Back to Journals » International Journal of Nanomedicine » Volume 13
Study of the mechanism of environmentally friendly translucent balsa-modified lysozyme dressing for facilitating wound healing
Authors Zhou D ![]() , Yang T
, Yang T ![]() , Qian W, Xing M, Luo G
, Qian W, Xing M, Luo G
Received 9 February 2018
Accepted for publication 24 April 2018
Published 17 July 2018 Volume 2018:13 Pages 4171—4187
DOI https://doi.org/10.2147/IJN.S165075
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Daijun Zhou, Tao Yang, Wei Qian, Malcolm Xing, Gaoxing Luo
Institute of Burn Research; State Key Laboratory of Trauma, Burn and Combined Injury; Southwest Hospital, Third Military Medical University (Army Medical University), Chongqing, China
Objective: This study aimed to prepare an eco-friendly dressing using a balsa-derived membrane with lysozymes designed for antibacterial purposes.
Methods: The groups included controls, balsa (group A), translucent balsa (group B), translucent balsa–lysozymes (group C), and translucent balsa-modified lysozymes (group D). Physical and chemical methods were used to characterize the materials, and the function of the materials was evaluated by in vivo and in vitro experiments.
Results: Antibacterial activity against Escherichia coli and Staphylococcus aureus was ordered D > C > B ≈ A (P<0.05). Healing rates in the control, A, B, C, and D groups were 30.6%, 48.3%, 56.7%, 70.9%, and 79.2%, respectively at 7 days after injury. The lengths of new epithelia of the wound surface were ordered D > C > B ≈ A > control (P<0.05). Reverse-transcription polymerase chain reaction showed that expression of Wnt3a, β-catenin, and PCNA mRNA were ordered D > C > B ≈ A > control (P<0.05). The order of expression of PCNA was D > C > B ≈ A > control (P<0.05). There were no differences in GSK3β expression (P>0.05). The order of expression of axin was D < C < B ≈ A < control (P<0.05). The cell-migration rate at 24 hours was ordered D > C > B ≈ A > control (P<0.05).
Conclusion: This translucent balsa-modified lysozyme dressing is characterized by strong antibacterial properties, stable and persistent release, no cytotoxicity, and capacity to promote antibacterial ability and epithelial growth, as well as cell proliferation and migration.
Keywords: balsa, lysozyme, wound dressing, wound healing
Introduction
Wood is a renewable resource characterized by low density, high modulus, strength, and toughness, and low heat-conductivity coefficients.1,2 Balsa is the lightest commercial timber in the world. It is used to make various structures, because of its low density, uniform texture, low heat-conductivity coefficient, ease of processing, and good volume stability.3,4 Studies have shown that balsa can be modified to form novel transparent lightweight structures, maintaining its cellulose structure while permitting light transmittance up to 90%.5 It is relatively inexpensive, with high air permeability and transparency. It also has functions relevant to wound-surface dressings.
Lysozymes are small monomeric proteins containing 129 amino acid residues with anti-inflammatory, antiviral, and antihistaminic activities.6,7 They have been used widely for antisepsis as well.8 Their bactericidal mechanism primarily derives from their biochemical functions. Specifically, catalytic hydrolysis of the peptidoglycan layer, the basic constituent of the cell wall, damages the cell wall of a microorganism, resulting in bacteriolysis. Therefore, differences in cell-wall structure among various microorganisms have significant effects on the action of lysozymes. For example, the cell wall of Gram-positive bacteria contains 80% peptidoglycan, whereas Gram-negative bacteria have only a small amount of peptidoglycan in the inner layer of the cell wall. In addition, Gram-negative bacteria are embedded with an adventitial lipopolysaccharide layer, preventing lysozyme molecules from effectively entering cells. Therefore, the actions of lysozymes differ substantially in terms of bacteriolytic actions in the two types of bacteria. Lysozymes effectively kill Gram-positive bacteria, but perform poorly against Gram-negative bacteria.9 Lysozymes are potentially ideal medical enzymes by virtue of their ability to deactivate viruses by interacting with negatively charged viral proteins.6,7 In an antibacterial drug, lysozymes participate in mucopolysaccharide metabolism, reducing inflammation. Moreover, lysozymes deactivate inflammatory responses induced by various acidoids. They also strengthen the efficacy of antibiotics and other drugs and improve tissue metabolism of mucopolysaccharide, thus reducing inflammation and repairing tissue.8,9 It is thus clear that lysozyme molecules perform various pharmacological functions, participating in immunoresponses and playing vital roles in maintaining physiological equilibrium.
Nevertheless, lysozymes have little activity against Gram-negative bacteria, limiting their applicability.8,9 To circumvent this limitation, lysozymes have been modified to gain antibacterial properties against Gram-negative bacteria.10 Currently, effective modification methods for lysozymes include physical, chemical, and biological modification methods.11 Chemical modification of enzyme molecules aims to change properties of an enzyme. By cutting and splicing the main chain and chemically modifying the side chain, lysozyme molecules may acquire expanded applications and achieve greater economic benefits.9,10 Recent studies have reported chemical modifications of side-chain groups of natural lysozymes,12 creating amphiphilic enzyme molecules, thus allowing modified lysozymes to penetrate the lipopolysaccharide layer of Gram-negative bacteria.10,11 Modified lysozymes with dithiothreitol showed that bacteriostatic activity of enzymes against Gram-negative bacteria increased.12 Modified the side chain groups of the lysozyme using short-chain and medium-chain saturated fatty acids (hexanoic acid, decanoic acid, and myristic acid).13 The modified enzymes had little activity loss, but had greater inhibitory effects on Gram-negative bacteria than lysozymes modified with long-chain fatty acids. The combination of lysozymes and dressing has become a popular field of research. The combination effectively improves the continuous bactericidal activity of lysozymes against Gram-negative bacteria.12,14,15
In this study, we used cinnamic acid, caffeic acid, and p-coumaric acid as modifiers, using dopamine cross-linking. Bacteriostatic functions were compared according to inhibition-zone diameter and minimal inhibitory concentration. Enzymatic properties were compared to find the optimal pH and temperature. Hydrophobicity and secondary-structure content were determined to screen for the optimal modifier. Translucent balsa was prepared and adhered to the optimal modifier with dopamine to design a translucent balsa-modified lysozyme group. The test results provide guidance for studies of dressings in clinical wound healing.
Methods
Materials
Dopamine hydrochloride and lysozymes were purchased from Solarbio Science and Technology (Beijing, China). Balsa was purchased from Lego. BALB/c mice (males, 20–25 g) were obtained from the Experimental Animal Department of the Third Military Medical University. Animals were raised in a room at 25°C with relative humidity of 50%, and a 12-hour circadian rhythm. Before the experiment, mice were fed in a single cage and adapted for 1 week. All trials were repeated more than three times. Methods are illustrated in Figure 1.
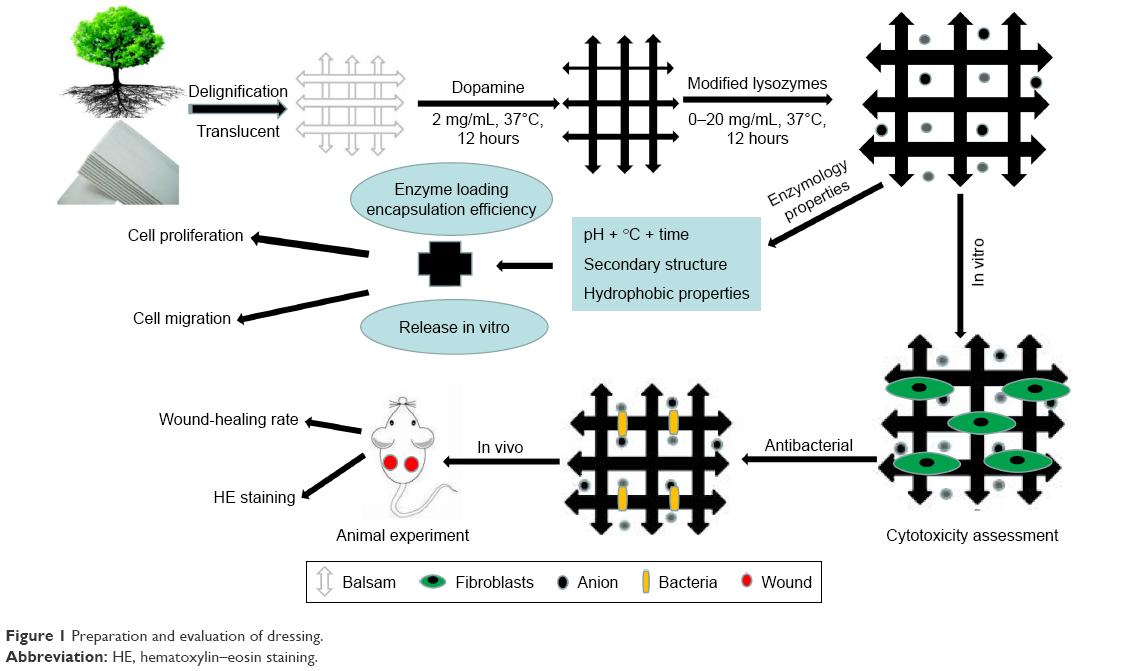 | Figure 1 Preparation and evaluation of dressing. |
Animal-care ethics statement
All in vivo experiments animal protocols that included primary cultures were approved by the institutional animal care and use committee of the Third Military Medical University, Chongqing, China. All animal experiments followed the regulations on the management of laboratory animals issued by the Chinese Association for Laboratory Animal Sciences.
