Back to Journals » Blood and Lymphatic Cancer: Targets and Therapy » Volume 11
Successful Non-Transplant Treatment of Double Hit Richter Transformation with Long-Term Remission
Authors Seegobin K ![]() , Alhaj Moustafa M
, Alhaj Moustafa M ![]() , Jiang L
, Jiang L ![]() , Tun HW
, Tun HW ![]()
Received 11 August 2021
Accepted for publication 15 September 2021
Published 22 September 2021 Volume 2021:11 Pages 67—72
DOI https://doi.org/10.2147/BLCTT.S330008
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Wilson Gonsalves
Karan Seegobin,1 Muhamad Alhaj Moustafa,1 Liuyan Jiang,2 Han W Tun1
1Department of Hematology and Medical Oncology, Mayo Clinic Hospital, Jacksonville City, FL, 32224, USA; 2Department of Pathology, Mayo Clinic Hospital, Jacksonville City, FL, 32224, USA
Correspondence: Karan Seegobin
Department of Hematology and Medical Oncology, Mayo Clinic Hospital, Jacksonville City, FL, 32224, USA
Email [email protected]
Abstract: Richter transformation (RT) is defined as the transformation of chronic lymphocytic leukemia (CLL) to a high-grade B cell lymphoma. It usually carries a dismal prognosis and represents an unmet need for novel therapeutic interventions. We report a case of an 80-year-old male who developed double-hit (DH) RT with translocations of MYC and BCL6 after 5 years of watchful waiting for standard-risk CLL. He was treated with induction therapy consisting of 4 cycles of anthracycline-based chemoimmunotherapy (CIT) and 2 cycles of platinum-based CIT with intrathecal methotrexate for CNS prophylaxis followed by continuous maintenance therapy with ibrutinib. He achieved complete remission after the induction therapy. At the time of writing, four and a half years after the diagnosis with DH-RT, he remains in complete remission without evidence of RT or CLL. The novel therapeutic approach used in successful treatment of this patient should be further explored.
Keywords: Richter transformation, RT, chronic lymphocytic leukemia, CLL, double hit lymphoma, ibrutinib maintenance
Plain Language Summary
Chronic lymphocytic leukemia and a high-grade B cell lymphoma are both different types of blood cancers. In some patients, chronic lymphocytic leukemia can transform into a high-grade B cell lymphoma in some patients, which behaves more aggressively, and this is known as Richter transformation. If the transformed lymphoma express MYC and BCL6/BCL2, it is termed double hit, and this implies a more aggressive lymphoma. The standard treatment for high-grade B cell lymphoma involves the use of chemotherapy and immunotherapy. After completing treatment with chemotherapy and immunotherapy, this transformed lymphoma is at high risk for returning. Bone marrow transplant is a potential option to reduce the risk of the lymphoma returning; however, not everyone is eligible for this. There is a lack of information and guidance on how to effectively prevent this transformed lymphoma from returning, especially in persons who are not eligible for bone marrow transplant. If the lymphoma does return, there are several treatment options available. This list continues to expand as the field moves forward. One of which is a new drug called Ibrutinib, which inhibits an enzyme in the cancer and prevents its growth. Incorporating these new agents in the upfront treatment setting has not been studied in clinical trials. We describe a case of continuous ibrutinib maintenance after response to initial chemoimmunotherapy that resulted in long-term remission in a patient with Richter transformation.
Introduction
Richter transformation (RT) is defined as the transformation of chronic lymphocytic leukemia (CLL) to a high-grade B cell lymphoma and is seen in approximately 2–8% of CLL patients.1,2 Double-hit/Triple-hit (DH/TH) RT has been sparsely reported in the literature. A lot more needs to be elucidated on this subset of RT, especially with regard to effective therapeutic approach. We report a case of a 76-year-old male who developed DH-RT after 5 years of watchful waiting for standard-risk CLL. He was successfully treated with induction CIT followed by continuous maintenance therapy with ibrutinib, achieving long-term continuous complete remission.
