human lung adenocarcinoma A549, which was derived from the human lung tumor of a 58-year-old white male patient with lung cancer, and 95D, which are highly metastatic cancer cells from human non-small cell lung cancer and human embryonic kidney 293T cells, were purchased from the Cell Bank of Chinese Academy of Sciences (Shanghai, People’s Republic of China). A549 and 293T cell lines were cultured in the Dulbecco’s Modified Eagle’s Medium (Hyclone; Thermo Fisher Scientific, Waltham, MA, USA) containing 10% (v/v) fetal bovine serum (S1810; Biowest, Shanghai, People’s Republic of China). All cell lines were cultured in the incubator at 37°C with 5% CO2.
Lentivirus package and transfection
A shRNA (5′-GAACTAGAACAGACCAAGCAACTCGAGTTGCTTGGTCTGTTCTAGTTCTTTTTT-3′) was designed against human SUN1 transcript variant 1 (NM_001130965), which represents the longest transcript and encodes the longest isoform. In order to confirm the specific knockdown of SUN1 gene, another shRNA (5′-GCTTTCCAAATAGTGGAACTTCTCGAGAAGTTCCACTATTTGGAAAGCTTTTTT-3′) was designed to repeat the experiments. A nontargeting shRNA (5′-GCGGAGGGTTTGAAAGAATATCTCGAGATATTCTTTCAAACCCTCCGCTTTTTT-3′) was used as control. Stem–loop–stem sequences corresponding to each shRNA construct were cloned into the pFH-L vector (Shanghai Hollybio, Shanghai, People’s Republic of China). Recombinant lentiviruses were produced by cotransfecting 293T cells with shRNA expression plasmid and two helper plasmids (pVSVG-I and pCMVΔR8.92) using Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacture’s instruction. Infectious lentiviruses were collected at 24, 48, and 72 hours after transfection and the pooled supernatants centrifuged to remove cell debris and filtered through 0.45 μm filters. Viral titer was determined by the method of end point dilution through counting the number of infected green fluorescent protein (GFP)-positive cells at 100× magnification under fluorescence microscope (Olympus Corporation, Tokyo, Japan). Titer in IU/mL = number of green fluorescent cells × dilution factor/volume of virus solution. Lentivirus solution was divided and put in separate microtubes, then stocked at −80°C. When we needed to use the lentivirus solution, we pulled out one of the microtubes, which did not affect the remaining lentivirus solution in the other microtubes. For lentivirus infection, A549 and 95D cells were seeded in six-well plates at a density of 50,000 cells/well and transduced with recombinant lentivirus (Lv-shSUN1 or Lv-shCon) at a multiplicity of infection of 20. Infection efficiency was determined by counting GFP-positive cells as described earlier.
Quantitative real-time polymerase chain reaction
Four days after lentivirus infection, A549 and 95D cells were washed by ice-cold phosphate-buffered saline (PBS) and harvested. Total RNA was extracted using Trizol (Thermo Fisher Scientific). cDNA was retrotranscribed using Moloney murine leukemia virus reverse transcriptase (Promega Corporation, Fitchburg, WI, USA) according to the manufacturer’s instructions. SUN1 mRNA level was then evaluated by quantitative real-time polymerase chain reaction with SYBR master mixture (Takara, Dalian, People’s Republic of China) on BioRad Connet real-time PCR platform. In brief, the 20 μL reaction mixture contained 10 μL 2× SYBR premix ex taq, 0.8 μL primers (2.5 μM), 5 μL cDNA, and 4.2 μL ddH2O. The qPCR amplification program is as follows: 1 minute at 95°C and 40 cycles of 5 seconds at 95°C, 20 seconds at 60°C. Actin was used as endogenous control. The primers were used as follows: SUN1: 5′-CGTTTCGCTCTCCTTGTAGG-3′ (forward) and 5′-GTCTTGCGCTCCCTATTCAG-3′ (reverse); Actin: 5′-GTGGACATCCGCAAAGAC-3′ (forward) and 5′-AAAGGGTGTAACGCAACTA-3′ (reverse). The experiments were repeated at least three times. Fold changes in expression were calculated using the 2−ΔΔCt method.