Material preparation
Balsa delignification
A block of balsa with a density of about 160 kg/m3 was penetrated with a puncher to create disks with a diameter of 0.6 mm and thickness of 0.8 mm. The wooden disks were dried for 24 hours at 105°C±3°C and were immersed in an NaClO2–acetate buffer solution (pH 4.6, 80°C) at a concentration of 1% for reaction for 12 hours. The extracted sample was carefully cleaned with deionized water and dried in ethanol 1:1 ethanol mixed with acetone–acetone for 10 minutes in each step. Steps were repeated three times.
Translucent balsa
The balsa was immersed in delignification solution (prepolymerized methyl methacrylate solution) and heated at 70°C for 4 hours. In other words, for the straight polymerization of methyl methacrylate monomer, azodiisobutyronitrile 0.3% served as an initiator in a double-neck round-bottom flask (reaction time 15 minutes at 75°C). Prepolymerization terminated when the monomer was cooled to room temperature in an ice-water bath. Then, the delignified wood was immersed in the prepolymerized methyl methacrylate solution for 30 minutes. This step was repeated three times for complete permeation. Finally, the wet wood was placed between two pieces of glass and wrapped with aluminum foil for further polymerization. The wet wood was heated to 70°C for 4 hours to complete the polymerization process.
Preparation of modified enzymes
We added 60 mg organic acid (cinnamic acid, caffeic acid, and p-coumaric acid). The three acids were dissolved in 3 mL 5 M NaOH. We added 5 M HCl to adjust the pH to 7.5. Sterilized deionized water was added to a total volume of 8 mL. We added 160 mg dopamine in this solution. The solution was allowed to stand at room temperature, and 60 mg lysozyme was added. The solution was mixed for 24 hours at 30°C in a constant-temperature bath. The insoluble portion was pelleted (6,000 rpm, 20 minutes) and discarded following the termination of reaction. The soluble portion was loaded on a Sephadex G-25 column (GE Lifescience). Eluting peaks with enzymatic activity were collected, dialyzed for 1 day at 4°C, concentrated, frozen, dried, and preserved.
Translucent balsa and dopamine cross-linked modified enzymes
We weighed 131.14 mg Tris–hydrochloric acid and dissolved it in 100 mL of deionized water, then added 200 mg dopamine powder. A Tris–dopamine solution at a concentration of 2 mg/mL at pH 8.5 was successfully prepared. Then, the balsa was immersed in the dopamine solution for 12 hours and placed on a shaking table at 37°C at 100 rpm. We weighed and dissolved 500 mg lysozyme and 500 mg optimal modified lysozyme powder in 50 mL deionized water for preparing a lysozyme solution to a final concentration of 10 mg/mL. The solution was divided into two groups – C and D. The extracted sample was carefully cleaned with the deionized water, immersed in group C and D lysozyme solutions for 12 hours, placed on a shaking table at 37°C, and rotated at 100 rpm. The sample was removed and preserved under aseptic conditions at room temperature after the termination of reaction.
Optimization and functional assessment of modified lysozymes
Determination of inhibition zone
Samples of lysozyme and modified-lysozyme were weighed and dissolved with 50 mM pH 7 PBS to prepare sample solutions accurately at 20 mg/mL. The cylinder-plate method was used to conduct an in vitro bacteriostatic test for the Gram-positive bacteria (Staphylococcus aureus) and Gram-negative bacteria (Escherichia coli). We placed 20 mL melted aseptic solid culture medium in a culture dish and allowed it to stand for solidification. We applied 0.2 mL diluted suspension of each bacteria at a concentration of 106–107 CFU/mL to each plate. An Oxford cup was placed on the surface of the culture dish 10 minutes later. Various sample solutions were added. Then, the culture dish was properly covered and cultured for 24 hours at a constant temperature of 37°C. The inhibition-zone diameter of each culture dish was measured. This test was repeated three times for each sample solution. The test results were averaged.
Coculture of bacteria
Cryopreserved S. aureus and E. coli bacterial strains were obtained from the Research Institute for Burns, Southwest Hospital, Third Military Medical University. Bacteria were amplified (shaken overnight) to 109 CFU/mL. The bacterial solution was diluted to 104 CFU/mL with lysogeny-broth culture medium. We pipetted 100 mL bacterial solution and detected OD values with a microplate reader. The standard was met when the OD was about 0.07. Two 96-well microplates were used. Lysozymes and modified enzymes in various groups were placed in the wells, with three duplicated wells for each group. To each well, we added 200 μL of the prepared bacterial solution. Solutions were incubated for 12 hours at 37°C on a shaking table. The solution in each group was tested for alterations. These steps were repeated three times.
Determination of minimal inhibitory concentration
Samples of lysozyme and modified-lysozymes were weighed and dissolved in 50 mM pH 7 PBS to prepare sample solutions accurately at concentrations of 0.25, 0.50, 1.00, 1.50, and 2.00 mg/mL. We placed 0.2 mL sample solution in each series in a sterile test tube. We added 0.2 mL S. aureus and E. coli at concentrations of 106–107 CFU/mL. The solution was well mixed and cultured for 2 hours at a constant temperature of 37°C. The mixed solution was diluted 1,000-fold with sterile deionized water. We applied 0.2 mL solution onto the plate with a spreading rod. The solution was allowed to stand, then poured to a culture dish, and cultured for 24 hours at a constant temperature of 37°C. Bacterial colonies were counted. The 50 mM pH 7 PBS was used to replace the sample solution. Similarly, a control group was created. The growth of bacterial colonies on the plate was observed. The concentration of the sample in the plate without growth of bacterial colonies gave the minimum inhibitory concentration.
Measurement of optimal pH, temperature, and time
Samples of lysozyme and modified-lysozyme were accurately weighed and dissolved in 50 mM PBS at various pH values (4, 5, 6, 7, and 8) for accurately preparing the solution at a concentration of 2 mg/mL. The solution was allowed to react in a bath at various constant temperatures (40°C, 45°C, 50°C, 55°C, and 60°C) for various periods (3, 15, and 30 minutes and 1 and 2 hours). The solution was rapidly cooled to room temperature. The enzyme activity of the lysozyme and modified-lysozymes was determined under various conditions. For easy comparison and determination, other variables would remain constant while a particular variable was changed. The highest enzyme activity was set at 100%. Relative enzyme activity under other conditions was the ratio (percentage) of enzyme activity to highest enzyme activity.
Determination of hydrophobicity
A certain amount of lysozyme and modified-lysozymes were accurately weighed and dissolved in 10 mM pH 7 phosphoric acid buffer solution for accurate preparation of sample solutions at concentrations of 0.1, 0.2, 0.3, 0.4, and 0.5 mg/mL. We withdrew 4 mL of each sample solution. We added 50 μL ANS solution (10 mM pH 7 phosphoric acid buffer solution was used to prepare the 8 mM solution). Excitation wavelength was 360 nm and emission wavelength 470 nm. Fluorescence intensity was determined with fluorophotometry.
Determination of secondary structure
The solid sample was well mixed with KBr, ground, and compressed into pellets. Fourier-transform infrared spectrometry (Nicolet iS10) was used to perform infrared spectrum scanning for the pellets. Spectral resolution was 4 cm−1, wave-number scan range 400–4,000 cm−1, and number of scans 32.
Functional assessment of translucent balsa-modified lysozymes
Drug-loading capacity and encapsulation efficiency
Determination of standard curve for lysozymes
We weighed 200 mg lysozymes and 200 mg modified lysozymes and dissolved them in 10 mL PBS to create a stock solution (20 mg/mL). Appropriate amounts of PBS were added to dilute the solution into three concentrations: 5 mg/mL, 10 mg/mL, and 20 mg/mL. Ultraviolet spectrophotometry was used to measure the absorbance of the lysozyme solution at various concentrations of 357 nm. The standard curve for lysozymes was plotted with the lysozyme concentration as the abscissa and the absorbance as the ordinate. Then, the linear regression equation was calculated.
Measurement of drug-loading capacity and encapsulation efficiency
We weighed 200 mg of the sample in group C and 200 mg of the sample in group D, froze these with liquid nitrogen, ground them to powder, and dispersed them into 10 mL PBS solution ultrasonically for 60 minutes. The solution was centrifuged for separation. The absorbance of the supernatant was measured with ultraviolet spectrophotometry at 357 nm. The concentration was computed according to the standard curve. Then, drug-loading capacity and encapsulation efficiency were computed:
|

|
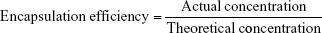
In vitro drug release
We weighed 200 mg of the sample in group C and 200 mg of the sample in group D, added 10 mL PBS, and cultured these on a constant-temperature shaking table (37°C, 70 rpm). The test tube was removed at specific time points (0.5, 12, 24, 48, and 72 hours). We removed 2 mL supernatant and measured absorbance with ultraviolet spectrophotometry. We added 2 mL fresh PBS to the test tube to maintain the total volume of the system. During the test, a cuvette filled with fresh PBS served as the control group. Concentration was computed according to the standard curve. The amount released in each period was computed. Finally, accumulated release was computed by summation.
Mechanical property measurements and determination of water-vapor-transmission rates
Samples were cut into cuboid (10×10×0.7 mm) shape and tested with a material-testing system (5567; Instron, Norwood, MA, USA). Samples were clamped, oriented vertically, and stretched to failure at 10 mm/min. Water-vapor-transmission rates (WVTRs) were measured in accordance with American Society for Testing and Materials standard 17. Samples were cut into disks with a diameter of 35 mm and mounted on the mouth of a cylindrical cup containing 10 mL distilled water. These assemblies were sealed with Teflon tape (Hengzhixin, Jiangsu, China) and then incubated at 37°C and 90% relative humidity. Assemblies were weighed every hour for 24 hours, and results were automatically recorded by a WVTR-testing system (W3/030; Labthink, Jinan, China).
