")
Back to Archived Journals » Cell Health and Cytoskeleton » Volume 7
Structure of, and functional insight into the GLUT family of membrane transporters
Authors Long W, Cheeseman C
Received 1 May 2015
Accepted for publication 19 June 2015
Published 6 October 2015 Volume 2015:7 Pages 167—183
DOI https://doi.org/10.2147/CHC.S60484
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Denis Wirtz
Wentong Long, Chris I Cheeseman
Department of Physiology, Faculty of Medicine and Dentistry, University of Alberta, Edmonton, AB, Canada
Abstract: This review examines the development of structure and function of the human GLUT proteins, gene family hSLC2A. These proteins are essential for moving the key metabolites, glucose, galactose, and fructose in and out of cells, as well as a number of other important substrates. Despite over five decades of research, it is still not fully understood how they work at the molecular level, although the recent publication of a crystal structure of GLUT1 suggests this may be resolved soon. The GLUT family is divided into three classes based on their sequence homology. The physiological roles of Class I GLUTs, ie, 1, 2, 3 (14), and 4 have been extensively studied for their contributions to metabolism and development. However, the other two classes have received far less attention. Genetic diseases associated with GLUTs are very rare, emphasizing their critical roles, but it is likely that as our understanding of these transporters increases, there may be more clinical conditions found to be associated with subtle changes in their activity. Another promising area of investigation is the changes in expression levels of GLUTs associated with, and likely in consequence of, disease processes. It has long been known that GLUT1 expression increases significantly in cancer cells, but it is now becoming appreciated that other GLUTs may also be involved. Determination of alterations in expression patterns may prove to be a useful diagnostic tool, and in some cases these are already being taken advantage of for the imaging of cancers. Finally, as we better understand how GLUTs bind and transport their substrates, it may be possible to design drugs that can be delivered into target cells for the treatment of a number of diseases.
Keywords: GLUT proteins, Facilitated hexose transporters, protein trafficking, cancer imaging, genetic diseases
Introduction
Hexoses are an essential carbon and energy source for the cells of the human body, and their entry and exit across the plasma membrane is critical for life.1 There are now known to be at least two distinct transporter gene families responsible for the handling of hexoses in mammalian cells. The first is the sodium/glucose co-transporters (SGLTs), which are secondary active transporters that use the sodium gradient across the cell membrane to drive glucose and galactose uptake.2,3 In contrast, GLUTs are passive transporters that use either chemical or electrochemical gradients to transport hexoses or other substrates.4,5
There are 14 mammalian facilitative glucose transporters (Table 1), and they belong to the sugar porter family in the major facilitator superfamily (MFS).4,5 They are encoded by the solute linked carrier family 2, subfamily A gene family, SLC2A, and the original name glucose transporter is somewhat misleading as many have substrates other than hexoses.6–8 They are subdivided into three classes based on their protein sequence and structural similarity. Class I, the “glucose transporters”, includes GLUT1, GLUT2, GLUT3, GLUT4, and GLUT14. Class II glucose transporters include GLUT5, GLUT7, GLUT9, and GLUT11, and are known to transport fructose as well. Class III is made up of GLUT6, GLUT8, GLUT10, GLUT12, and GLUT13 (HMIT1); this class is believed to be the most ancient group, with largely as yet unknown substrates.