Western blot analysis
After washing by ice-cold PBS, A549 and 95D cells were harvested and lysed using radioimmunoprecipitation assaybuffer for 1 hour at 4°C. After centrifuging at 13,000 rpm for 15 minutes, supernatant were collected, mixed with 4× protein loading buffer and treated for 10 minutes at 95°C. Protein samples were then separated by sodium dodecyl sulfate polyacrylamide gel electrophoresisand transferred to the polyvinylidene difluoride membrane. The membrane was incubated with primary antibody against SUN1 (#ab124770, 1:4,000 dilution; Abcam, Cambridge, UK), Cyclin D1 (#MD-17-3, 1:1,000 dilution; Medical & Biological Laboratories, Nagoya, Japan), CDK6 (#19117-1-AP, 1:500 dilution; Proteintech, Chicago, IL, USA), CDK2 (#2546, 1:1,000 dilution; Cell Signaling Technology, Danvers, MA, USA), CDK4 (#2906, 1:500 dilution; Cell Signaling Technology) or, glyceraldehyde 3-phosphate dehydrogenase (#10494-1-AP, 1:50,000 dilution; Proteintech) overnight at 4°C, followed by incubation of anti-rabbit or anti-mouse horseradish peroxidase-linked secondary antibody (1:5,000 dilution; Santa Cruz Biotechnology Inc., Dallas, TX, USA) for 1 hour at room temperature. Enhanced chemiluminescence reaction was performed using kit from the manufacturer (Amersham, Marlborough, MA, USA). Experiments were repeated at least three times.
MTT proliferation assay
After lentivirus infection, A549 and 95D cells were seeded in a 96-well plate at an initial density of 2,500 cells/well. After incubating with 10 μL 3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) for 3 hours, 100 μL acidic isopropanol containing 10% sodium dodecyl sulfate, 5% isopropanol, and 0.01 mol/L HCl was added into each well. After the formazan precipitate dissolved, the plates were read using a microplate reader at a wavelength of 595 nm. Experiments were repeated at least three times.
Colony formation assay
After lentivirus infection, A549 cells were seeded in a 6-well plate at an initial density of 500 cells/well and cultured at 37°C for 10 days. Then the cells were washed with PBS and fixed with 4% paraformaldehyde for 30 minutes at room temperature. The fixed cells were then stained with freshly prepared diluted crystal violet for 10 minutes, washed with water, and air-dried. Colony formation was measured according to the usual criterion of 50 cells or more per colony.19 The total number of colonies was counted using a microscope. Experiments were repeated at least three times.
Cell cycle analysis
After lentivirus infection, A549 cells were seeded in the 6 cm dish at an initial density of 100,000 cells/dish. After the density reached around 80%, A549 cells were collected, resuspended in cold PBS and fixed with precold 75% ethanol for 30 minutes at 4°C. After removing the ethanol out, the fixed cells were resuspended subsequently with DNase-free RNase at 37°C for 30 minutes. Samples were washed by PBS and incubated with PBS containing RNAase and propidium iodide at 4°C overnight in dark. Cell cycles were then analyzed by flow cytometry. Experiments were repeated at least three times. The data were analyzed by the software Flowjo (Ashland, OR, USA).
Statistical analysis
Data were evaluated by Student’s t-test, and differences were considered statistically significant at P<0.05. Results are shown as the mean ± SD from three independent experiments.
Results
Knockdown of SUN1 in lung adenocarcinoma cells by lentivirus-based RNAi
To investigate the function of SUN1 in lung adenocarcinoma cells, the shRNA against SUN1, shSUN1, and shSUN1#2, was designed and packaged into the lentivirus (Lv-shSUN1 and Lv-shSUN#2). Nontargeting shRNA expression lentivirus (Lv-shCon) was used as control. Four days after lentivirus infection, more than 80% of A549 cells were positively infected by both lentiviruses as assessed by GFP fluorescence (Figures 1A and S2A). Similarly, a satisfying infection efficiency was also observed in 95D cells (Figure S1A). The knockdown efficiency of SUN1 was then detected using quantitative real-time polymerase chain reaction and Western blot. We found that the transcription level of SUN1 in A549 cells was reduced by 61.8% and 55.4% after Lv-shSUN1 and Lv-shSUN1#2 infection, respectively, compared with the Lv-shCon group (Figures 1B and S2B). The translation level of SUN1 was concomitantly reduced after SUN1 silencing (Figure 1C). Lv-shSUN1 infection also downregulated the expression of SUN1 in 95D cells at both mRNA and protein levels (Figures S1B and 1C). These results suggest that the constructed Lv-shSUN1 could significantly decrease endogenous SUN1 expression in lung adenocarcinoma cells.