Coculture of bacteria
Methods were the same as in the previous “Coculture of bacteria” section. Changes in OD values were measured at 12 and 24 hours after samples in the control, A, B, C, and D groups had been incubated on the shaking table at 37°C. Steps were repeated three times.
Inhibition of cell proliferation
A newborn mouse was sacrificed for preparation of primary fibroblasts. Cells were usable until passages 2–3. Cells were counted. The DMEM culture was determined according to the 96-well microplate with 2,000 cells per well. The materials in each group were placed in the wells. We set 12 duplicated wells for each group (measured at 1, 3, 5, and 7 days). The biocompatibility of the materials was assessed using CCK8 (Dojindo, Kumamoto, Japan) proliferation assays on days 1, 3, 5, and 7 after seeding. In brief, at each time point, the medium was replaced with DMEM and 10 mL of CCK8 solution added to each well, followed by incubation at 37°C for 2 hours. Mean OD values were determined at 450 nm using an enzyme-linked immunosorbent assay reader (Varioskan Flash; Thermo Fisher Scientific, Waltham, MA, USA), and cell proliferation is expressed as percentages relative to the control group.
Establishment of mouse models with surface wounds induced by full-thickness skin infection
A total of 25 BALB/c mice (male, weighing about 250 g) were purchased from the Animal Research Institute, Third Military Medical University. Five mice were assigned to each group. The animals were raised in a specific pathogen free chamber at room temperature (25°C), relative moisture 50%, and circadian rhythm 12 hours. Experimental mice were raised in separate cages for 1 week for adaptation before the experiment. All experimental operations conformed to the requirements of the ethics committee for laboratory animals at the Third Military Medical University.
Each mouse was intraperitoneally injected with 1% pentobarbital sodium (70 μL/g), shaved for skin preparation, and raised in a separate cage 1 day before the experiment. The next day, the back of each mouse was anesthetized and sterilized. Then, a puncher was used to create two horizontally symmetrical circular full-skin-thickness defects with a diameter of about 0.6 mm in the lower-middle segment of the mouse. Then, 5 μL S. aureus and 5 μL E. coli prepared in the previous steps were added dropwise to the wound surface, each at a concentration of 108/mL. The material was sterilized with 75% ethanol and rinsed with PBS for complete removal of ethanol. The material was applied to the wound surface and fixed with an adhesive operation towel. The wound surface was photographed and the material replaced with new material of the same type at 0, 1, 3, 5, and 7 days after injury.
Wound-healing rate and hematoxylin–eosin (HE) staining in mice
The original area of the wound surface and its area at various time points were measured with IPP6.0 software (Image-Pro Plus). The surface was selected with the area-of-interest function. The pixel area of wound surface was measured with “count size”. The wound-surface area was obtained by scale conversion.
|

For HE staining, wound-surface specimens from each mouse were taken at 3 and 7 days after injury for preparation of paraffin sections. High-quality images were selected for HE staining. Several pathological experts measured the length of new epithelium using the blind method.
Research on mechanisms related to wound healing
Detecting the effects of modified dressing on expression of Wnt3a, β-catenin, and PCNA mRNA with RT-PCR
Cells were inoculated into the membrane patch as described earlier, digested into the cell suspension, and collected at 3 days. Total RNA of cells was extracted. Reverse transcription was performed in accordance with the reaction system 50°C for 30 minutes, 99°C for 5 minutes, and 4°C for 5 minutes. The resulting cDNA was preserved at −20°C for later use after the reaction terminated. Reaction conditions for real-time polymerase chain reaction (RT-PCR) were denaturation for 5 minutes at 95°C, 95°C for 30 seconds, 55°C for 30 seconds, and 72°C for 40 seconds for 35 cycles. The Applied Biosystems 7500 RT-PCR system (Thermo Fisher Scientific) was used to acquire signals and output data. The inherent internal reference GAPDH of the sample was used for standardization. The results were computed and analyzed with the ΔΔCT method on the basis of relevant literature.
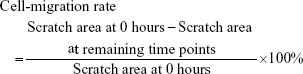
Studying cell migration at 6, 12, and 24 hours based on scratch test
The scratch-wound experiment was used to measure primary fibroblast cell-migration ability. Cell concentration was adjusted to 105/mL using liquid culture medium. Cells were inoculated into a 24-well microplate with 1 mL per well. Cells were cultured for 2 hours with RPMI 1640 medium liquid culture medium containing mitomycin at a concentration of 4 μg/mL. Cell adherence was good, and cell fusion reached 80%–90%. A vertical scratch was made at the center of the microplate base with the tip of a 10 μL pipette. The liquid culture medium was discarded. Cells were washed three times. There were three wells for each group. Observation was performed at the living-cell working station. ImageJ software was used to measure scratch area at 0, 6, 12, 24 hours after the scratch had been made.
|
Western blot detection of Wnt–β-catenin signaling pathway and migrated F-actin and vinculin proteins
Cell concentration was adjusted to 2×106/mL with the liquid culture medium. Cells were inoculated into a six-well microplate, with 1 mL per well and three wells for each group. Radioimmunoprecipitation-assay lysis buffer was added to lyse the cells completely after each group had been routinely cultured for 24 hours. Total cell protein was extracted and quantified. Protein was incubated overnight at 4°C after addition of Wnt3a, β-catenin, PCNA, p-GSK3β, GSK3β, axin 2, F-actin, vinculin, β-actin, and GAPDH. The next day, the membrane was washed five times for 5 minutes each with Tris-20 buffer solution. Protein was incubated for 1 hour at room temperature after addition of antirabbit and mouse-derived horseradish peroxidase-linked IgG secondary antibody. A chemiluminescence agent was added. Proteins were developed, subjected to gel electrophoresis, and images obtained. β-Actin and GAPDH proteins served as the internal reference. Quantity One software (Biorad) was used for gray analysis. The final concentration of the Wnt-signaling-pathway inhibitor XAV939 was 10 μM. Proteins on the membrane were digested and collected at 3 days after inoculation. The protein was extracted as described earlier. Changes in expression of Wnt3a, β-catenin, axin 2, PCNA, and β-actin were measured.
Statistical analysis
All data are presented as means ± SD. Student’s t-test and one-way analysis of variance were used to evaluate statistical significance, followed by post hoc least-significant-difference tests. Values of P<0.05 were considered significant.
Results
Determination of inhibition zone, cell coculture, and minimal inhibitory concentration
The three acids were arranged in the order of p-coumaric acid > caffeic acid > cinnamic acid (Figure 2) in terms of antibacterial effects and minimal inhibitory concentrations. p-Coumaric acid-modified lysozymes had the strongest activity when pH was 6, temperature 45°C, and reaction time 30 minutes (Figure 3). The order was p-coumaric acid > cinnamic acid > caffeic acid for surface-hydrophobicity index and secondary-structure stability (Figure 4).
 | Figure 2 Antibacterial properties of different acids and modified enzymes. |
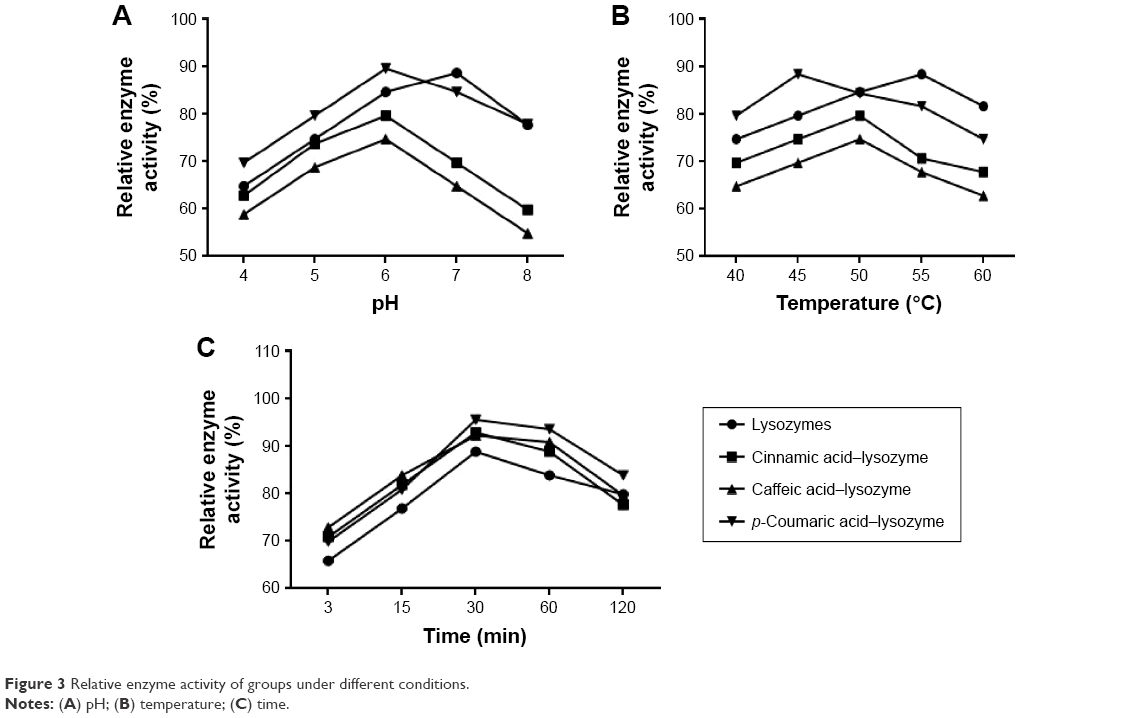 | Figure 3 Relative enzyme activity of groups under different conditions. |
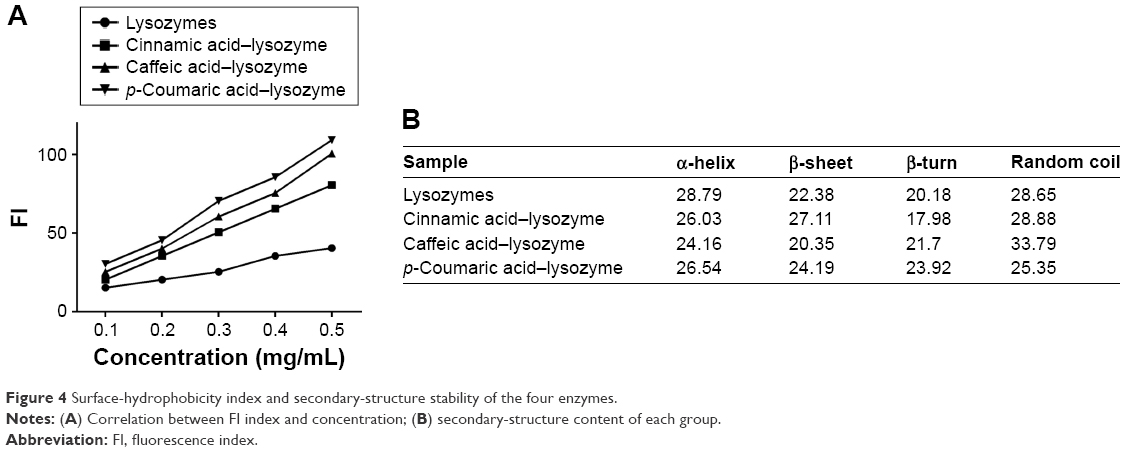 | Figure 4 Surface-hydrophobicity index and secondary-structure stability of the four enzymes. |
Drug-loading capacity, encapsulation efficiency, and in vitro drug release
The drug-loading capacity and encapsulation efficiency in group D were superior to that of group C (P<0.05). As can be seen from the release curve, the release in groups C and D tended to be stable, reaching a maximum at 48 hours. Accumulated release at 72 hours was 85.2% and 93.8%, respectively (Figure 5).
