Back to Journals » International Medical Case Reports Journal » Volume 18
Successful Local Control of Orbital ASPS Using VMAT-Based Adjuvant Radiotherapy with Simultaneous Integrated Boost: A 3-Year Follow-Up Case Report
Authors Albadrani HM ![]() , Abduljabbar L
, Abduljabbar L
Received 3 August 2025
Accepted for publication 6 December 2025
Published 10 December 2025 Volume 2025:18 Pages 1585—1592
DOI https://doi.org/10.2147/IMCRJ.S557971
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tanvi Dhere
Hind Muteb Albadrani,1 Lulwah Abduljabbar2
1Department of Clinical Laboratory Sciences, College of Applied Medical Sciences, Imam Abdulrahman Bin Faisal University, Dammam, Saudi Arabia; 2Department of Radiation Oncology, King Fahad Specialist Hospital, Dammam, Saudi Arabia
Correspondence: Hind Muteb Albadrani, Department of Clinical Laboratory Sciences, College of Applied Medical Sciences, Imam Abdulrahman Bin Faisal University, Dammam, Saudi Arabia, Email [email protected]
Abstract: Orbital Alveolar Soft Part Sarcoma (ASPS) is an extremely rare malignancy, with limited case reports. Diagnosis is challenging owing to its slow-growing nature and non-specific symptoms. Furthermore, standardized treatment protocols are lacking, and the long-term outcomes remain poorly understood. We aimed to address these gaps by presenting a rare case of orbital ASPS and outlining the diagnostic challenges, treatment approach, and long-term follow-up to inform the clinical management. A 28-year-old male presented with diplopia, restricted ocular movements, and orbital swelling. Magnetic resonance imaging revealed a 3.81 × 1.99-cm mass adjacent to the left medial rectus muscle, which was histopathologically confirmed as ASPS. A personalized therapeutic approach involving surgery and radiotherapy achieved stable disease, preserved vision, and no recurrence over a 3-year follow-up period. These findings contribute to the existing body of knowledge regarding the clinical management of orbital ASPS, underscoring the importance of individualized treatment strategies. Routine long-term surveillance and multidisciplinary care are essential in the management of rare cases. This case further emphasizes the need for continued research and documentation to enhance the diagnostic accuracy and refine the treatment guidelines for ASPS.
Plain Language Summary: Alveolar Soft Part Sarcoma (ASPS) is a rare cancer that can appear in the soft tissues of the body. It is even more uncommon and difficult to diagnose when it occurs in the eye socket (orbit) because the tumor grows slowly, and the symptoms are vague, such as double vision and eye swelling. There are very few reports on this condition and there is no standard treatment plan. In this report, we describe the case of a 28-year-old man who developed orbital ASPS. The patient experienced double vision, limited eye movement, and swelling in one eye. A magnetic resonance imaging (MRI) scan showed a tumor near the eye muscle, which was confirmed to be ASPS through tissue testing. The patient was treated with a combination of surgery and targeted radiotherapy. Over a 3-year follow-up period, his vision remained stable and the cancer did not return. This case shows that careful planning using advanced radiotherapy techniques, along with long-term follow-up and regular eye examinations, can help to control this rare cancer while protecting vision. Our experience highlights the importance of a personalized approach involving a team of medical specialists. Improved radiotherapy methods, such as proton therapy, may further reduce side effects and improve outcomes. More research is needed to better understand this rare disease and develop clearer treatment guidelines.