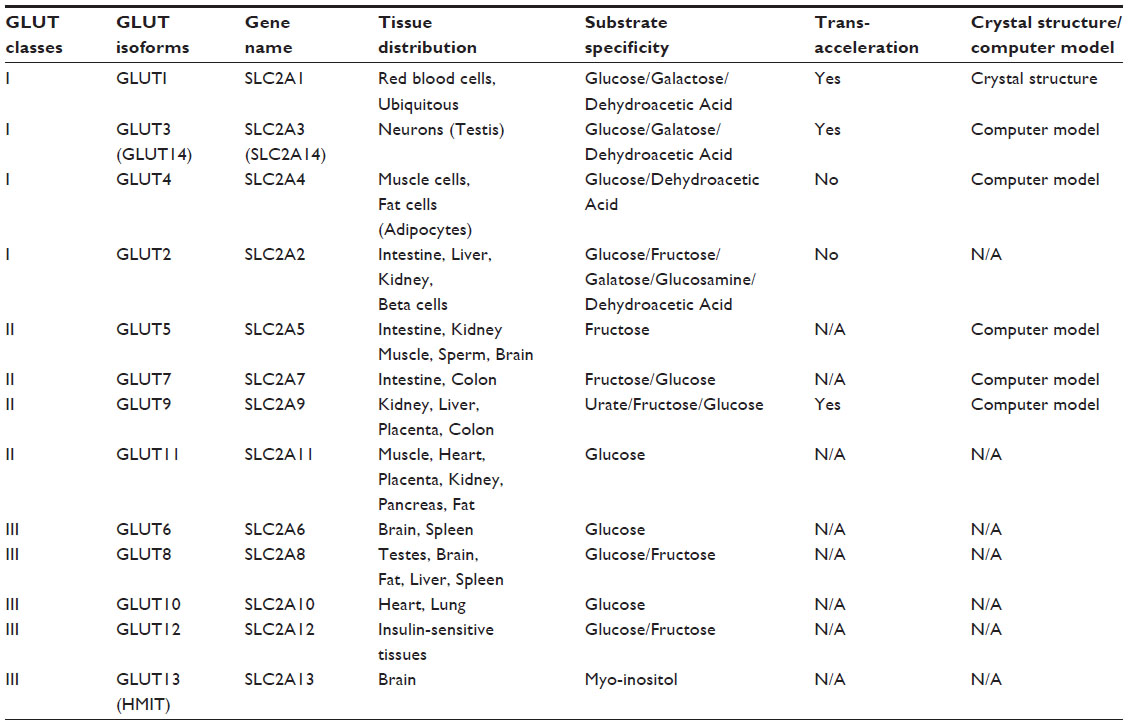 | Table 1 Summary of the GLUT family proteins and their characteristics |
Molecular characterization of Class I, II, and III GLUTs
Class I GLUTs were all cloned in the 1980s. Human GLUT1 protein was purified by Kasahara and Hinkle in 1977 from human erythrocytes, in which GLUT1 has very high expression levels. The molecular weight of the transporter protein was identified as a 55,000 Dalton band on sodium dodecyl sulfate-polyacrylamide gel, sometimes referred to as band 3.9,10 After reconstitution into liposomes, this protein was able to show the transport activities for d-glucose but not l-glucose. Furthermore, 90% of the d-glucose transport activity was inhibited by low concentrations of cytochalasin B, a cell-permeable mycotoxin. In 1985, Mueckler et al obtained the complete gene and protein sequence of GLUT1 from the human HepG2 hepatoma cell line.11 Northern blot analysis demonstrated that GLUT1 is expressed in most tissues and cell types in mammals.6,11 Consisting of 492 amino acids, it was predicted to have 12 putative membrane spanning domains with both the C-terminus and N-terminus in the cytoplasm, and a putative glycosylation site Asn45 on the first extracellular loop.11
Human GLUT2 was cloned from rat and human liver cDNA libraries in 198812,13 having 524 amino acids and an 80% homology and 55% identity with GLUT1. It is found mainly in the intestine, liver, kidney, and islet cells, and has been shown to transport both glucose and fructose with relatively low affinities, an important feature of liver hexose transport.14–16 Kayano et al identified human GLUT3 in 1988.15 GLUT3 is a 496 amino acid protein and was cloned from a human fetal skeletal muscle cDNA library.15 It has 64.4% and 51.6% identity with GLUT1 and GLUT2, respectively, and is expressed mostly in the brain, in particular in the frontal lobe of the cerebrum.15,17–19 Its primary substrate is also glucose.15 GLUT14 was identified by Wu and Freeze as a duplicon of GLUT3 with 94.5% identity.20 It appears to be exclusively expressed in the human testis. Human GLUT4 was cloned from the human small intestine and skeletal muscle by Fukumoto et al in 1989.13 It is a 509 amino acid protein that has 65.3%, 54.3%, and 57.5% identities with GLUT1, GLUT2, and GLUT3, respectively. The majority of human GLUT4 is expressed in cardiac and skeletal muscle and in adipose tissue, and glucose transport activity is highly insulin-sensitive.21
Class II GLUTs comprise GLUT5, GLUT7, GLUT9, and GLUT11, and they all transport fructose and to some degree glucose. Human GLUT5 was cloned from small intestinal cDNA, and was proposed as a second hexose transport system in the brush border (apical) membrane of intestinal epithelial cells along with SGLT1.22 GLUT5 is a 501 amino acid protein expressed mostly in the human small intestine and at low levels in kidney, skeletal muscle, and adipose tissues. GLUT5 has 41.7%, 40.0%, 38.7%, and 41.6% identities with GLUT1, GLUT2, GLUT3, and GLUT4, respectively. Use of 2-deoxy-d-glucose as a substrate indicated that GLUT5 was also a very low affinity glucose transport system in the brush border membrane. However, its primary substrate is fructose and it is responsible for much of the fructose uptake at the apical surface of the intestine.23
Initially, Waddell described GLUT7 in 1992 as an hepatic microsomal transporter protein that had 68% identity with GLUT2.24 Subsequently, it was reported to be a cloning artifact, and its mRNA could not be found in either the human or rat liver.25 In 2004, human GLUT7 was the last member of the GLUTs to be cloned using an intestinal cDNA and a polymerase chain reaction-based strategy.26 Having 528 amino acids, GLUT7 shares 68% similarity and 53% identity with GLUT5 and is predominantly expressed at the apical membrane of the enterocytes in the small intestine. This GLUT can transport both glucose and fructose when expressed in Xenopus oocytes with high affinity, but with very low transport capacity, and it is not sensitive to cytochalasin B.26,28 It is likely that its correct physiological substrate has yet to be identified (see GLUT9 below).