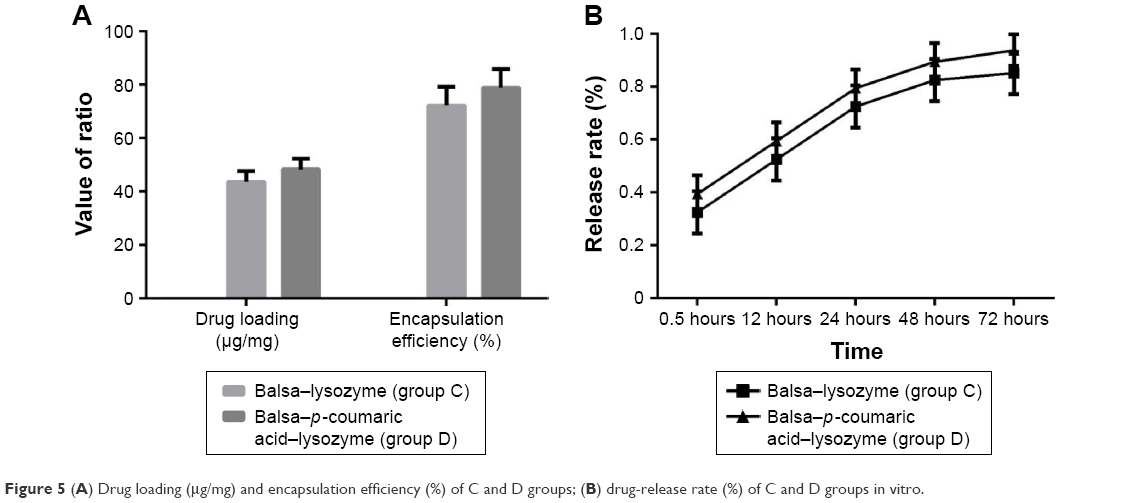 | Figure 5 (A) Drug loading (μg/mg) and encapsulation efficiency (%) of C and D groups; (B) drug-release rate (%) of C and D groups in vitro. |
Mechanical strength and WVTR
Tensile strength, elongation at break, and Young’s modulus of membranes are summarized in Table 1. As seen, the strength (tensile strength and Young’s modulus) of group A was obviously lower than group D (P<0.05). As shown in Table 2, the average WVTR was D ≈ C ≈ B > A (P<0.05).
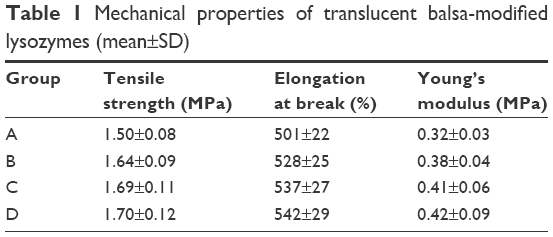 | Table 1 Mechanical properties of translucent balsa-modified lysozymes (mean±SD) |
 | Table 2 Water-vapor-transmission rates of translucent balsa-modified lysozymes (mean±SD) |
In vitro antisepsis and cell-proliferation inhibition
The growth of S. aureus and E. coli in the control group conformed to the normal trend at 24 hours. The groups’ order in terms of their antibacterial activity against E. coli and S. aureus was D > C > B ≈ A (P<0.05). Proliferation of fibroblasts was not inhibited in any group at 1–7 days (P>0.05, Figure 6).
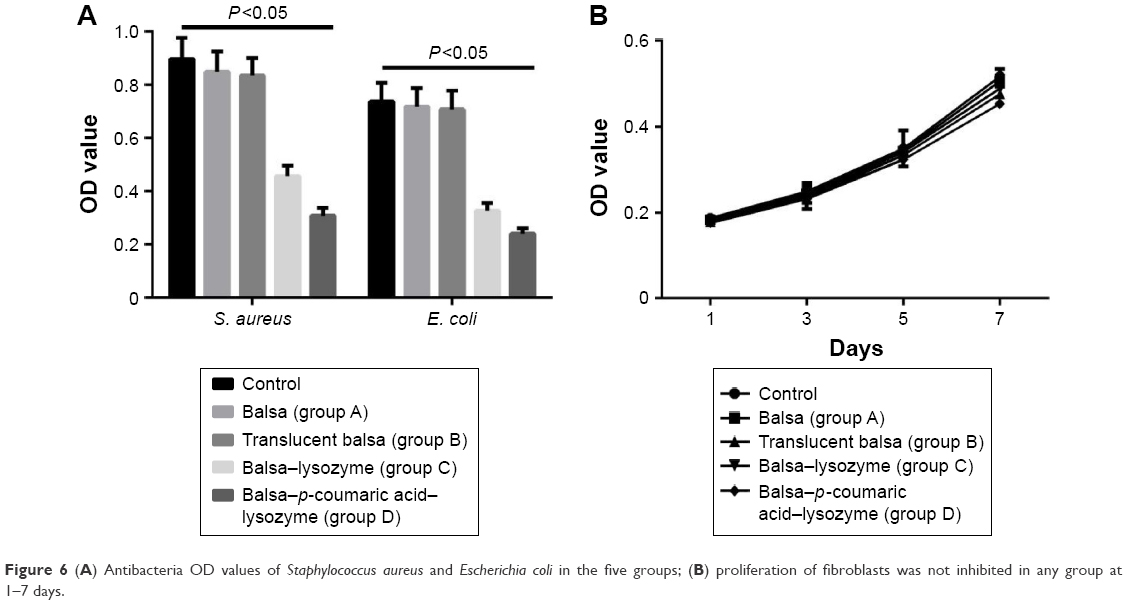 | Figure 6 (A) Antibacteria OD values of Staphylococcus aureus and Escherichia coli in the five groups; (B) proliferation of fibroblasts was not inhibited in any group at 1–7 days. |
Effects on wound healing and length of new epithelium
Healing rates in the control, A, B, C, and D groups were 18.5%, 26.3%, 28.5%, 36.0%, and 39.8%, respectively, at 3 days after injury. Wound-healing rates were 30.6%, 48.3%, 56.7%, 70.9%, and 79.2%, respectively, at 7 days after injury. The five groups had the order D > C > B ≈ A > control in terms of wound-healing rates at both 3 and 7 days after injury (P<0.05, Figure 7). There were no differences in new-epithelium length among the groups at 3 days after injury (P>0.05). Lengths of new epithelia of the wound surface in the control, A, B, C, and D groups were 618.0 μm, 687.3 μm, 726.5 μm, 819.8 μm, and 886.7 μm, respectively, at 7 days after injury (D > C > B > A > control; P<0.05; Figure 8).