Keywords: long-term surveillance, orbital ASPS, personalized treatment, radiotherapy, recurrence management, TFE3 gene
Introduction
Alveolar soft part sarcoma (ASPS) is a rare malignant soft tissue neoplasm with a global incidence of < 1 per million individuals annually. ASPS accounts for <1% of all soft tissue sarcomas.1,2 It typically occurs in adolescents and young adults but may also be observed in children as young as two years of age.2 ASPS is characterized by distinctive histological features and clinical behavior, presenting as slow-growing tumors located in deep soft tissues, particularly in the lower extremities and the head and neck region. Its insidious growth and absence of specific clinical symptoms often complicate diagnosis.3 Despite its indolent course, ASPS possesses a high metastatic potential, most commonly in the lungs, brain, and bones. Prognosis varies significantly, with a 5-year survival rate of 60–88% for localized disease, dropping to 20–62% in cases with metastases.2
Although ASPS is rare, orbital ASPS is exceptionally uncommon, accounting for approximately 5–15% of all reported ASPS cases.4,5 Orbital tumors exert pressure on adjacent structures, leading to complications, such as progressive proptosis, pain, ophthalmoplegia, and visual disturbances. Lesions in the retroorbital space present additional diagnostic challenges owing to their slow-growing nature, which often delays the onset of noticeable symptoms. Consequently, tumors may remain undetected until they produce significant pressure effects. Imaging modalities, including magnetic resonance imaging (MRI) and computed tomography (CT), are essential for identifying intraorbital tumors. Furthermore, histopathological analysis is critical for definitive diagnosis, as ASPS can mimic other soft tissue sarcomas. Features such as polygonal tumor cells with abundant eosinophilic cytoplasm and prominent nucleoli may resemble those observed in clear cell sarcomas or metastatic renal cell carcinomas. Moreover, the alveolar growth pattern typical of ASPS may resemble that of other tumors with similar architectural patterns, further contributing to diagnostic uncertainty and the delayed initiation of appropriate treatment.
In this case report, our primary aim was to enhance the current understanding of orbital ASPS, a rare and underreported malignancy. Due to its rarity, diagnostic challenges persist, standardized treatment protocols remain undefined, and long-term outcomes and recurrence rates are insufficiently characterized, complicating optimal clinical management. We sought to provide valuable insights by documenting the clinical presentation, diagnostic complexities, and therapeutic strategies in challenging cases. Our goals were to raise awareness, promote early recognition, and support the development of improved treatment approaches to enhance patient outcomes.
Case Report
Overview of the Case
A 28-year-old male, with no significant medical or family history presented to a private hospital with left eye diplopia and ptosis, which raised clinical suspicion of multiple sclerosis. Magnetic resonance imaging (MRI) of the brain and orbit revealed no pathological findings. The patient was managed conservatively with local steroids, but no improvement was observed. Five months later, the patient developed additional symptoms including redness, swelling, and restricted eye movement in the left eye. These findings prompted a referral to a specialized ophthalmology hospital. Magnetic resonance imaging (MRI) of the orbit revealed an enhancing mass (measuring 3.81×1.99 cm) along the left medial rectus muscle, with no direct invasion of the optic nerve (Figure 1).
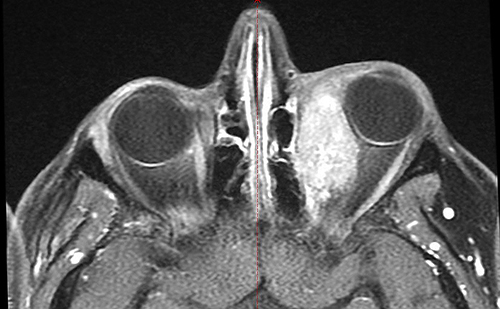 |
Figure 1 Contrast-enhanced T1-weighted fat-saturated magnetic resonance imaging (MRI T1 FS+C) of the orbits. An enhancing mass is visible in the left orbit, originating from the left medial rectus muscle, without evidence of direct optic nerve invasion. |
Owing to the lack of availability of neurosurgeons at the ophthalmology center, the patient was referred to a tertiary institution. Unfortunately, a delay of 3 months before the neurosurgical consultation was obtained. At this tertiary center, the patient underwent intranasal biopsy of the orbital mass under general anesthesia. A staging workup, including CT tomography of the chest, abdomen, and pelvis, revealed no evidence of metastatic disease.