Case
The patient is an 80-year-old male with a past medical history of Rai stage I standard-risk CLL characterized by 13q deletion by Fluorescent in-situ hybridization analysis (FISH), mutated status of immunoglobulin heavy chain variable region (IGHV), and negative expression of CD38 and ZAP-70 on peripheral blood flow cytometry. TP53 was negative. He initially presented with atypical lymphocytosis, and bilateral axillary lymphadenopathy, retroperitoneal lymphadenopathy, and pelvic lymphadenopathy. Bone marrow biopsy (Figure 1A) showed nodular proliferation of neoplastic lymphocytes, immunohistochemistry (Figure 1B) revealed the lymphocytes positive for CD23, flow cytometry analysis (Figure 1C) confirmed lymphocytes positive for CD20 and predominantly negative for CD38, FISH study (Figure 1D) revealed loss of D13S319 signal, which indicated the deletion of 13q. He was asymptomatic from CLL and was put under watchful waiting. After about 5 years of watchful waiting, he presented with progressive worsening of pelvic pain over a period of 3 weeks. Laboratory workup showed white cell count 10.8 [3.4–9.6x10ʌ9/L], Absolute lymphocyte count 1.28 [0.95–3.07 x10ʌ9/L], Hb 13.2 [13.2–16.6g/dL], platelet 153 [135–317x10ʌ9/L], lactate dehydrogenase 256 [122–222U/L]. Renal function and liver function were within normal limits. CT imaging showed a new left pelvic sidewall mass 7.6 cm in maximum transverse dimension (Figure 2A), hypermetabolic on PET however it was unable to accurately measure SUVs in this mass due to concentrated radiotracer within the involved left ureter (Figure 2B), there was also associated left hydronephrosis (Figure 2C), and increased pelvic mesenteric lymphadenopathy and external iliac lymphadenopathy. Other areas of lymphadenopathy were unchanged compared to previous scans. CT-guided core biopsy of the pelvic mass showed a diffusely infiltrating large atypical cells (yellow arrow) with intermixed clusters of small lymphocytes (black arrow) (Figure 3A). Immunohistochemical studies showed both small and large lymphocytes were strongly positive for CD20 (Figure 3B). While the small lymphocytes showed aberrant expression of CD5 (Figure 3C) and CD23 (Figure 3D), the large lymphocytes were negative for CD5 and CD23 but positive for BCL6 (Figure 3E) with a high proliferative rate by ki-67 (Figure 3F). FISH analysis with break-apart probes demonstrated MYC gene rearrangement (Figure 3G), arrows indicate separation of 5ʹMYC and 3ʹMYC) and BCL6 gene rearrangement (Figure 3H), arrows indicate separation of 3ʹBCL6 and 5ʹBCL6), which confirmed the diagnosis of “double-hit” large cell lymphoma. Pathologic findings are consistent with high grade B cell lymphoma coexisting with CLL in the same tumour. FISH for CLL showed persistence of 13q deletion and no new additional cytogenetic abnormality. These features supported a diagnosis of DH-RT with MYC and BCL6 rearrangements. CSF analysis (cytology and flow cytometry) was negative. Left ureteric stent was placed to relieve hydronephrosis. He was initially treated with one cycle RCHOP (Rituximab 375 mg/m2, cyclophosphamide 750 mg/m2, hydroxydoxorubicin 50 mg/m2, vincristine 1.4 mg/m2, and prednisone 100 mg/m2) without dose adjustment or GCSF support; and then switched to dose adjusted REPOCH (Rituximab 375 mg/m2, etoposide 50 mg/m2/day, prednisone 60 mg/m2/day, vincristine 0.4 mg/m2/day, cyclophosphamide 750 mg/m2, and hydroxydoxorubicin 10 mg/m2/day), intrathecal methotrexate 15mg for CNS prophylaxis (with each cycle) and GCSF support with Neulasta 6mg subcutaneous. Restaging following 3 cycles of REPOCH showed marked partial response. He tolerated the first two cycles well without any hematologic toxicity, however after the third cycle of REPOCH, ECHO performed for shortness of breath showed EF of 52% compared to 62% pre-treatment. He also had grade 2 anemia (hemoglobin of 9.6 g/dL) and grade 1 thrombocytopenia with platelet of (97 x10 ʌ9/L). He did not have any neutropenia. He was switched to treatment with two cycles of RICE chemoimmunotherapy (Rituximab 375 mg/m2, ifosfamide 5000 mg/m2/day (with 20% dose reduction), carboplatin AUC = 5 (with 20% dose reduction), and etoposide 100 mg/m2/day (with 20% dose reduction)), with Neulasta support 6mg subcutaneous. Dose reduction was done in light of the prior reduction in ejection fraction and cytopenias. PET scan after completing cycle 6 showed complete remission with no evidence of disease activity (Figure 2D). Three months after completion of induction chemoimmunotherapy, he was started on maintenance therapy with ibrutinib 420 mg daily. About a month before the writing of this report, he was diagnosed with COVID-19 pneumonia requiring hospitalization for one month. He was recently discharged from the hospital and has been recovering well. Ibrutinib has been on hold with the plan to resume soon. The last imaging scans (CT chest, abdomen, and pelvis) performed during recent hospitalization did not show any lymphadenopathy or splenomegaly. Peripheral blood tests have not shown any evidence of CLL. At the time of writing of this manuscript, he remains in complete remission at four and a half years after the diagnosis of DH-RT.