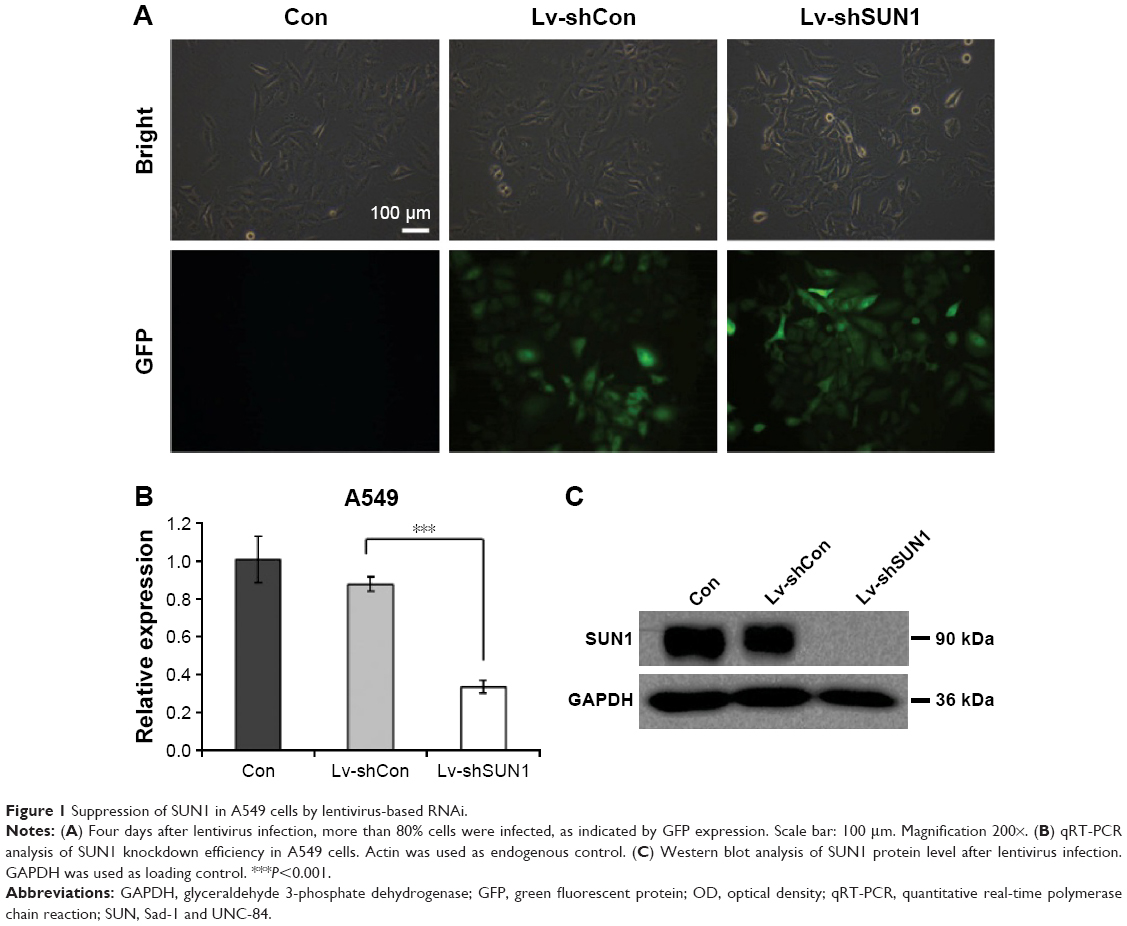 | Figure 1 Suppression of SUN1 in A549 cells by lentivirus-based RNAi. |
Suppression of SUN1 reduces cell proliferation and colony formation
To study the functions of SUN1 in lung adenocarcinoma cells, MTT assay was performed after SUN1 knockdown. As shown in Figures 2A, S1D, and S2C, OD595 nm of Lv-shSUN1- and Lv-shSUN1#2-infected A549 cells was significantly decreased compared with that of control groups (P<0.001), suggesting that the proliferation ability of A549 cells was inhibited by SUN1 silencing. Similarly, a diminution of proliferation was also observed in 95D cells after Lv-shSUN1 infection (P<0.001, Figure S1A). Furthermore, the tumorigenicity of A549 cells in vitro was then evaluated by colony formation assay. We found that after Lv-shSUN1 infection, the size of single colony and the number of colonies formed were significantly reduced (Figure 2B). There were 82±2 and 73±4 colonies in Con and Lv-shCon groups, while only 28±3 colonies in the Lv-shSUN1 group (Figure 2C). These results suggest that knockdown of SUN1 alleviates the proliferation and colony formation ability of lung adenocarcinoma cells.