Histopathological Findings
Hematoxylin and eosin-stained sections of the orbital mass revealed a malignant mesenchymal tumor, characteristic of ASPS. The tumor sections were composed of large polygonal cells with abundant eosinophilic cytoplasm arranged in a vaguely nested pseudo-alveolar growth pattern (Supplement 1A and B). The cells were separated using thin fibrovascular septa to create a pseudo-alveolar architecture. Periodic acid–Schiff (PAS) staining revealed intracytoplasmic glycogen and characteristic PAS-positive, diastase-resistant rhomboid or rod-shaped crystals within the tumor (Supplement 1C), a hallmark feature of ASPS. This detailed examination highlighted the cellular morphology and further confirmed the diagnosis of ASPS, consistent with clinical findings. No evidence of atrophy, metaplasia, or dysplasia was found.
Molecular Cytogenetic Staining
Fluorescence in situ hybridization for TFE3 was performed to confirm the diagnosis of ASPS. Abnormal rearrangement was observed in the TFE3 gene region, which has been noted in ASPS and Xp11.2 translocation-associated renal cell carcinoma.
Treatment and Management
Diagnosis of orbital ASPS requires a comprehensive, cross-disciplinary approach to formulate an optimal treatment plan. The patient presented to a multidisciplinary tumor board comprising neurosurgeons, oncologists, ophthalmologists, and radiation oncologists. Two primary treatment strategies were evaluated: complete enucleation and surgical resection followed by adjuvant radiotherapy. A critical consideration in the decision-making process is the high likelihood of achieving a positive surgical margin.
The patient ultimately underwent surgical resection followed by adjuvant radiotherapy. Surgery was successfully performed with uneventful postoperative recovery. However, postoperative positron emission tomography (PET) scan revealed an enlarged cervical lymph node, necessitating close clinical monitoring. Owing to the social circumstances, the patient was subsequently referred to our center for radiotherapy.
Postoperative Clinical Course and Follow-up
The patient underwent CT simulation in the supine position with the head in neutral alignment. Immobilization was achieved using an S-frame mask and the patient was asked. A planning CT scan with 1.3 mm slice thickness was acquired and co-registered with preoperative and postoperative MRI for accurate target delineation.
Target volumes were defined according to standard guidelines. The high-risk clinical target volume (CTV60) included the tumor bed and regions suspected of microscopic residual disease. The low-risk clinical target volume (CTV54) encompasses the entire surgical cavity and the adjacent orbital tissues. Planning target volumes (PTVs) were generated by expanding each CTV isotropically by 3 mm to account for setup uncertainties.
Adjuvant radiotherapy was delivered using a Volumetric Modulated Arc Therapy (VMAT) technique with a simultaneous integrated boost (SIB) approach. A total dose of 60 Gy in 30 fractions (2 Gy/fraction) was prescribed to PTV60, while 54 Gy in 30 fractions (1.8 Gy/fraction) was concurrently delivered to PTV54. Treatment planning was performed on Eclipse Treatment planning system. Two coplanar partial arcs were used with gantry rotation spanning 0o → 179o and 179o → 0o in opposite directions, optimized to minimize the dose to the contralateral orbit and optic structures. Collimator angels were set at 30 and 330 to reduce tongue and groove effects and enhance conformity near critical structures. Dose calculation was performed using Anisotropic Analytical Algorithm (AAA) with a grid resolution of 2.5 mm. The plan achieved good target coverage while maintaining dose constraints to the organ at risks. (Figure 2). Organs-at-risk (OARs) were delineated and evaluated per QUANTEC and institutional constraints (Table 1).