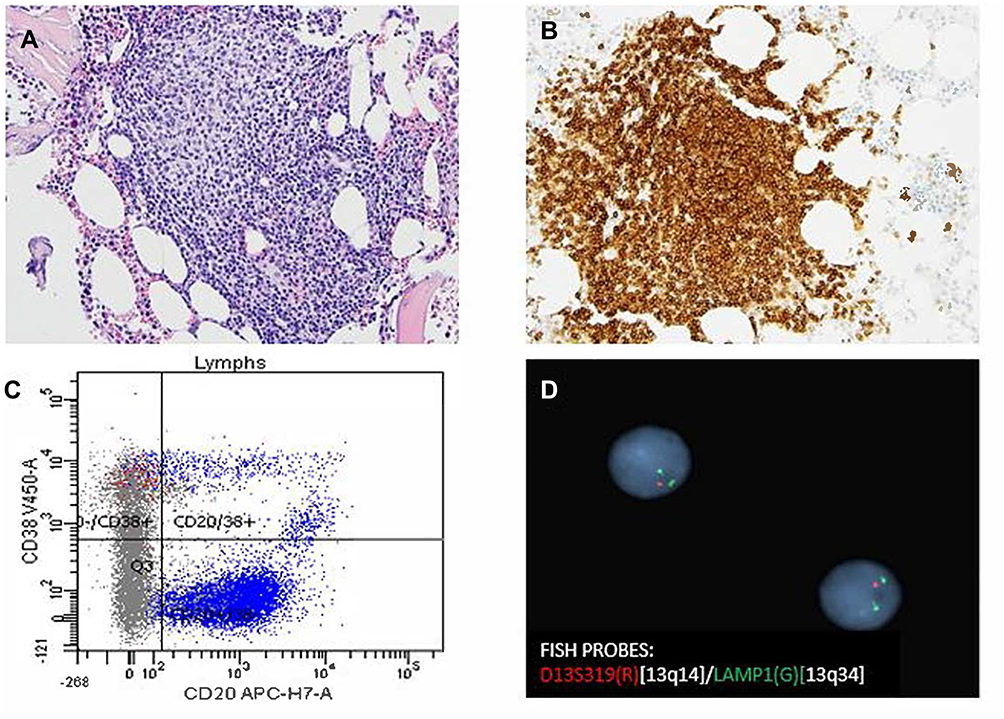 |
Figure 1 The bone marrow biopsy showed nodular proliferation of neoplastic lymphocytes (A, H&E x 20). Immunohistochemistry revealed the lymphocytes positive for CD23 (B, x 20). Flow cytometry analysis confirmed lymphocytes positive for CD20 and predominantly negative for CD38 (C). FISH study revealed loss ofD13S319 signal, which indicated the deletion of 13q (D). |
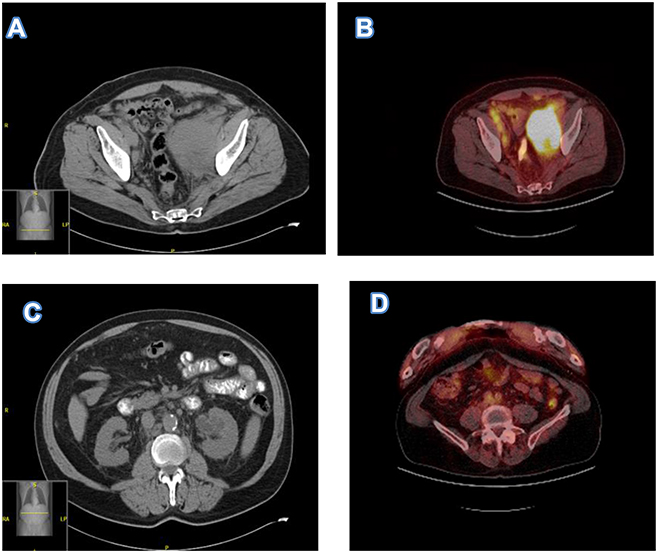 |
Figure 2 (A) Pretreatment CT imaging showed a left pelvic sidewall mass, pelvic mesenteric lymphadenopathy, and external iliac lymphadenopathy. (B). Pretreatment PET shows hypermetabolic pelvic lesion however unable to accurately measure SUVs in this mass due to concentrated radiotracer within the involved left ureter. (C). Pretreatment CT imaging showed associated left hydronephrosis. (D) Post treatment PET scan after 6 cycles of induction therapy showed low level metabolic activity within the left pelvic sidewall mass, SUV max 2.1, no evidence for recurrence. |
 |
Figure 3 The core needle biopsy of the pelvic mass showed a diffusely infiltrating large atypical cells (yellow arrow) with intermixed clusters of small lymphocytes (black arrow) (A, H&E x 20). Immunohistochemical studies showed both small and large lymphocytes were strongly positive for CD20 (B, x 20). While the small lymphocytes showed aberrant expression of CD5 (C, x 20) and CD23 (D, x 20), the large lymphocytes were negative for CD5 and CD23 but positive for BCL6 (E, x 20) with a high proliferative rate by ki-67 (F x 20). FISH analysis with break-apart probes demonstrated MYC gene rearrangement (G, arrows indicate separation of 5ʹMYC and 3ʹMYC) and BCL6 gene rearrangement (H, arrows indicate separation of 3ʹBCL6 and 5ʹBCL6), which confirmed the diagnosis of “double-hit” large cell lymphoma. |
Discussion