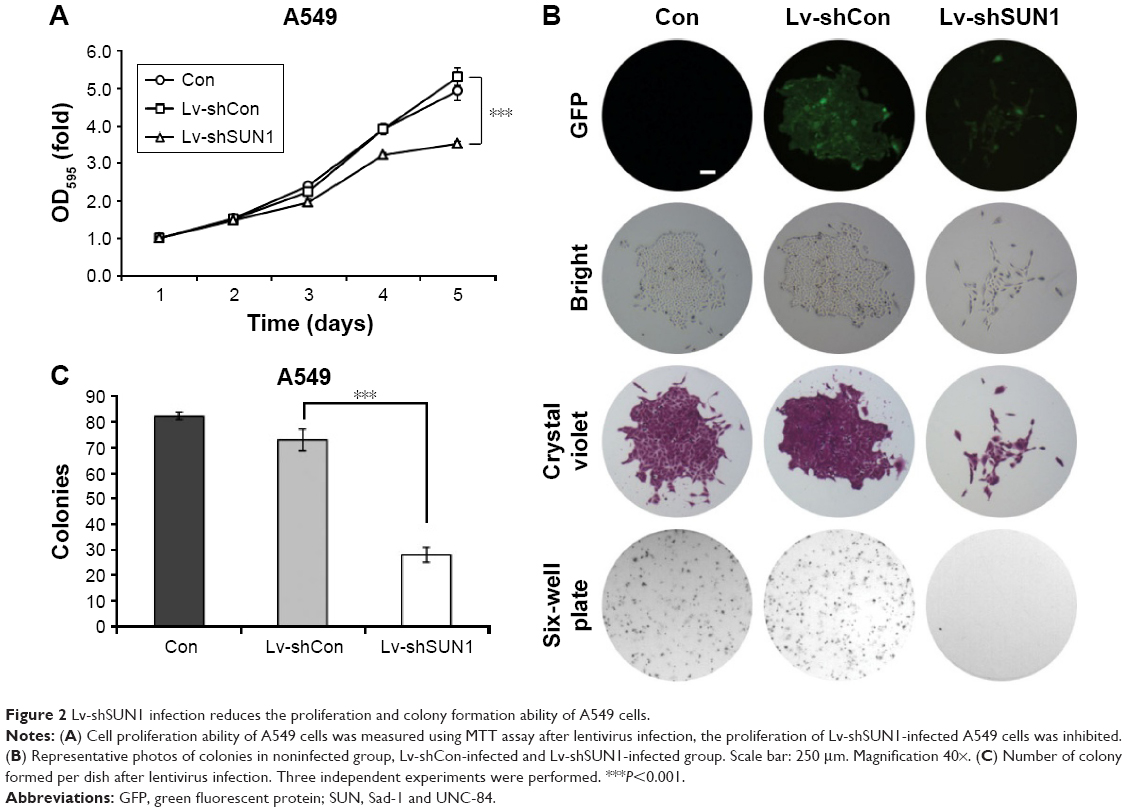 | Figure 2 Lv-shSUN1 infection reduces the proliferation and colony formation ability of A549 cells. |
Suppression of SUN1 leads to cell cycle arrest at G0/G1 phase
To find out the underlying mechanisms of SUN1 silencing induced cell growth inhibition, we analyzed the cell cycle distribution of A549 cells after lentivirus infection (Figure 3A). We found that more cells accumulated in G0/G1 phase of cell cycle (86.39%±0.69%) after Lv-shSUN1 infection, compared with the Con group (56.71%±1.42%) and Lv-shCon group (55.67%±1.34%). The percentage of cells in the S phase and G2/M phase was dramatically decreased after Lv-shSUN1 infection (Figure 3B). To illuminate the molecular basis for G0/G1 phase arrest caused by SUN1 knockdown, we detected the expression alterations of some cell cycle-associated protein. As shown in Figure 3C, Cyclin D1, CDK6, and CDK2 expressions were obviously reduced in A549 cells after Lv-shSUN1 infection, and CDK4 was unaffected. These results suggest that knockdown of SUN1 in A549 cells leads to cell cycle arrest at G0/G1 phase probably via suppression of Cyclin D1, CDK6, and CDK2.
 | Figure 3 Lv-shSUN1 infection induces cell cycle arrest at G0/G1 phase. |
Discussion
In the present study, we investigated the functions of SUN1 in human lung adenocarcinoma A549 and 95D cell lines and found that SUN1 regulates cell proliferation of A549 and 95D cells, colony formation and cell cycle progression of A549 cells. Previous studies suggest that SUN–KASH complexes are associated with cancer.1,14–16 However, there is no evidence that SUN proteins are directly related with cancer. For the first time, we proved that SUN1 plays important roles in human lung adenocarcinoma progression.
Previous studies in yeast and C. elegans showed that SUN proteins are tethered to telomeres and specific chromosomal loci. SUN–KASH protein complexes connect chromosomes to cytoskeleton, thus promote chromosome movement and pairing during meiosis.20 In mice, SUN1 is also involved in meiosis.21 It is concentrated