In 2000, Phay et al cloned human GLUT9 from cancer tissues using polymerase chain reaction-based methods.27 GLUT9 has two splice variants, a 540 amino acid length transporter (hSLC2A9a) targeted to basolateral membranes in the liver and kidney and an N-terminal truncated form (hSLC2A9b) found in the renal apical membrane, apparently in the distal convoluted tubule.28 It shares 38% and 44% identity with GLUT1 and GLUT5, respectively. Human GLUT9 was initially shown to be a high affinity/low capacity glucose and fructose transporter, but more recently as a consequence of a series of genome-wide association studies (GWAS) was identified as a high capacity urate transporter.29–32
Human GLUT11 was cloned from the human heart in 2001 and shares 35% and 41.9% identity, respectively, with GLUT1 and GLUT5.33 To date, it has only been detected in heart and skeletal muscle and has three splice variants of unknown function. GLUT11 transports glucose and fructose when expressed in Cos-7 cells.
Class III GLUTs include GLUT6, GLUT8, GLUT10, GLUT12, and GLUT13 (HMIT). A non-functional pseudo gene called GLUT6 with high sequence identity to GLUT3 was initially identified in 1990.22 Subsequently, the functional human GLUT6 was cloned in 2000 from leucocytes but was initially named GLUT9.30 Later, the gene was designated as SLC2A6 (GLUT6) by the HUGO Gene Nomenclature committee.31 Human GLUT6 is a 507 amino acid protein, and has high sequence identity with GLUT8 (44.8%) and has 28.5% identity with GLUT1. It appears to be exclusively expressed in the brain and lymphoid tissues and transports glucose when reconstituted into Cos-7 membrane vesicles. GLUT8, another glucose transporter, was cloned from the human testis, its primary expression site, and has 477 amino acids with 29.4% sequence identity to GLUT1.30,34,35
GLUT10 was cloned from human liver cDNA as a 541 amino acid protein and has 29.7% and 33.6% sequence identity with GLUT3 and GLUT8, respectively.36 GLUT10 is expressed in most human tissues, with the highest level found in the liver and pancreas. GLUT10 displays very low transporter activity and shows high affinity for 2-deoxyglucose when expressed in Xenopus oocytes.37 Human GLUT12 was identified using a malignant breast cancer cell line (MCF-7) as a 617 amino acid protein.38 GLUT12 shares 40% and 29% sequence identity with GLUT10 and GLUT4, respectively. Immunocytochemistry assays indicated that GLUT12 is expressed in skeletal muscle, the small intestine, and adipose tissue. Other studies indicated that GLUT12 is also present in the heart, prostate, brain, placenta, and kidney tissues.39–42 GLUT12 shows glucose transporter activity when expressed in Xenopus oocytes and this activity is inhibited by other hexoses, including fructose, galactose and 3-O-methyl-D-glucose (3-OMG), suggesting that they may also be substrates.43
Human GLUT13, is also known as the H+-myo-inositol transporter, HMIT, for its proton-coupled activity. Both human and rat GLUT13 were cloned at the same time.44 The rat GLUT13 cDNA encoding a 618 amino acid protein, and the human GLUT13 has 629 amino acids. GLUT13 is expressed mainly in the brain, in particular in ganglion cells and some neurons.44 GLUT13 is the only GLUT that has specific electrogenic transport activity for myo-inositol. To date, no glucose transport activity has been reported for GLUT13, although it does appear to mediate inositol-3-phosphate (IP3) movement.