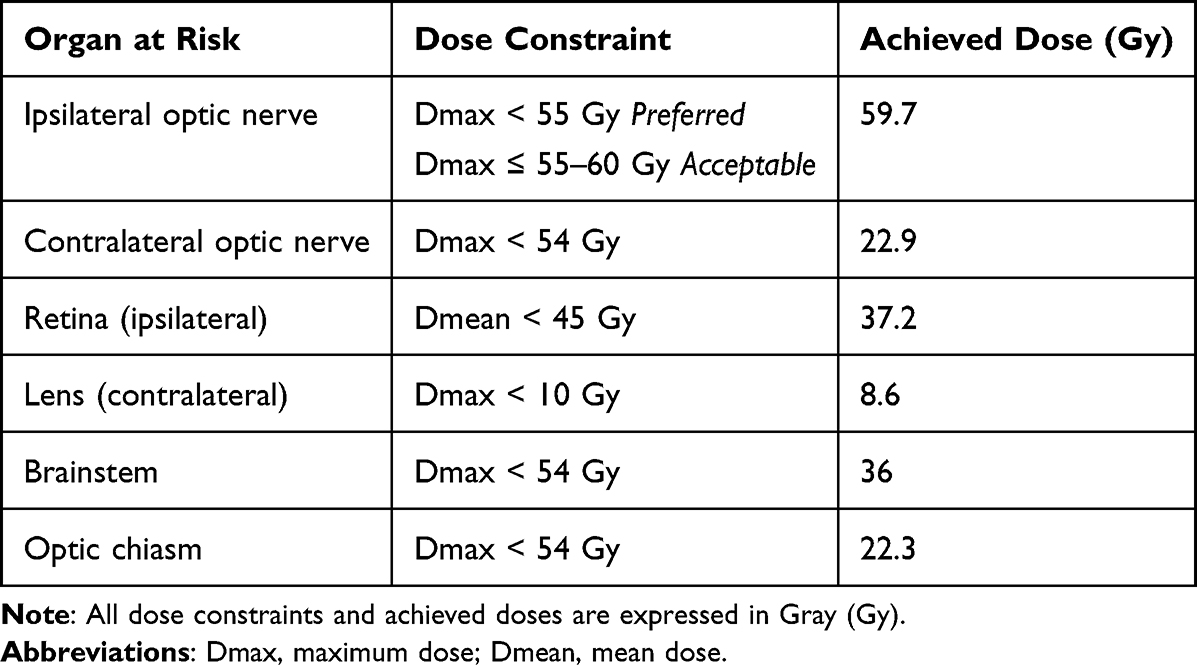 |
Table 1 Dose Constraints and Achieved Doses for Organs at Risk (OARs) |
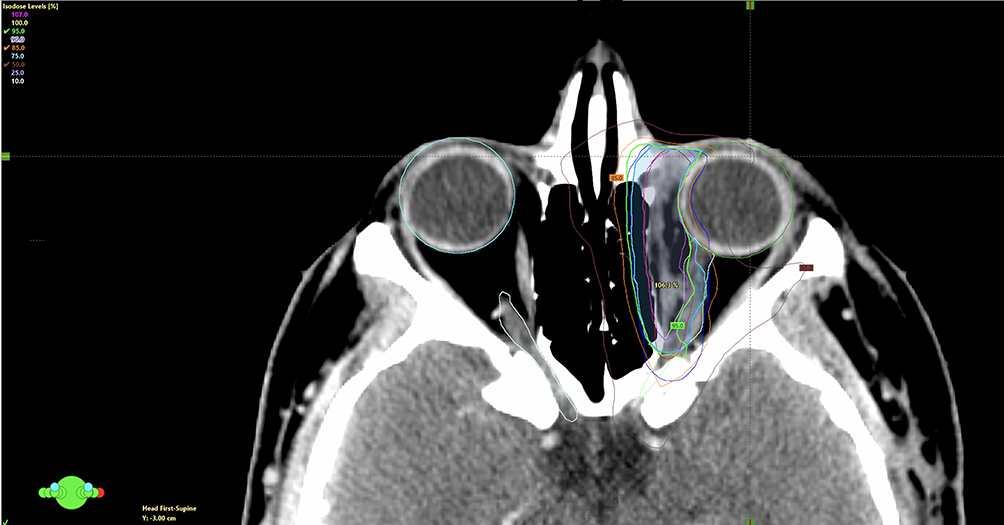 |
Figure 2 The image illustrates the radiation treatment plan for the case, demonstrating the simultaneous integrated boost (SIB) technique. The 95%, 85%, and 50% isodose lines, calculated relative to the prescribed 60 Gy dose, are clearly delineated. The planning target volume (PTV) for 54 Gy is shown in dark blue, encompassing the low-risk areas, while the PTV for 60 Gy is highlighted in cyan blue, covering the high-risk tumor bed and areas of suspected positive margins. The green circle at the bottom left (with red, blue, and green dots) represents the patient’s orientation in the scanner. |
The patient was able to tolerate the treatment satisfactorily. Acute toxicities were limited to grade 1 conjunctivitis and excessive tearing, which were resolved with conservative management. No Grade ≥2 acute toxicity was observed.