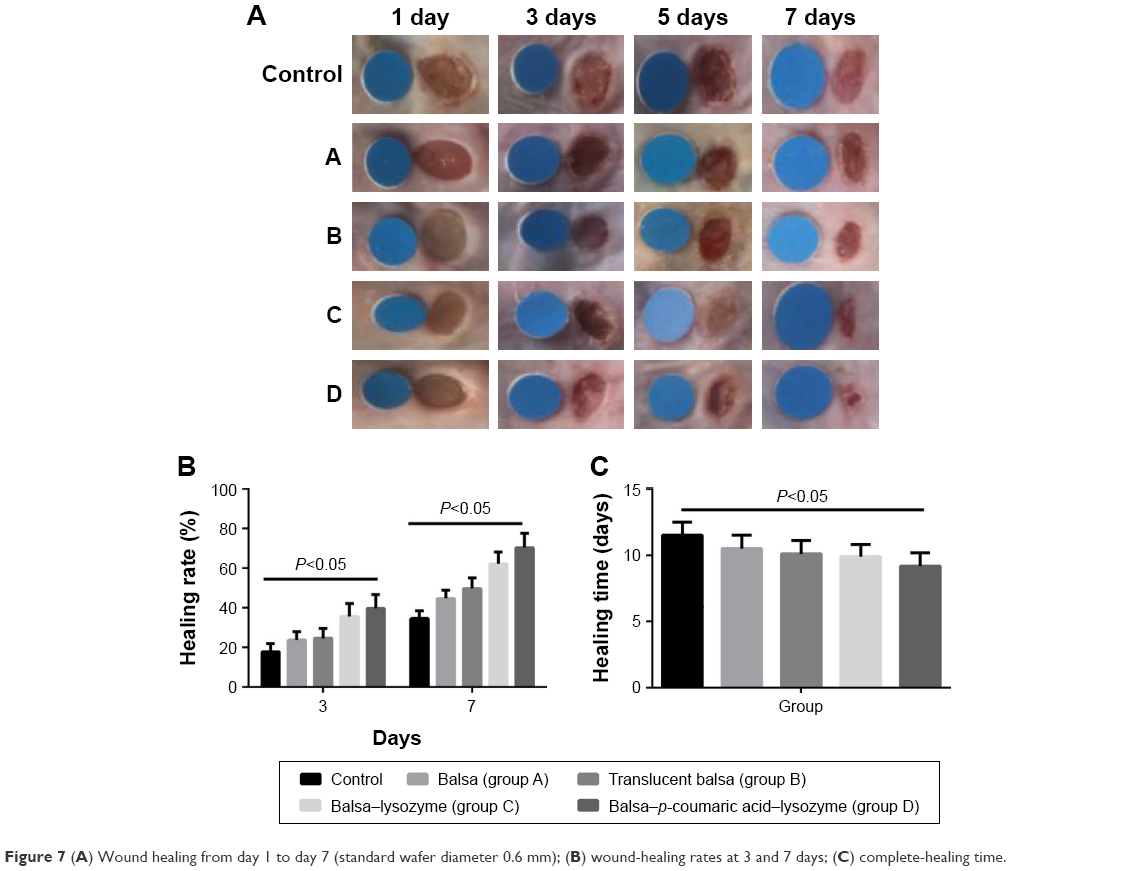 | Figure 7 (A) Wound healing from day 1 to day 7 (standard wafer diameter 0.6 mm); (B) wound-healing rates at 3 and 7 days; (C) complete-healing time. |
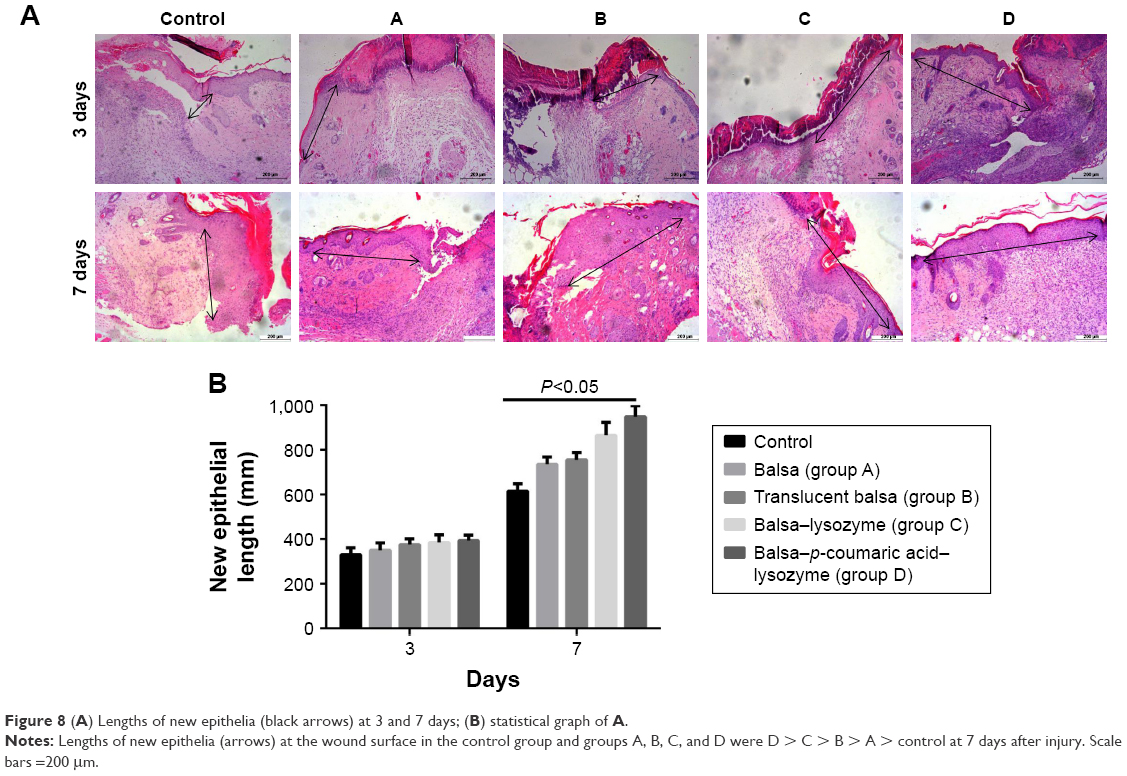 | Figure 8 (A) Lengths of new epithelia (black arrows) at 3 and 7 days; (B) statistical graph of A. |
Effects on cell proliferation and relevant pathways
RT-PCR showed that the five groups had the order D > C > B ≈ A > control (P<0.05) in terms of increase in expression of Wnt3a, β-catenin, and PCNA mRNA (Figure 9). Western blot (WB) was used to detect changes in the Wnt–β-catenin signaling pathway and Wnt3a, β-catenin, p-GSK3β, and p-GSK3β:GSK3β ratios at 7 days. The five groups could be arranged in descending order, D > C > B ≈ A > control in terms of expression of PCNA (P<0.05). There were no differences in GSK3β expression (P>0.05). The five groups could be arranged in ascending order, D < C < B ≈ A < control (P<0.05) in terms of expression of axin (Figure 10). The CCK8 method showed that cell growth was significantly inhibited (P<0.05) after the Wnt–β-catenin signaling-pathway inhibitor XAV939 had been added to group D. Expression of Wnt3a, β-catenin, and PCNA was inhibited, but the expression of axin 2 increased (P<0.05, Figure 11).
 | Figure 9 Reverse-transcription polymerase chain reaction showed that the five groups could be arranged in descending order. |
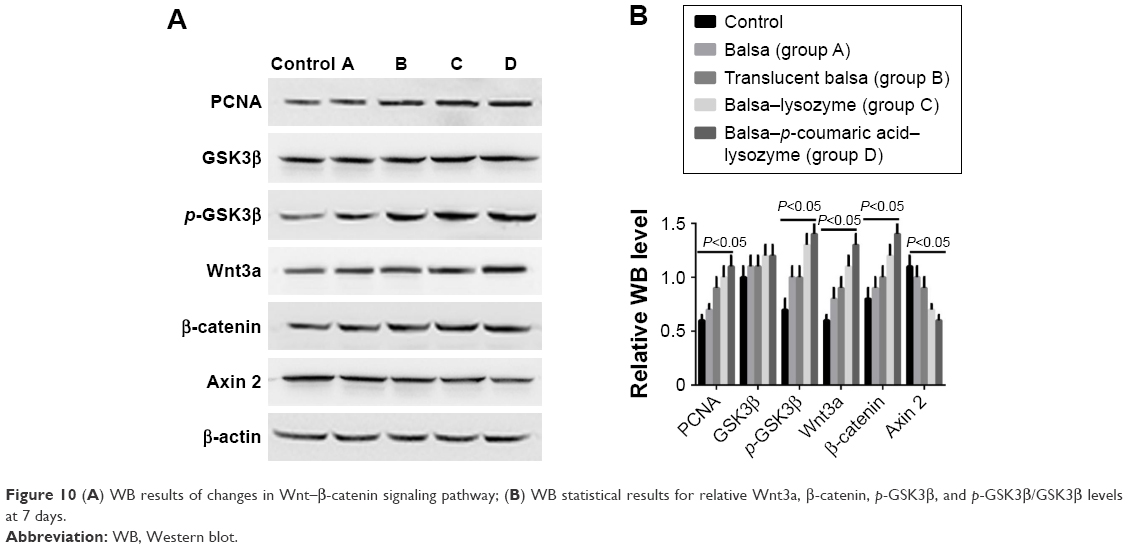 | Figure 10 (A) WB results of changes in Wnt–β-catenin signaling pathway; (B) WB statistical results for relative Wnt3a, β-catenin, p-GSK3β, and p-GSK3β/GSK3β levels at 7 days. |
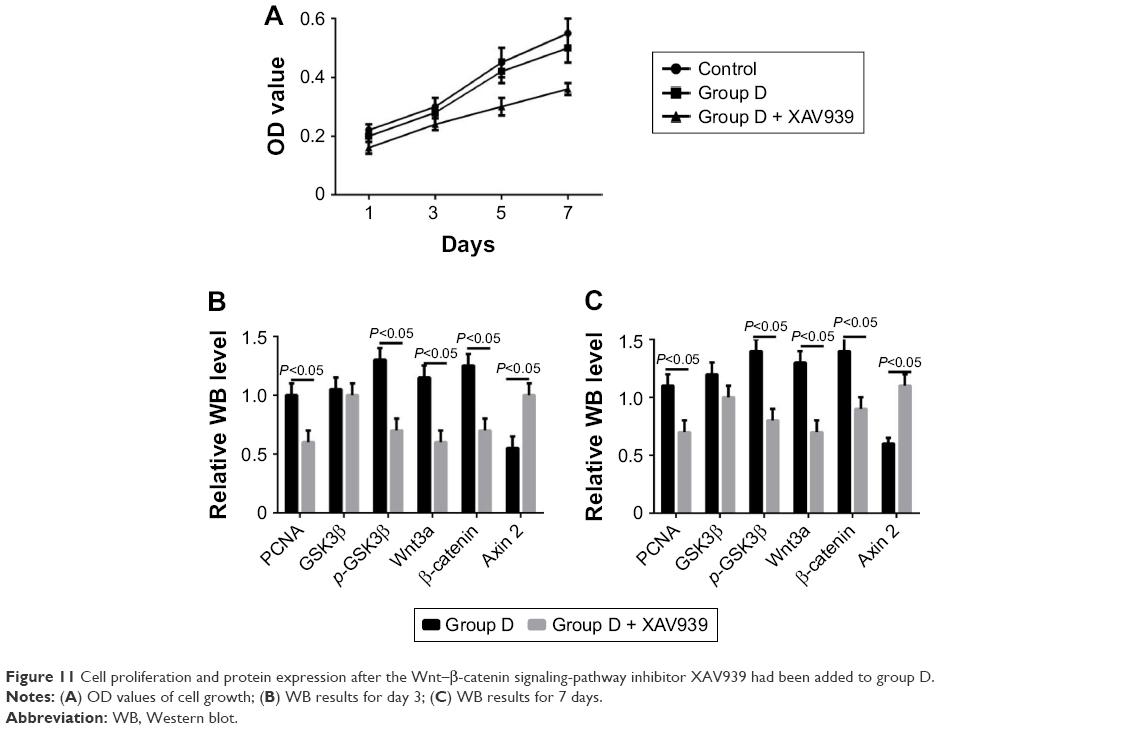 | Figure 11 Cell proliferation and protein expression after the Wnt–β-catenin signaling-pathway inhibitor XAV939 had been added to group D. |
Effects on cell migration and relevant pathways
The five groups could be arranged in descending order, D > C > B ≈ A > control (P<0.05) in terms of cell-migration rate at 24 hours after the scratch test (Figure 12), and D > C > B ≈ A > control (P<0.05) in terms of the expression of migrated proteins detected on WB, including F-actin and vinculin (Figure 13).