Structural comparisons among Class I, II, and III GLUT transporters
Hydropathy analysis of the GLUTs reveals that they all have 12 predicted transmembrane domains (TMs) with both the C-terminus and N-terminus and a long loop connecting the TM6 and TM7 present on the cytosolic side of the membrane.45–47 Protein sequence alignments of all GLUTs reveal that many amino acids are conserved among these proteins. Good examples are PMY in TM4, PESPRY/FLL in TM7, GRR in Loop8, GPGPIP/TW in TM10, and VPETKG in the C-terminus (Figure 1).48 These motifs are considered to be the signature characteristics of glucose transporters. Other residues, like R92 in Loop2, E146 and R153 in Loop4, Y293 in Loop7, E329, R333, and R334 in Loop8, E393 and R400 in Loop10, and P385 in TM10, are also highly conserved in all the GLUT members. (Note that all amino acid residue numbers are assigned according to the corresponding residues in GLUT1). Mutations of these residues in GLUT1 reduced or abolished glucose transport activity with or without affecting cytochalasin B binding.48–51
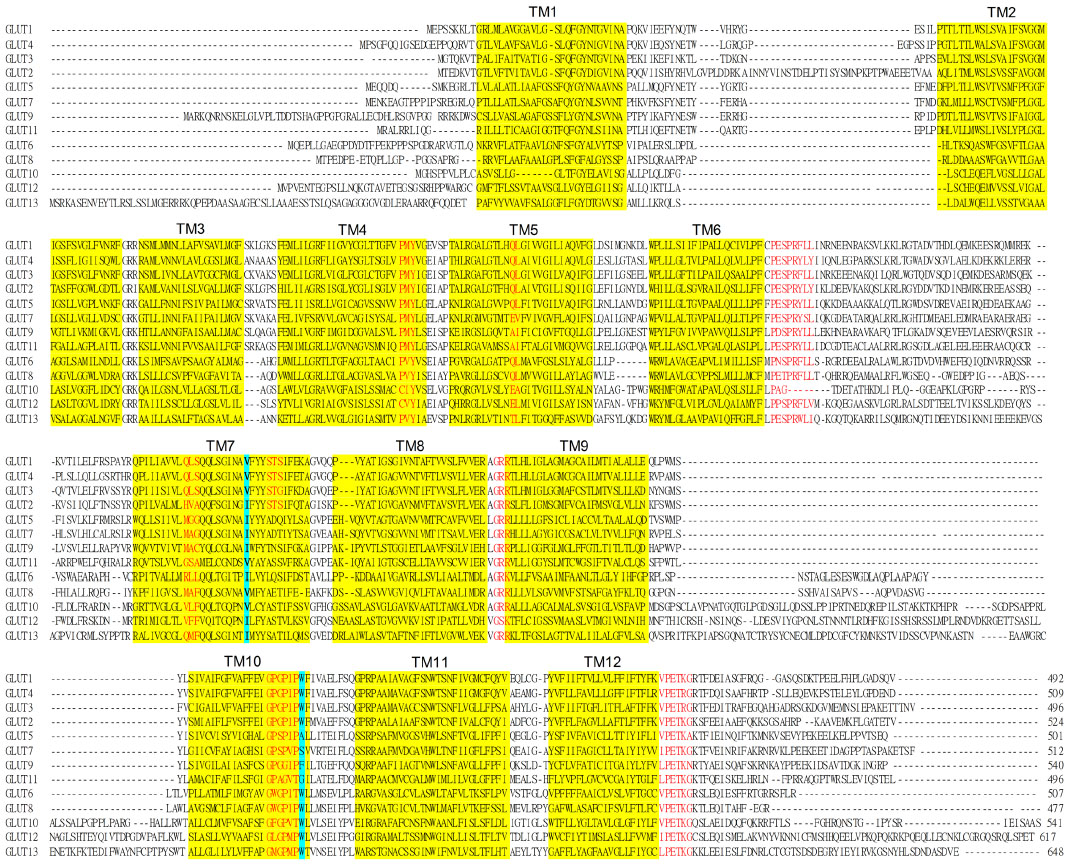 | Figure 1 Amino acid sequences alignments of the GLUT family of proteins. |
Class I GLUTs
Although the Class I GLUTs have a similar putative topology, only 38% of all the amino acids are conserved in GLUT1–4.52 They all appear to have a single N-linked glycosylation site on Loop1, which connects TM1 and TM2.11,13–15 A glutamine residue (part of the QL motif) in TM5 is shared among the Class I GLUTs, which suggests this motif is important for glucose recognition.53 The serine-threonine-serine motif in extracellular Loop7 between TM7 and TM8 is also conserved in Class I GLUTs.54 Mutation of these serine or threonine residues locks the protein conformation, suggesting this is a critical site for conformation change. Another highly conserved motif is GPXXXP in TM10, where a tryptophan also appears immediately after this sequence.55,56 It has been proposed that this tryptophan is a crucial residue for binding the two inhibitors, cytochalasin B and forskolin, without directly affecting glucose transport. In addition, a QLS motif in TM7 is only present in transporters like GLUT1, GLUT3, and GLUT4 which carry glucose, but not in those that transport fructose, including GLUT2, suggesting that this motif is involved in glucose and fructose selectivity.57
Class II GLUTs