We report a case of DH-RT with MYC and BCL6 translocations successfully treated with induction CIT followed by continuous maintenance therapy with ibrutinib resulting in continuous complete remission at four and a half years so far after the diagnosis of DH-RT. DH/TH RT is rather uncommon and has not been well characterized. In the largest series on RT with a total of 204 patients, it represents 12.1% of RT cases (8/66) in which gene rearrangement data on MYC, BCL2, and BCL6 are available.3 In this series, median overall survival for DH/TH RT is about 14 months with no survivor at 24-month follow-up. In one case report, a patient with DH-RT with MYC and BCL6 translocation survived for 9 months following the diagnosis of DH-RT.2 Our patient has done quite well compared to those survival outcomes.
The median time to transformation from CLL to RT is 4 years, with a rate of 0.5%/year.1 Our patient developed DH-RT five years after the initial diagnosis of CLL. RT is characterized by rapid disease kinetics,1 and clinically presents with rapidly enlarging lymphadenopathy, constitutional symptoms, elevated LDH, and frequent extra nodal tissue involvement.3 The clinical presentation of DH-RT in our case is classic with sudden onset of severe and progressive pelvic pain related to rapid enlargement of pelvic lymph nodes. The gold standard for diagnosis is tissue biopsy and should be performed in those with suspected RT and an SUV max of 5 or greater on PET, with excisional biopsy being preferred.4,5 The median survival after transformation is about 12 months; patients who had no prior CLL treatment have better overall survival (OS), with a median OS of approximately four years.6 Untreated status in our case before the onset of DH-RT may have contributed to excellent treatment outcome.
We were not able to perform molecular studies to establish a clonal relationship between CLL and DH-RT. However, the biopsy showed coexistence of DH-RT and CLL in the same pelvic tumor. As such, it is quite likely that CLL and DH-RT are clonally related in our case.