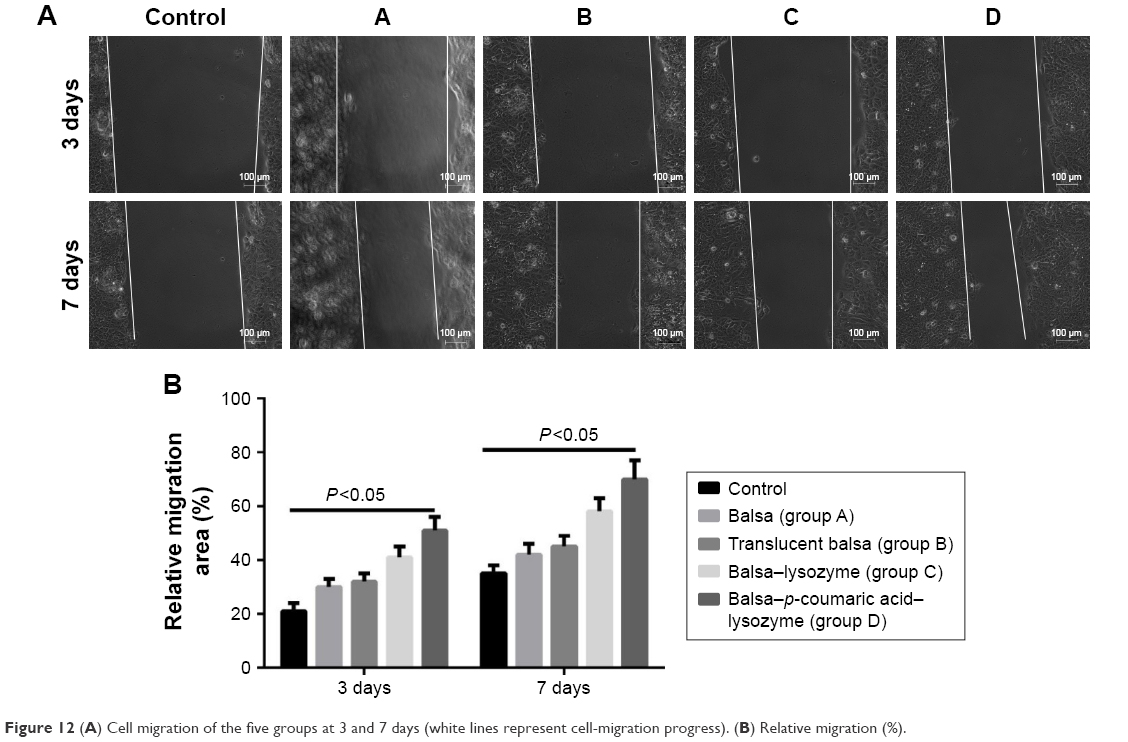 | Figure 12 (A) Cell migration of the five groups at 3 and 7 days (white lines represent cell-migration progress). (B) Relative migration (%). |
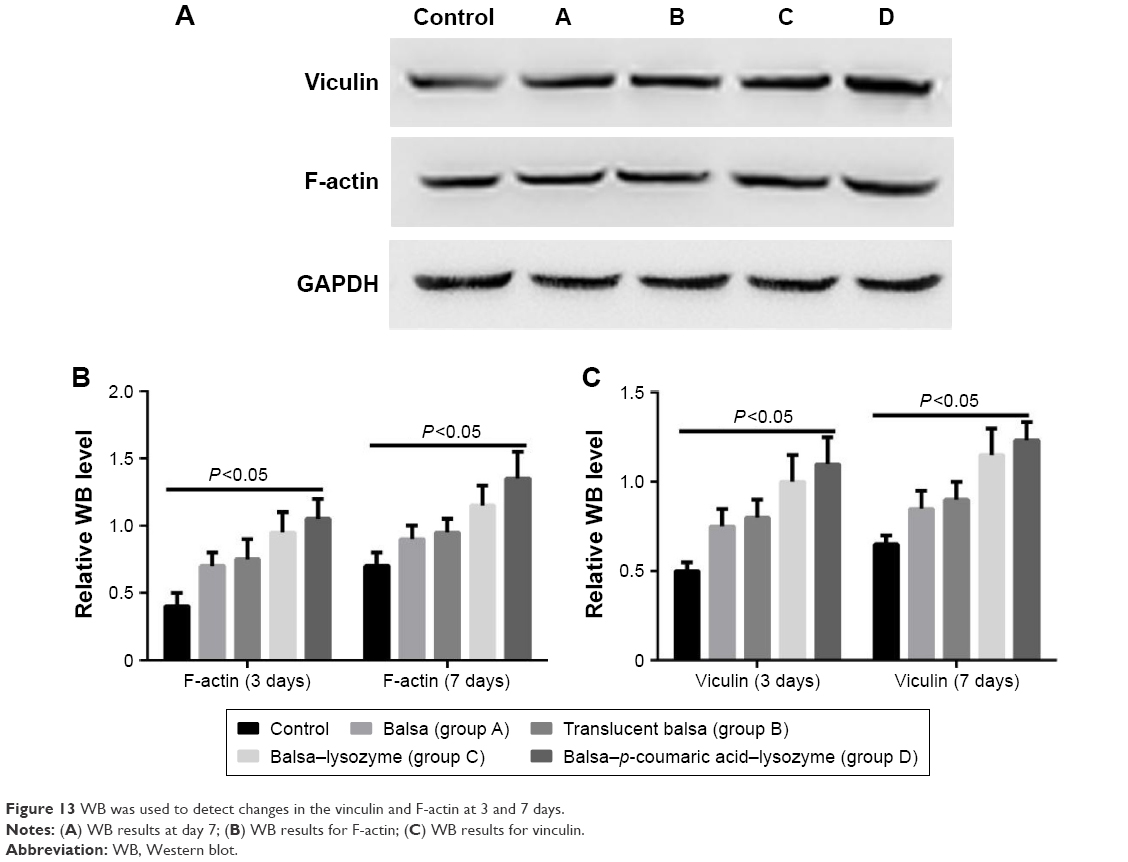 | Figure 13 WB was used to detect changes in the vinculin and F-actin at 3 and 7 days. |
Discussion
Patients with large skin defects urgently require effective coverage of the wound surface to prevent bacterial invasion and loss of mucus, water electrolytes, and energy. A wound dressing can reconstruct the skin barrier and accelerate wound healing, thus creating conditions appropriate for subsequent surgery.16 An ideal wound dressing should have excellent mechanical properties, proper water and air permeability, and outstanding biological compatibility. Infections and imbalance in inflammatory responses retard or prevent wound healing. An ideal wound dressing should also provide an aseptic microenvironment for the wound.17,18 Natural materials, including chitosan, chitin, glucans, celluloses, alginate, and fibroin, are thought to be ideal materials for preparing wound dressings, because of their high bioavailability and good biological compatibility. As such, promoting healing of wound infections by a combination of the natural materials and various antibacterial agents, growth factors, or other chemical substances has become a focus of research in the field of wound repair.19,20
Three acids can be arranged in descending order (p-coumaric acid > caffeic acid > cinnamic acid) in terms of their antibacterial effects and minimal inhibitory concentration. This suggests that modified enzymes have greater antibacterial effects than natural enzymes (Figure 2). According to studies,21,22 modified enzymes are amphiphilic molecules. Enzymes can penetrate the lipopolysaccharide layers of Gram-negative bacteria and may possess bactericidal effects against these bacteria as their hydrophobicity increases. Therefore, modified enzymes have significantly more inhibitory effects on these bacteria than natural enzymes. According to the bactericidal mechanism of lysozymes and the structural characteristics of the bacterial cell wall, the inhibitory effects of enzymes on Gram-positive bacteria depend primarily on enzyme activity, whereas the inhibitory effects of enzymes on Gram-negative bacteria are jointly determined by the ability of enzymes to penetrate the lipopolysaccharide layer and the enzymatic activity. Therefore, variations in the inhibitory effects of enzymes on various bacteria may be different under the same heat-treatment conditions, possibly accounting for the greatest inhibitory effects of p-coumaric acid.12,14 p-Coumaric acid-modified lysozymes had the highest activity at pH 6 and temperature 45°C, with reaction time of 30 minutes (Figure 4). We believe that changes possibly occurred at the optimal pH for the chemically modified lysozymes because the covalent binding of various acids by natural enzymes led to changes in isoelectric point as well as enzyme structure. The three acids can be arranged in descending order p-coumaric acid > cinnamic acid > caffeic acid in terms of surface-hydrophobicity index of the four enzymes (Figure 3). This may be due to the fact that the modified lysozymes experienced changes in their structure, thus exposing the hydrophobic groups within the natural enzymes and increasing the surface hydrophobicity index of natural enzymes.10,15 According to the structural characteristics and covalent binding characteristics of the three acids, the covalent binding of the natural enzymes and the three acids is the covalent binding of the amidogen of the enzyme molecules and the carboxyl of the organic acids. The hydrophobic benzene rings contained in the three organic acids remained intact. The existence of hydrophobic benzene rings may also enable the surface-hydrophobicity index of the modified enzymes to increase.6,9 The three acids can be arranged in descending order p-coumaric acid > cinnamic acid > caffeic acid in terms of secondary-structure stability of the four enzymes (Figure 3). The content of random coils in the remaining two modified enzymes increased somewhat compared with that of the remaining p-coumaric acids. The molecular structure of modified enzymes changed. The ordered structure of enzymes decreased, whereas disordered structure increased. The peptide-chain structure of the enzyme molecules tended to be loose, affecting enzyme stability.8,13 As seen, the strength (tensile strength and Young’s modulus) of group A was obviously lower than group D (P<0.05). This result indicates that translucent balsa helps significantly to improve tractility and flexibility of the balsa, thereby adding to the application for wound dressing under high stresses. As shown in Table 2, average WVTR was D ≈ C ≈ B > A (P<0.05), which was very suitable to supply a moist environment for wound healing without wound-exudate accumulation. Collectively, compared with conventional products, our translucent balsa-modified lysozymes had the advantage of bionic performance.
Drug-loading capacity and encapsulation efficiency in group D was superior to group C (P<0.05). As can be seen from the release curve, the release in groups C and D tended to be stable, reaching the maximum at 48 hours. Accumulated release at 72 hours was 85.2% and 93.8%, respectively (Figure 5). This may be due to the fact that the p-coumaric acid-modified lysozymes were more likely to cover the balsa surface during preparation of pellets. In addition, the excessively high concentration of pure lysozymes led to a low content of the cross-linked lysozymes and decreased encapsulation efficiency. In terms of drug release, the release in group D was rapid during the first 24 hours. The significant abrupt release in the early stage may be due to the fact that the p-coumaric acid-modified lysozymes were embedded in the balsa structure using the cross-linking method. The embedding structure had poor compactness. Lysozymes themselves are antibacterial agents with high water solubility. Lysozymes were very likely to have penetrated the surface structure of balsa and release in PBS, thus increasing the amount released when lysozymes were cultured in PBS. The amount of lysozymes released remained unchanged at 48 hours, indicating that their release tended to be stable, reaching maximum and fluctuating at about 80%–90%.