Class II GLUTs also have 12 TMs and a single N-linked glycosylation site on exofacial Loop1 between TM1 and TM2.22,26,27,30 A major difference between Class I and II is that Class II lacks the tryptophan residue after the GPXXXP motif.58 Another striking difference is that all Class II GLUTs have a single hydrophobic residue, isoleucine, in TM7 associated with fructose/glucose selectivity.59 It was speculated that this residue was facing the aqueous pore at the exofacial vestibule of GLUT7 using a computer model of the GLUT7 protein based on the GlpT crystal structure.60 However, analysis using a GLUT9 computer model based on the recent GLUT1 crystal structure indicates that this isoleucine is more likely to be facing away from the aqueous pore and to be acting as a structural regulator through hydrophobic interactions with adjacent TMs.61 At the equivalent position in Class I GLUTs (except for GLUT2 which can also transport fructose) there is a valine residue.7 This suggests that subtle interactions between TMs mediated by hydrophobic residues can affect substrate specificity within this family of transporters. Another important structural variation between the Class I and II GLUTs is that Class II do not contain the QLS motif.57
Class III GLUTs
Class III GLUTs have similar topology to classes I and II and many of the motifs found in the other classes are conserved, such as PESPR in TM6 (GLUT6 and GLUT8), GRR in Loop2 and Loop8, PETKGR in TM12, and arginine and glutamate residues in Loop4 and Loop10.30,34,36,38,41 Class III GLUTs also have a tryptophan residue after the GPXXXP motif in TM10. However, one significant difference is that Class III GLUTs do not have an N-linked glycosylation site in Loop1 between TM1 and TM2. Instead, Loop1 is shorter, and the predicted glycosylation sites are in the long Loop9 between TM9 and TM10.30,34,36,38 GLUT13 (HMIT) is predicted to have more than one N-linked glycosylation site within this loop.44 In addition, GLUT6 has two arginine residues present in TM7 and TM8, which are absent in other GLUTs.31 Again, Class III GLUTs do not contain the QLS motif, which is unique to the Class I proteins.8
Structure of human GLUT1
Despite the amino acid sequence differences among all the GLUTs, the overall structural protein arrangements are predicted to be very similar. Extensive studies have been carried out to elucidate the structure–function relationship, primarily using GLUT1.