Three months after radiation therapy, small enhancing tissue was observed at the posterolateral aspect of the medial rectus muscle (Figure 3). At the 1-year follow-up, significant interval regression of this tissue was noted (Figure 4A). By 3 years post-treatment, imaging demonstrated further reduction of the enhancing tissue, consistent with postoperative changes (Figure 4B). The most recent orbital MRI, performed 40 months after treatment, showed no evidence of local recurrence. A PET scan conducted 21 months post-treatment revealed stable but persistent fluorodeoxyglucose (FDG) activity in the left medial rectus muscle and resolution of the cervical lymph nodes previously identified in the postoperative scan, with no new FDG-avid lesions elsewhere. During treatment, the patient experienced mild conjunctivitis and excessive tearing, which were conservatively managed. Annual ophthalmological examinations revealed no late-onset ocular complications.
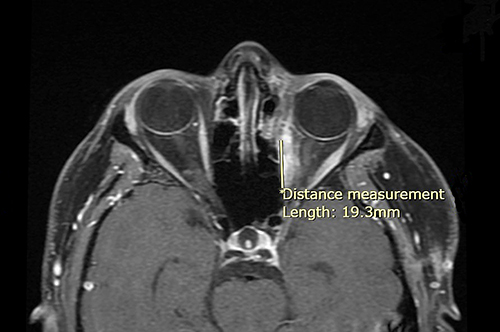 |
Figure 3 Contrast-enhanced T1-weighted fat-saturated MRI (T1 FS+C) obtained 3 months after radiation therapy. A small enhancing focus is noted along the posterolateral aspect of the left medial rectus muscle. (*) Indicates the beginning of the measurement. |
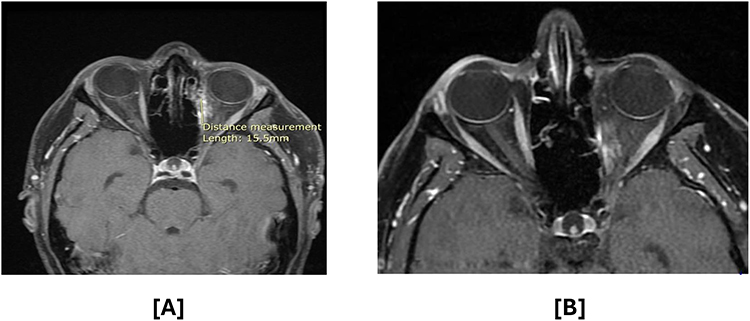 |
Figure 4 Contrast-enhanced T1-weighted fat-saturated MRI (T1 FS+C) at (A) 1 year and (B) 3 years following radiation therapy. Interval regression of the ill-defined enhancing tissue along the left medial rectus muscle is observed over time. (*) Indicates the beginning of the measurement. |
The follow-up protocol included orbital MRI and chest CT every four months for the first two years, followed by scans every six months for the subsequent three years. Three years post-treatment, the patient continued to demonstrate excellent outcomes, with preserved visual function, good quality of life, and no evidence of local recurrence or distant metastasis.