The growth of S. aureus and E. coli in the control group tended to be normal at 24 hours. The four groups can be arranged in descending order, D > C > B ≈ A (P<0.05, Figure 6) in terms of their antibacterial activity against E. coli and S. aureus. At present, it is well known that lysozymes have inhibitory effects on Gram-positive bacteria. The mechanism lies in hydrolysis of the chemical bond between N-acetylmuramic acid and peptidoglycans of the bacterial cell wall.23 This was verified in our results. According to recent studies,23,24 the exposure of hydrophobic groups in enzyme molecules is the major cause of increased activity against Gram-negative bacteria. The increase in hydrophobicity index of the modified enzymes provided a further theoretical basis for the increase in function against Gram-negative bacteria. In terms of cytotoxicity, the proliferation of fibroblasts in different groups was not inhibited (P>0.05, Figure 6). This may be due to the fact that lysozymes originate from egg white and are relatively safe enzymes. It is believed that the safety of lysozymes allows for their wide application in food preservation.24
The in vitro and in vivo defense barriers of burn patients are damaged, and immunity declines significantly. Extensive tissue necrosis and invasion by exogenous and endogenous bacterial colonies lead to wound infections.25 Wound infection is one of the major complications and causes of death in burn patients, accounting for approximately 52%–70% of deaths following burns.26
We added the Gram-positive bacterium S. aureus and the Gram-negative bacterium E. coli to the infectious burn-wound surface. The bacterial concentration was 108/mL.27–29 Healing rates in the control, A, B, C, and D groups were 18.5%, 26.3%, 28.5%, 36.0%, and 39.8%, respectively, at 3 days after injury. Wound-healing rates were 30.6%, 48.3%, 56.7%, 70.9%, and 79.2%, respectively, at 7 days after injury. The five groups could be arranged in descending order, D > C > B ≈ A > control in terms of wound-healing rates at both 3 days and 7 days after injury (P<0.05, Figure 7). With the increase in antibacterial action, particularly antibacterial action against Gram-negative bacilli, p-coumaric acid-modified lysozymes effectively altered the infectious wound surface and promoted healing. The process of wound healing consisted of two periods: wound contraction and wound epithelization. For species with compact skin (eg, humans), wound healing is achieved primarily by wound epithelization.30 There were no differences in length of new epithelium among the groups at 3 days after injury. This was primarily because the wound-edge epidermis was in the preparation phase of cell proliferation and migration, and the new epithelium was not significant.31 The lengths of new epithelia of the wound surface in the control, A, B, C, and D groups were 618.0 μm, 687.3 μm, 726.5 μm, 819.8 μm, and 886.7 μm, respectively, at 7 days after injury (D > C > B > A > control; P<0.05; Figure 8). These results suggested that p-coumaric acid-modified enzymes significantly promoted reepithelization of the wound and thus accelerated healing of the wound by controlling infections at the late stage of injury (at 7 days after injury). To study the mechanism of wound healing further, we conducted a study of cell proliferation and migration.
PCNA is a downstream target of the Wnt–β-catenin signaling pathway. The Wnt–β-catenin signaling pathway itself also has important effects on cell proliferation and cell fate in different tissue types.32 Therefore, we also measured mRNA levels of Wnt3a and β-catenin in cells. RT-PCR revealed that the five groups could be arranged in descending order, D > C > B ≈ A > control (P<0.05) in terms of increased expression of Wnt3a, β-catenin, and PCNA mRNA (Figure 9). According to these results, we propose that p-coumaric acid-modified lysozymes influenced the adherence and proliferation of cells via the Wnt-signaling pathway. The protein in cells was extracted for WB detection. The Wnt–β-catenin signaling pathway altered at 7 days. We found that the five groups could be arranged in descending order, D > C > B ≈ A > control (P<0.05) in terms of expression of Wnt3a, β-catenin, p-GSK3β, and p-GSK3β:GSK3β ratios and with respect to the expression of PCNA. There were no differences in GSK3β expression (P>0.05). The five groups could be arranged in ascending order, D < C < B ≈ A < control (P<0.05) in terms of expression of axin (Figure 10).
Protein expression of Wnt3a, β-catenin, and PCNA was significantly higher in the macroporous silica-membrane group. This result coincided with the RT-PCR detection result. In the current literature, it is generally accepted that intracellular β-catenin protein is regulated by the degraded complex APC–axin–CK1–GSK3β. GSK3β is an active form and can degrade β-catenin. Phosphorylated GSK3β is inactivated and thus cannot degrade β-catenin. Axin 2 is closely associated with the negative feedback of the Wnt–β-catenin signaling pathway.32 We found that protein expression in β-catenin and p-GSK3β increased in group D, while axin 2 expression decreased, suggesting that the β-catenin-degraded complex was deactivated,33 thus leading to accumulation of β-catenin within cells. Generally, only a small amount of β-catenin is present in the cytoplasm and nuclei. β-Catenin enters the nuclei and activates the expression of related downstream genes with accumulation of β-catenin within the cells. These genes include the proliferation-related PCNA.34
XAV939 is a small molecule compound that maintains the integrity and activity of the β-catenin-degraded complex by stabilizing the structure of axin 2. β-Catenin is degraded and the Wnt–β-catenin signaling pathway inhibited.32 In the present study, we clarified the important role of the Wnt-signaling pathway in this process, where the pore structure regulated cell proliferation using the Wnt–β-catenin signaling-pathway inhibitor. As the effects on cell proliferation in group D were the most significant, we added the Wnt–β-catenin signaling-pathway inhibitor XAV939 in group D. The CCK8 result showed that cell growth was significantly inhibited (P<0.05). Meanwhile, the expression of Wnt3a, β-catenin, and PCNA was inhibited, while the expression of axin increased (P<0.05, Figure 11). This suggested that p-coumaric acid-modified lysozymes regulated cell proliferation via the Wnt–β-catenin signaling pathway.