The secondary structure of GLUT1 was first proposed by Mueckler et al11 to have 12 transmembrane α-helical domains. They also noted that over half of the residues are hydrophobic and proposed that amphipathic helices TMs 3, 5, 7, 8, and 11 might form a central aqueous channel allowing glucose to be translocated through the membrane.11 Later, this structure was confirmed by Fourier transform infrared spectroscopy, circular dichroism spectroscopy analysis, scanning glycosylation mutagenesis, and mass spectrometric analysis.45,62,63 Cysteine scanning mutagenesis was subsequently employed to probe which residues lined this proposed central pore.64–74 The resulting model indicated that TMs 2, 4, 5, 7, 8, 11, and possibly 1 and 10 formed a central aqueous transport channel for glucose, whereas TMs 3, 6, 9, and 12 formed a structural scaffold on the outside of the protein.70 A possible substrate binding site was also proposed, involving Q161, Q282, and W412.70
Subsequently, a three-dimensional inwar facing computer model of GLUT1 was created based on the newly generated GlpT crystal structure.75 Remarkably, this model agreed with most of the previous data for GLUT1, in which the 12 transmembrane helices an be viewed as two symmetrical six α-helical bundles connected by a long intracellular loop between TM6 and TM7.70 This also supports the hypothesis that the 12-TM GLUT proteins arose from a gene duplication event from a protein with six TMs.70,76,77 The new model also indicated that the central transport cavity was formed by TMs 2, 4, 5, 7, 8, and 10, and that most of the important residues previously described as crucial for substrate/inhibitor transport/binding functions faced the pore. However, docking studies for the binding site of the GLUT inhibitor, cytochalasin B, predicted that it was on the intracellular side, a positioning that contradicts the experimental data.75,78
Other models for GLUT3, GLUT4, GLUT5, and GLUT9 were also constructed based on computer models of the mechanosensitive ion channels of the Mscl homolog from Mycobacterium tuberculosis, GlpT, and an Escherichia coli homolog of GLUTs 1–4 (XylE).79–85 The GLUT4 model was very similar to that for GLUT1 (also based on the GlpT) structure with the same TM arrangement with conserved residues lining the transport cavity, and both ATP-binding and cytochalasin B-binding sites in the cytosolic side. The GLUT3 computer was based on a Mscl crystal structure. However, the authors need to manipulate the gaps of GLUT3 model due to the TMs differences between the two proteins.79,83 The authors also suggested that glucose is transported by the key flexible TM segments and a network of polar and aromatic residues with two different glucose binding sites on each side of the transport pathway, rather than by an alternating access mechanism with one stable binding site, as proposed based on the GlypT crystal structure.79
In 2014, new GLUT1 and GLUT5 computer models were developed based on the XylE crystal structure, which is a bacterial d-xylose transporting homolog sharing a high degree of similarity and identity with GLUT1–4.81 In parallel with other MFS protein transporters, the topology of these models fitted into the common structural fold with 12 TMs. More important, this study proposed that the 12 TM topology of MFS may have arisen by two gene duplication events through an initial triple-helix bundle into the six-helix bundle, then into two linked pseudo symmetrical six-helix bundles.4 Thus, the alignment of GLUT1 and GLUT5 sequences to XylE is in the triple-helix array. This modeling also predicted that Asn411 in GLUT1 and His419 in GLUT5 are the corresponding substrate binding sites for glucose and fructose, respectively.
More recently, a GLUT9 model was also constructed based on XylE.82 The resulting GLUT9 model topology is similar to the XylE structure, with 12 TMs arranged in a double six-helical bundle arrangement. In the same study, single monomer GLUT9 protein was purified from the GLUT9 overexpressing oocyte. Consequently, it allowed the construction of a low resolution crystal structure of GLUT9. The structure revealed a transport cavity, which contains the plausible urate binding sites. These included amino acids: H23, R31, L182, Q203, A206, Q328, L332, N333, F426, W459, and N462.
Finally, almost four decades after the first purified GLUT, a high resolution (3.2 å) crystal structure was reported in 2014. The purified protein was generated using a baculovirus transfection system with High Five insect cells.84 This GLUT1 model reveals a partially open inward facing conformation that is locked due to a single missense mutation, E329Q, and confirms a 12-TM transporter protein formed of two six-helical domains, with cytoplasmic N-domains and C-domains. One additional feature was the presence of a helical bundle, an intracellualr coiled heical, ICH, domain, within the long intracellular loop. This ICH domain has also been observed in other sugar transporter crystals, such as XylP and GlcP,85,86 and is believed to be unique to the sugar porter family and not in other MFS. This ICH domain may function as a latch to ensure the closing of an intracellular gate in the inward facing conformation of GLUT1. In addition, only a single sugar binding site was identified, located toward the C-domain of the transporter, which substrate could access from either side of the protein. The residues forming this proposed binding site are Q282/Q283/N288 from TM7, N317 from TM8, and N415 from TM11. The N-domain of the structure is believed to play a role in regulation of the conformational change during transport. Another notable feature of this GLUT1 crystal is the extracellular gate formed by TM1 and TM7. N34 is an essential residue in TM1 that serves as the central coordinator for hydrogen bonding between S294, T295 in TM7, and T310 in TM8. Note added in proof: a new GLUT3 crystal structure was published with a D-glucose bound to the outward-occluded conformation and with maltose bound to outward-open and outward-occluded conformations.87
Importance of transmembrane domain 7 in GLUTs
The new GLUT1 crystal structure indicated that TM7 is critical for both structural regulation and substrate binding. TM7 forms an extracellular gate in the inward facing conformation of GLUT1,84 and three of the potential binding residues were observed within the substrate-bound crystal structure.84 Interestingly, Q282, Q283, and N288 match the molecular data observed in Mueckler and Makepeace’s original cysteine scanning mutagenesis studies.70 Substitution of these residues into cysteines reduced the glucose transport substantially. However, only Q282 and Q283 were shown to be facing the aqueous environment, but not N288.