at telomeres in meiotic prophase I, to promote telomere movement and homolog pairing. Most recently, Lei et al found that SUN proteins are involved in mitotic cell division and DNA damage response.22 SUN1/SUN2 double knock-out mouse embryonic fibroblasts (MEFs) display slower proliferation rate with increased apoptosis and DNA damage compared with wild type MEFs, and the percentage of MEFs stored at G0/G1 phase increased and the percentage at S phase and G2/M phase decreased after SUN1 and SUN2 were both knocked out. This is consistent with our results from MTT and flow cytometry analyses of SUN1-silenced A549 cells. Interestingly, SUN1 and SUN2 play redundant roles in cell proliferation and DNA damage response in mice MEFs. Either SUN1−/− or SUN2−/− alone does not show significant phenotypes.22 However, in our study, knockdown of SUN1 itself led to a significant decrease in cell growth and cell cycle arrest. This implies that SUN proteins might play different roles in different cell types.
It is known that Cyclins and CDKs are two kinds of crucial regulatory molecules determining cell cycle progression.23 CDK2, CDK4, and CDK6 are activated in association with the D-type Cyclins or Cyclin E during G1 progression and G1-S transition.24 In this study, G0/G1 phase arrest by SUN1 silencing in A549 cells was found to be associated with marked downregulation of Cyclin D1, CDK6, and CDK2. The reason why SUN1 silencing could reduce the expression of Cyclin D1, CDK4, and CDK6 may be because more cells were arrested in the G0/G1 phase and so less cells went through the G1-S transition. Since Cyclin D1, CDK4, and CDK6 function during G1 progression and G1-S transition, their expression were also affected by the changed cell cycle. Therefore, we suggest that SUN1 modulates the growth of A549 lung adenocarcinoma cells via cell cycle control. Inhibition of SUN1 may become a potential therapy for lung cancer in the future through inhibiting the growth of lung adenocarcinoma cells. Further studies are required to know more about the function of SUN1 in vivo.