Wound reepithelization relies on the migration of epithelial cells from the wound periphery to the wound center. Therefore, the migration of epithelial cells is thought to be the key to wound healing.35 In our study, the five groups had the order D > C > B ≈ A > control (P<0.05) in terms of cell-migration rate at 24 hours after the scratch (Figure 12). This demonstrated that group D effectively promoted migration of epithelial cells and thus accelerated wound healing. The five groups could be arranged in descending order, D > C > B ≈ A > control (P<0.05) in terms of expression of migrated proteins, including F-actin and vinculin (Figure 13). It is generally accepted that the recombination dynamic structure of F-actin protein plays a vital role in cell migration.36 As a highly conservative intracellular protein, vinculin also plays a vital role in maintaining and regulating cell adherence and migration.35,36 Therefore, we believe that p-coumaric acid-modified lysozymes promoted cell migration by influencing the expression of migrated proteins, including F-actin and vinculin.
Conclusion
We successfully optimized the design of modified lysozymes and prepared a translucent balsa-modified lysozyme dressing characterized by strong antibacterial properties, with stable and persistent release, no cytotoxicity, and capacity to promote antibacterial ability, epithelial growth, and cell proliferation and migration.
Disclosure
The authors report no conflicts of interest in this work.
References
Vay O, de Borst K, Hansmann C, Teischinger A, Müller U. Thermal conductivity of wood at angles to the principal anatomical directions. Wood Sci Technol. 2015;49(3):577-589. | ||
Trey S, Jafarzadeh S, Johansson M. In situ polymerization of polyaniline in wood veneers. ACS Appl Mater Interfaces. 2012;4(3):1760-1769. | ||
Nejat P, Jomehzadeh F, Taheri MM, Gohari M, Majid MZ. A global review of energy consumption, CO2 emissions and policy in the residential sector (with an overview of the top ten CO2 emitting countries). Renew Sustain Energy Rev. 2015;43:843-862. | ||
Wand S, Lu A, Zhang L. Recent advances in regenerated cellulose materials. Prog Polym Sci. 2016;53:169–206. | ||
Li Y, Fu Q, Yu S, Yan M, Berglund L. Optically transparent wood from a nanoporous cellulosic template: combining functional and structural performance. Biomacromolecules. 2016;17(4):1358–1364. | ||
Jing J, Chang Z, Zeng GM, et al. The disinfection performance and mechanisms of Ag/lysozyme nanoparticles supported with montmorillonite clay. J Hazard Mater. 2016;317:416–429. | ||
Jing M, Song W, Liu R. Binding of copper to lysozyme: spectroscopic, isothermal titration calorimetry and molecular docking studies. Spectrochim Acta A Mol Biomol Spectrosc. 2016;164:103–109. | ||
Zheng L, Wan Y, Yu L, Zhang D. Lysozyme as a recognition element for monitoring of bacterial population. Talanta. 2016;146:299–302. | ||
Zhang T, Zhou P, Zhan Y, et al. Pectin/lysozyme bilayers layer-by-layer deposited cellulose nanofibrous mats for antibacterial application. Carbohydr Polym. 2015;117:687–693. | ||
Amara CB, Eghbal N, Degraeve P, Gharsallaoui A. Using complex coacervation for lysozyme encapsulation by spray-drying. J Food Eng. 2016;183:50–57. | ||
Dekina S, Romanovska I, Ovsepyan A, Tkach V, Muratov E. Gelatin/carboxymethyl cellulose mucoadhesive films with lysozyme: development and characterization. Carbohydrate Polymers. 2016;147:208–215. | ||
Liang H, Wang B, Wang H, et al. Inhibition of lysozyme fibrillation by gold nanorods and nanoparticles. J Nanosci Nanotechnol. 2018;18(5):3087–3094. | ||
Touch V, Hayakawa S, Saitoh K. Relationships between conformational changes and antimicrobial activity of lysozyme upon reduction of its disulfide bonds. Food Chem. 2004;84(3):421–428. | ||
Liu ST, Sugimoto T, Azakami H, Kato A. Lipophilization of lysozyme by short and middle chain fatty acids. J Agric Food Chem. 2000;48(2):265–269. | ||
Jing M, Song W, Liu R. Binding of copper to lysozyme: spectroscopic, isothermal titration calorimetry and molecular docking studies. Spectrochim Acta A Mol Biomol Spectrosc. 2016;164:103–109. | ||
Guo S, Dipietro LA. Critical review in oral biology and medicine: factors affecting wound healing. J Dent Res. 2010;89(3):219–229. | ||
Morsy R, Hosny M, Reicha F, Elnimr T. Development and characterization of multifunctional electrospun ferric oxide-gelatin-glycerol nanofibrous mat for wound dressing applications. Fiber Polym. 2016;17(12):2014–2019. | ||
Chan LW, Kim CH, Wang X, Pun SH, White NJ, Kim TH. PolySTAT-modified chitosan gauzes for improved hemostasis in external hemorrhage. Acta Biomater. 2015;31:178–185. | ||
Dreifke MB, Jayasuriya AA, Jayasuriya AC. Current wound healing procedures and potential care. Mater Sci Eng C Mater Biol Appl. 2015;48:651–662. | ||
Wang Y, Xu R, Luo G, et al. Biomimetic fibroblast-loaded artificial dermis with “sandwich” structure and designed gradient pore sizes promotes wound healing by favoring granulation tissue formation and wound re-epithelialization. Acta Biomater. 2016;30:246–257. | ||
Morshedi D, Ebrahim-Habibi A, Moosavi-Movahedi AA, Nemat-Gorgani M. Chemical modification of lysine residues in lysozyme may dramatically influence its amyloid fibrillation. Biochim Biophys Acta. 2010;1804(4):714–722. | ||
Evran S, Yasa I, Telefoncu A. Modification of lysozyme with oleoyl chloride for broadening the antimicrobial specificity. Prep Biochem Biotechnol. 2010;40(4):316–325. | ||
Yuan SJ, Yin J, Jiang W, Liang B, Pehkonen SO, Choong C. Enhancing antibacterial activity of surface-grafted chitosan with immobilized lysozyme on bioinspired stainless steel substrates. Colloids Surf B Biointerfaces. 2013;106(6):11–21. | ||
Tiwari BK, Valdramidis VP, O’Donnell CP, Muthukumarappan K, Bourke P, Cullen PJ. Application of natural antimicrobials for food preservation. J Agric Food Chem. 2009;57(14):5987–6000. | ||
Fournier A, Eggimann P, Pagani JL, et al. Impact of the introduction of real-time therapeutic drug monitoring on empirical doses of carbapenems in critically ill burn patients. Burns. 2015;41(5):956–968. | ||
Ito K, Saito A, Fujie T, et al. Sustainable antimicrobial effect of silver sulfadiazine-loaded nanosheets on infection in a mouse model of partial-thickness burn injury. Acta Biomater. 2015;24:87–95. | ||
Costley D, Nesbitt H, Ternan N, et al. Sonodynamic inactivation of Gram-positive and Gram-negative bacteria using a rose bengal-antimicrobial peptide conjugate. Int J Antimicrob Agents. 2016;49(1):31–36. | ||
Sarhan WA, Azzazy HM, el-Sherbiny IM. Honey/chitosan nanofiber wound dressing enriched with Allium sativum and Cleome droserifolia: enhanced antimicrobial and wound healing activity. ACS Appl Mater Interfaces. 2016;8(10):6379–6390. | ||
Chadha P, Katare OP, Chhibber S. In vivo efficacy of single phage versus phage cocktail in resolving burn wound infection in BALB/c mice. Microb Pathog. 2016;99:68–77. | ||
Jangpromma N, Preecharram S, Srilert T, et al. In vitro and in vivo wound healing properties of plasma and serum from Crocodylus siamensis blood. J Microbiol Biotechnol. 2016;26(6):1140–1147. | ||
Liang D, Lu Z, Yang H, Gao J, Chen R. Novel asymmetric wettable AgNPs/chitosan wound dressing: in vitro and in vivo evaluation. ACS Appl Mater Interfaces. 2016;8(6):3958–3968. | ||
Wang C, Zhu H, Sun Z, et al. Inhibition of Wnt/β-catenin signaling promotes epithelial differentiation of mesenchymal stem cells and repairs bleomycin-induced lung injury. Am J Physiol Cell Physiol. 2014;307(3):C234–C244. | ||
Huang Y, Zhao K, Hu Y, et al. Wogonoside inhibits angiogenesis in breast cancer via suppressing Wnt/β-catenin pathway. Mol Carcinog. 2016;55(11):1598–1612. | ||
Cai C, Zhu X. The Wnt/β-catenin pathway regulates self-renewal of cancer stem-like cells in human gastric cancer. Mol Med Rep. 2012;5(5):1191–1196. | ||
Le CC, Carlier MF. Regulation of actin assembly associated with protrusion and adhesion in cell migration. Physiol Rev. 2008;88(2):489–513. | ||
Kirby GT, Mills SJ, Cowin AJ, Smith LE. Stem cells for cutaneous wound healing. Biomed Res Int. 2015;2015:2858–2869. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.