Based on the GLUT1 crystal structure, a revised model of GLUT9 and a new GLUT5 model have also been generated.61 These models indicate that this hydrophobic residue (I335 in GLUT9 and I296 in GLUT5) is not projecting toward the aqueous transport pathway as previously proposed; instead, it appears to interact with residues on the adjacent TMs via an complex hydrophobic network (Figure 2). Additional functional studies confirmed that replacement of I335 in GLUT9 with valine resulted in a loss of trans-stimulation between fructose and urate. It has been hypothesized that I335 in GLUT9 affects the rigid body movement of one of the two six-helix bundles, and subsequently the orientation of helix 7 (TM7) in the translocation pore of the transporter.
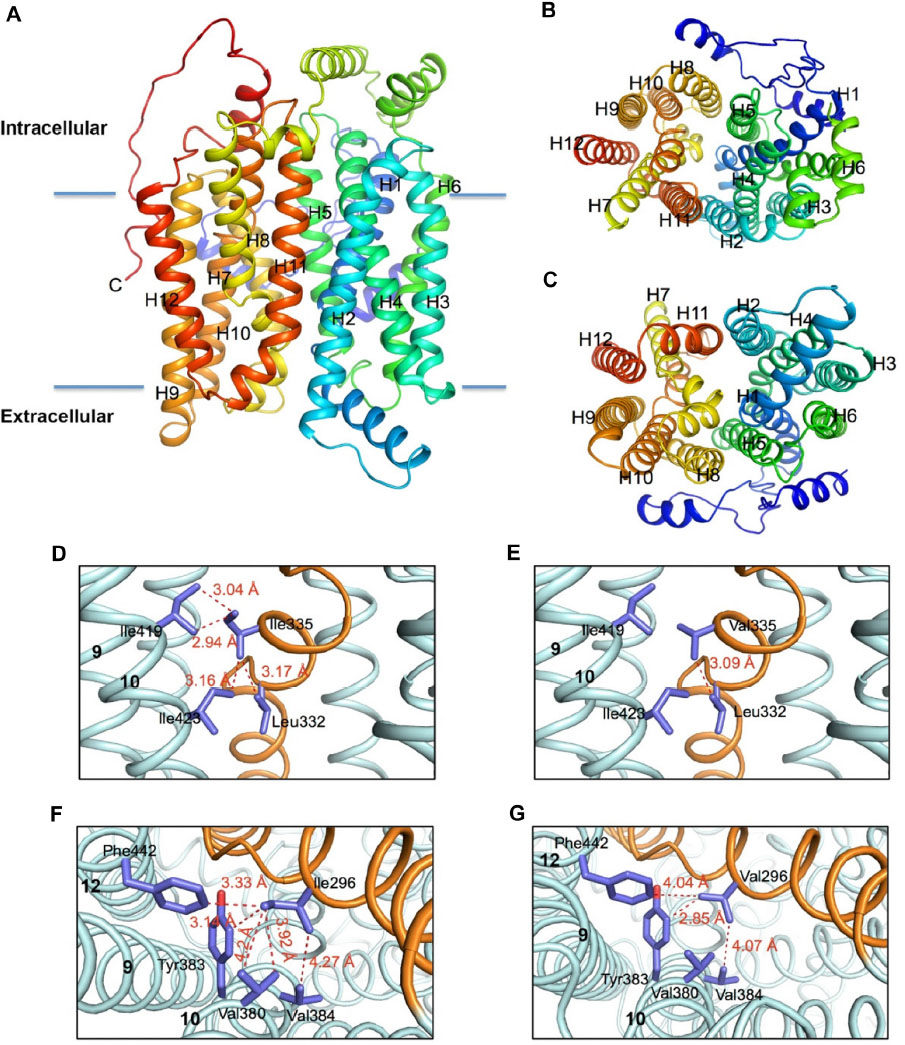 | Figure 2 Molecular models of the human hGLUT9 and hGLUT5 transporters comparing possible hydrophobic interactions. |
Mechanisms of substrate transport by GLUTs
The mechanism of how glucose is transported has mainly been investigated using GLUT1. Initial explanations of how glucose is transported across the cell membrane were based on mathematical analysis of the transport activities of hexose utilizing erythrocyte preparations. The fluxes were best fit with the Michaelis–Menten enzyme kinetics model89–90 which predicts that the rate of hexose absorption depends on both the initial hexose concentrations and the binding affinity of the protein for the substrate and that there is a maximum rate of transfer (rate of transfer is saturable).91 The earliest mechanistic theory, ie, the “simple carrier model”, was proposed by Widdas and assumes four transport stages: an empty carrier that opens to one side of the membrane (cis side), to which glucose can bind; the substrate binding carrier has to translocate to the other side of the membrane (trans side); the carrier releases the glucose on the trans side; and the empty carrier switches back to the cis side. Moreover, Widdas postulated that both phosphorylation and metabolism of glucose were the two possible mechanisms providing the necessary energy for glucose transfer. However, due to the complexities of the erythrocyte system, differences between experimental methods and variations in kinetic data, many other models have been proposed over the same time period.92 These models include the following: the Regen–Tarpley asymmetrical carrier model,93 the Eilam model,94 the lattice-pore model,95 the tetramer model,96 and the introversion model.97 Despite the differences between these models, they all tried to explain two principal scenarios that occur during glucose transfer: the asymmetry of the transport affinities (KM, substrate concentration at half the maximal transport rate) of zero trans between hexose influx and efflux; and the trans-acceleration that occurs when hexose is present on the trans side.92,98
Purification and cloning of GLUT1 subsequently permitted a better picture of how glucose is transported to be proposed. Today there are two popular models still under consideration. One is the two-site/fixed sites transporter, in which both substrate binding sites are simultaneously available from either side of the membrane.99–101 After binding, hexoses can then exchange between these sites and accelerate the binding process. In another words, this model suggests that GLUT1 can work as an antiporter. However, human GLUTs (all except GLUT13) were