Discussion
The diagnosis of orbital ASPS remains challenging owing to its rarity and non-specific clinical presentation. In the present case, the orbital mass was initially identified via MRI and subsequently confirmed via intranasal biopsy. Histopathological examination revealed hallmark features of ASPS, including pseudo-alveolar architecture and PAS-positive diastase-resistant crystals, consistent with previous reports.6,7 Notably, this case demonstrated subtle variations in the degree of pseudo-alveolar patterning and prominence of cytoplasmic glycogen deposition compared with previously reported cases. These histological differences may reflect anatomical specificity or intertumoral heterogeneity, as orbital ASPS is exceptionally rare and presents unique interpretive challenges distinct from ASPS at other anatomical sites.
Molecular cytogenetic analysis confirmed the TFE3 gene rearrangement, an established diagnostic hallmark of ASPS, further substantiating the diagnosis. Prior studies, such as those by Schoolmeester et al, have emphasized the critical role of molecular testing in distinguishing ASPS from morphologically similar neoplasms, underscoring the robustness of the diagnostic approach in this case.7
Management of orbital ASPS presents unique challenges given the proximity of the tumor to critical orbital structures. In the present case, the treatment involved surgical resection followed by adjuvant radiotherapy, which is consistent with the existing literature.8,9 Achieving negative surgical margins is particularly difficult because the tumor is located adjacent to the medial rectus muscle. Despite the presence of positive margins, adjuvant radiotherapy effectively reduced the risk of recurrence while preserving ocular function. The use of VMAT with SIB allows optimized dose delivery, a technique rarely described in previous orbital ASPS reports.
Radiation-related complications such as retinal edema, telangiectasia, hemorrhage, and neovascularization typically develop within 6 months to 3 years of treatment. It has been shown that doses exceeding 45 Gy mean retinal dose (Dmean) or 65 Gy maximum point dose (D10) are associated with increased risk, particularly under conventional fractionation regimens. Parsons et al reported retinopathy in nearly all patients receiving 45–55 Gy, whereas et al demonstrated that hyperfractionated schedules can reduce this risk even at higher doses.10 In the current case, the patient received a mean retinal dose of 35 Gy and maximum dose of 59.9 Gy. Notably, no signs of radiation retinopathy were observed over the follow-up period of > 3 years. These findings support prior evidence indicating that Dmean doses below 45 Gy are associated with lower toxicity, especially in patients without comorbidities such as diabetes.
During the 3-year follow-up period, the patient adhered to a standard structured surveillance plan involving orbital MRI and chest CT every four months for the first two years, followed by biannual imaging. This approach enables a comprehensive assessment of tumor stability and early detection of potential recurrence or metastasis. The follow-up strategy aligns with the NCCN guidelines, which recommend imaging every 3–6 months during the first 2–3 years, every 6–12 months for the subsequent 2 years, and annually thereafter, based on individual risk.
This case highlights several novel aspects, including the application of advanced radiotherapy techniques and a rigorous follow-up protocol, illustrating the significant advancements in the management of orbital ASPS. These findings contribute to the growing body of literature advocating individualized treatment approaches tailored to the unique anatomical and clinical challenges of orbital sarcomas.
Limitations
This case report is limited by its single-patient design, 3-year follow-up period, and splitting of treatment across two different hospitals, with surgery performed at a separate facility. Furthermore, although effective, advanced radiotherapy techniques may not be accessible in all settings. Larger, multi-institutional studies with longer follow-up periods are required to confirm these findings and establish safe dose thresholds for ocular structures to further enhance treatment safety and efficacy.
Conclusion
This case report underscores the diagnostic and therapeutic complexities associated with managing orbital ASPS, highlighting the critical role of surgical resection and radiotherapy in achieving local disease control even in the presence of microscopically positive surgical margins. The use of an SIB radiotherapy technique enables precise dose escalation to high-risk regions while sparing adjacent healthy tissues, thereby demonstrating the safety and efficacy of this approach in treating orbital tumors.
Furthermore, the incorporation of long-term follow-up, featuring regular imaging and annual ophthalmologic evaluations, offers valuable insights into the ongoing management and surveillance of ASPS in this anatomically sensitive region.