Conclusion
We provide new evidence that SUN1 plays an important role in the growth of human lung adenocarcinoma cells, which opens a possibility to treat lung adenocarcinoma through limitation of SUN1, such as RNAi.
Disclosure
The authors report no conflicts of interest in this work.
References
Starr DA, Fridolfsson HN. Interactions between nuclei and the cytoskeleton are mediated by SUN-KASH nuclear-envelope bridges. Ann Rev Cell Dev Biol. 2010;26:421–444. | ||
Tapley EC, Starr DA. Connecting the nucleus to the cytoskeleton by SUN-KASH bridges across the nuclear envelope. Curr Opin Cell Biol. 2013;25(1):57–62. | ||
Razafsky D, Hodzic D. Bringing KASH under the SUN: the many faces of nucleo-cytoskeletal connections. J Cell Biol. 2009;186(4):461–472. | ||
Sosa BA, Kutay U, Schwartz TU. Structural insights into LINC complexes. Curr Opin Struct Biol. 2013;23(2):285–291. | ||
Sosa BA, Rothballer A, Kutay U, Schwartz TU. LINC complexes form by binding of three KASH peptides to domain interfaces of trimeric SUN proteins. Cell. 2012;149(5):1035–1047. | ||
Zhou Z, Du X, Cai Z, et al. Structure of Sad1-UNC84 homology (SUN) domain defines features of molecular bridge in nuclear envelope. J Biol Chem. 2012;287(8):5317–5326. | ||
Crisp M, Liu Q, Roux K, et al. Coupling of the nucleus and cytoplasm: role of the LINC complex. J Cell Biol. 2006;172(1):41–53. | ||
Fridolfsson HN, Starr DA. Kinesin-1 and dynein at the nuclear envelope mediate the bidirectional migrations of nuclei. J Cell Biol. 2010;191(1):115–128. | ||
Horvitz HR, Sulston JE. Isolation and genetic characterization of cell-lineage mutants of the nematode Caenorhabditis elegans. Genetics. 1980;96(2):435–454. | ||
Malone CJ, Fixsen WD, Horvitz HR, Han M. UNC-84 localizes to the nuclear envelope and is required for nuclear migration and anchoring during C. elegans development. Development. 1999;126(14):3171–3181. | ||
Starr DA. Laminopathies: too much SUN is a bad thing. Curr Biol. 2012;22(17):R678–R680. | ||