R-CHOP-like therapy is most frequently used in the management of RT.2 However, it has limited efficacy, and higher intensity chemotherapy has not been shown to improve outcomes.6,7 In select cases, autologous stem cell transplant is associated with relative long-term survival, where the median post-transplant survival is 55.4 months.6 Novel therapeutic agents such as Bruton’s tyrosine kinase (BTK) inhibitors, B-cell lymphoma 2 (BCL-2) inhibitor, and checkpoint inhibitors have shown activity against RT, but the survival benefit is limited.8
The best therapeutic approach for DH/TH RT is not known. Our patient was induced with anthracycline-based CIT (one cycle of RCHOP and three cycles of dose-escalated REPOCH) followed by platinum-based CIT (two cycles of RICE). REPOCH has been used as the standard CIT for DH/TH lymphomas.9 He responded well-achieving complete remission following anthracycline-based CIT.
Ibrutinib has been shown to be effective in both CLL and RS.10 There have been reports of ibrutinib being used upfront in patients with RS with good response.5 In one study of ibrutinib in 85 pretreated CLL patients, the overall response rate (ORR) was 71%.10 In another study of 3 patients with RS who did not respond well to initial rituximab-based chemoimmunotherapy regimens and subsequently treated with ibrutinib due to lack of response; the median duration of ibrutinib therapy was 6.1 months, all patients experienced an improvement in constitutional symptoms, 1 patient had a complete response and 2 patients had a partial response.10 Ibrutinib was well tolerated, and no patient required discontinuation because of adverse events.
Despite these data, the role of ibrutinib maintenance after induction CIT in the management of RT has not been tested in clinical trials. It is quite likely that ibrutinib maintenance plays an important role in achieving an excellent survival outcome in our case.
In conclusion, we report a case of DH-RT successfully treated with induction CIT followed by ibrutinib maintenance achieving an excellent survival outcome. This novel non-transplant therapeutic approach should be further explored for management of RT.
Ethics Approvall
Institutional approval was not required to publish the case details.
Consent
Written and informed consent was obtained from the patient for publication of this case report.
Authorship Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
There is no funding to report.
Disclosure
All authors have no conflicts of interest to declare.
References
1. Parikh SA, Rabe KG, Call TG, et al. Diffuse large B-cell lymphoma (Richter syndrome) in patients with chronic lymphocytic leukaemia (CLL): a cohort study of newly diagnosed patients. Br J Haematol. 2013;162(6):774–782. doi:10.1111/bjh.12458
2. Rogers TS, Gardner JA, Devitt KA. High-grade B-cell lymphoma with MYC and BCL6 rearrangements associated with Richter transformation of chronic lymphocytic leukemia. Autops Case Rep. 2019;9(3):e2019090. doi:10.4322/acr.2019.090
3. Rossi D, Spina V, Gaidano G. Biology and treatment of Richter syndrome. Blood. 2018;131(25):2761–2772. doi:10.1182/blood-2018-01-791376
4. Wang YC, Rabe KG, Bold MS, et al. The role of F-18-FDG-PET in detecting Richter transformation of chronic lymphocytic leukemia in patients receiving therapy with a B-cell receptor inhibitor. Haematologica. 2020;105(11):2675–2678. doi:10.3324/haematol.2019.240564
5. Wang YC, Ding W. Richter transformation of chronic lymphocytic leukemia in the era of novel agents. Clin Adv Hematol Onc. 2020;18(6):348–357.
6. Wang YC, Tschautscher MA, Rabe KG, et al. Clinical characteristics and outcomes of Richter transformation: experience of 204 patients from a single center. Haematologica. 2020;105(3):765–773. doi:10.3324/haematol.2019.224121
7. Rogers KA, Huang Y, Ruppert AS, et al. A single-institution retrospective cohort study of first-line R-EPOCH chemoimmunotherapy for Richter syndrome demonstrating complex chronic lymphocytic leukaemia karyotype as an adverse prognostic factor. Br J Haematol. 2018;180(2):259–266. doi:10.1111/bjh.15035
8. Ding W. Richter transformation in the era of novel agents. Hematology Am Soc Hematol Educ Program. 2018;2018(1):256–263. doi:10.1182/asheducation-2018.1.256
9. Phuoc V, Sandoval-Sus J, Chavez JC. Drug therapy for double-hit lymphoma. Drugs Context. 2019;8:1–13. doi:10.7573/dic.2019-8-1
10. Tsang M, Shanafelt TD, Call TG, et al. The efficacy of ibrutinib in the treatment of Richter syndrome. Blood. 2015;125(10):1676–1678. doi:10.1182/blood-2014-12-610782
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.