Future research should emphasize the refinement and clinical integration of advanced radiotherapy modalities, such as proton therapy and individualized fractionation schedules, to further optimize the treatment outcomes. Personalized treatment planningguided by multidisciplinary collaborationshould be prioritized to address the unique anatomical and functional challenges posed by orbital ASPS. Further research into evidence-based dose constraints and their application may enhance therapeutic precision and reduce toxicity, ultimately preserving ocular function, while minimizing long-term complications.
Data Sharing Statement
The original contributions of this study are included in the article/supplementary material, and further inquiries can be directed to the corresponding author.
Ethical Approval
This study was reviewed and approved by the Institutional Review Board (IRB) of King Fahad Specialist Hospital, Dammam (IRB No. Pub-024-012). Informed consent was obtained from the patient for publication of clinical details and images.
Informed Consent
Informed consent was obtained from the patient before treatment and again for the publication of this case report. All personally identifiable information was removed or altered to protect patient privacy and to ensure confidentiality.
Acknowledgments
The authors would like to express their sincere gratitude to Dr. Marwah M Abdulkader and Dr. Haitham A. Alamer for their invaluable contributions in providing the histopathological images used in this study. Their expertise and support were instrumental in enhancing the clarity and depth of case presentation.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
No funding was received for this study’s conduct or the manuscript’s preparation.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Brahmi M, Vanacker H, Dufresne A. Novel therapeutic options for alveolar soft part sarcoma: antiangiogenic therapy, immunotherapy and beyond. Curr Opin Oncol. 2020;32(4):295–300. doi:10.1097/CCO.0000000000000652
2. Fujiwara T, Kunisada T, Nakata E, et al. Advances in treatment of alveolar soft part sarcoma: an updated review. Jpn J Clin Oncol. 2023;53(11):1009–1018. doi:10.1093/JJCO/HYAD102
3. Lin YK, Wu PK, Chen CF, et al. Alveolar soft part sarcoma: clinical presentation, treatment, and outcome in a series of 13 patients. J Chin Med Assoc. 2018;81(8):735–741. doi:10.1016/J.JCMA.2018.01.006
4. Ordóñez NG. Alveolar soft part sarcoma: a review and update. Adv Anat Pathol. 1999;6(3):125–139. doi:10.1097/00125480-199905000-00001
5. McCarville MB, Muzzafar S, Kao SC, et al. Imaging features of alveolar soft-part sarcoma: a report from Children’s Oncology Group Study ARST0332. AJR Am J Roentgenol. 2014;203(6):1345–1352. doi:10.2214/AJR.14.12462
6. Kaur K, Gami A, Shah A, Gandhi J, Trivedi P. Clinico-pathological spectrum of alveolar soft part sarcoma: case series from a tertiary care cancer referral centre in India with a focus on unusual clinical and histological features. Turk Patoloji Derg. 2024;40(2):89–100. doi:10.5146/TJPATH.2023.01605
7. Kenneth Schoolmeester J, Carlson J, Keeney GL, et al. Alveolar soft part sarcoma of the female genital tract. A morphologic, immunohistochemical, and molecular cytogenetic study of 10 cases with emphasis on its distinction from morphologic mimics. Am J Surg Pathol. 2017;41(5):622–632. doi:10.1097/PAS.0000000000000796
8. Ogura K, Beppu Y, Chuman H, et al. Alveolar soft part sarcoma: a single-center 26-patient case series and review of the literature. Sarcoma. 2012;2012(1):907179. doi:10.1155/2012/907179
9. Sherman N, Vavilala M, Pollock R, Romsdahl M, Jaffe N. Radiation therapy for alveolar soft-part sarcoma. Med Pediatr Oncol. 1994;22(6):380–383. doi:10.1002/MPO.2950220605
10. Monroe AT, Bhandare N, Morris CG, Mendenhall WM. Preventing radiation retinopathy with hyperfractionation. Int J Radiat Oncol Biol Phys. 2005;61(3):856–864. doi:10.1016/j.ijrobp.2004.07.664
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.