Haque F, Mazzeo D, Patel JT, et al. Mammalian SUN protein interaction networks at the inner nuclear membrane and their role in laminopathy disease processes. J Biol Chem. 2010;285(5):3487–3498. | ||
Chen CY, Chi YH, Mutalif RA, et al. Accumulation of the inner nuclear envelope protein Sun1 is pathogenic in progeric and dystrophic laminopathies. Cell. 2012;149(3):565–577. | ||
Marme A, Zimmermann HP, Moldenhauer G, et al. Loss of Drop1 expression already at early tumor stages in a wide range of human carcinomas. Int J Cancer. 2008;123(9):2048–2056. | ||
Doherty JA, Rossing MA, Cushing-Haugen KL, et al. ESR1/SYNE1 polymorphism and invasive epithelial ovarian cancer risk: an Ovarian Cancer Association Consortium study. Cancer Epidemiol Biomarkers Prev. 2010;19(1):245–250. | ||
Sjoblom T, Jones S, Wood LD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314(5797):268–274. | ||
Zhou L, Pante N. The nucleoporin Nup153 maintains nuclear envelope architecture and is required for cell migration in tumor cells. FEBS Lett. 2010;584(14):3013–3020. | ||
Collins LG, Haines C, Perkel R, Enck RE. Lung cancer: diagnosis and management. Am Fam Physician. 2007;75(1):56–63. | ||
Puck TT, Marcus PI. Action of x-rays on mammalian cells. J Experimen Med. 1956;103(5):653–666. | ||
Hiraoka Y, Dernburg AF. The SUN rises on meiotic chromosome dynamics. Dev Cell. 2009;17(5):598–605. | ||
Ding X, Xu R, Yu J, Xu T, Zhuang Y, Han M. SUN1 is required for telomere attachment to nuclear envelope and gametogenesis in mice. Dev Cell. 2007;12(6):863–872. | ||
Lei K, Zhu X, Xu R, Xu T, Zhuang Y, Han M. Inner nuclear envelope proteins SUN1 and SUN2 play a prominent role in the DNA damage response. Curr Biol. 2012;22(17):1609–1615. | ||
Israels ED, Israels LG. The cell cycle. Oncologist. 2000;5(6):510–513. | ||
Malumbres M, Barbacid M. Cell cycle kinases in cancer. Curr Opin Genet Dev. 2007;17(1):60–65. |
Supplementary materials
 | Figure S1 Lv-shSUN1 infection decreases the proliferation of 95D cells. |
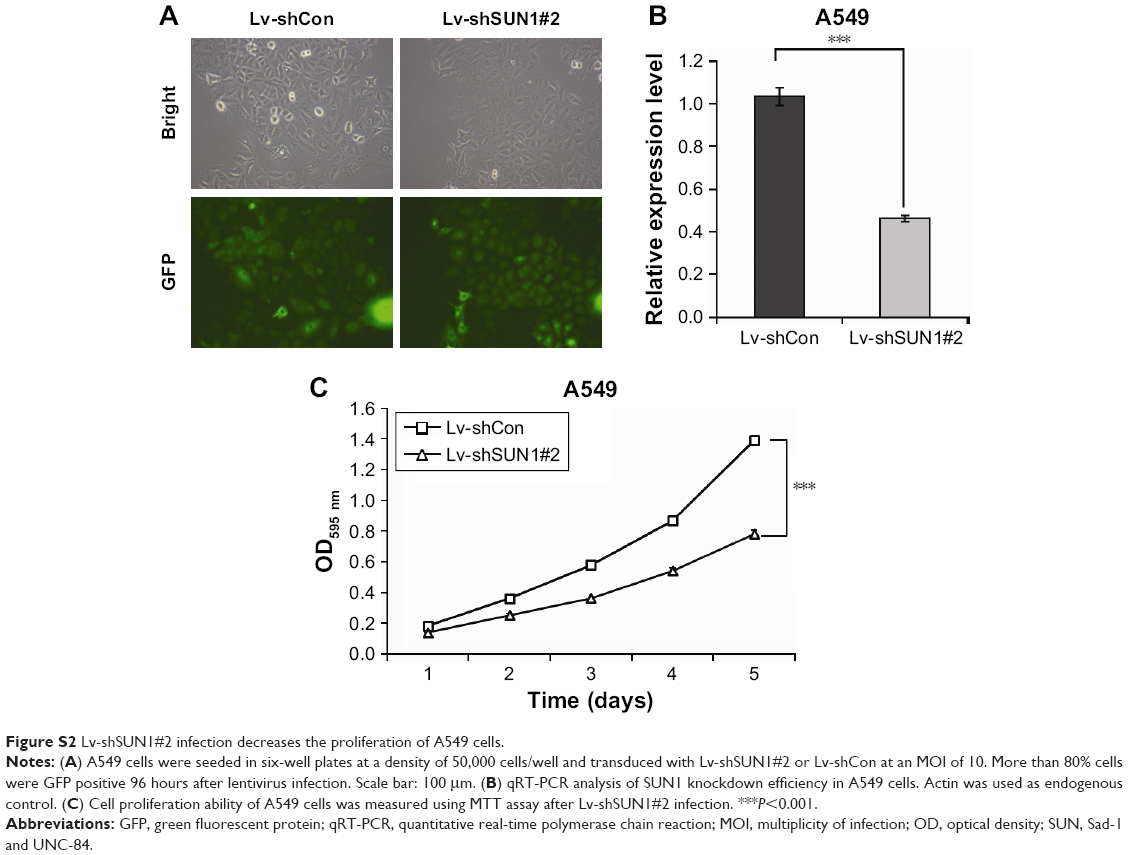 | Figure S2 Lv-shSUN1#2 infection decreases the proliferation of A549 cells. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.