thought to be uniporters,100 in which they transport one hexose molecule at a time in a unidirectional approach, whereas an antiporter (or exchanger) will simultaneously transport two molecules in opposite directions. It is argued that this two-site model can explain the complex asymmetry and multiphasic transport kinetics, while the simple carrier model cannot sufficiently account for these incidences without violating the energy conservation law.98,99 However, asymmetric glucose transport activities are not seen in hepatocytes or adipocytes, which are mediated by GLUT2 and GLUT4, respectively.16,102,103 Several recent investigations also indicate that GLUT1 can form oligomers, such as dimers or tetramers, which could account for the three phases and asymmetric kinetics.104–107 Cloherty et al106 and De Zutter et al107 proposed that the glucose transporters in mammalian cells present as cooperative dimers or tetramers of GLUT1, and they hypothesized that one (or two in a tetramer) exofacial and one (or two in a tetramer) endofacial hexose binding site(s) present simultaneously. In both dimers and tetramers of GLUT1, cis-allosteric hexose transport was observed, ie, hexose binding to one oligomer subunit induces the transport by other subunit(s). Therefore, the GLUT1 dimers are able to transport cis- hexose in exchange with the trans side substrate, which is similar to the two-state model assumption; the tetramer GLUT1 cooperative transporter could also further explain the observed multiphasic transport.
Another proposed mechanism is the alternating access model, and this appears to be supported by the available MFS crystal structures.77,84–86,108,109 Jardetzky explained the assumptions of the mechanism as early as 1966.110 They are the following three premises: the transporter has a cavity to admit a small substrate; the transporter contains a substrate binding site; and the transporter has two different configuration openings to one side of the cell membrane or the other.110 This alternating access mechanism supports the presumption that GLUT1 is an uniporter, and has only one binding site that substrate can alternately access from either side of the membrane in the course of conformational changes. The newly crystallized GLUT1 also fits with the alternating access mechanism,84 which predicts four conformational states during a complete hexose transport cycle: an empty outward-open transporter state, a ligand-bound and occluded transporter state, an inward-open state, and a ligand-free and occluded state. These four states appear as a slightly more detailed version of the original simple carrier theory that Widdas proposed in 1952.88 Additionally, the crystal structure of GLUT1 suggests that the transporter has a favorable conformation when it is substrate-free. Note added in proof: The recently reported GLUT3 crystal structure shows strong evidence to support the alternating access mechanism with a bound D-glucose in an outward-occluded conformation.87
Mechanism of trans-acceleration
Another often observed property of GLUT-dependent glucose transport is that of trans-acceleration. This was first observed in erythrocytes as uphill hexose counterflow (other terms include countertransport acceleration and trans-stimulation transport).111,112 In these experiments, hexose was detected flowing apparently against its concentration gradient from the cis side into the trans side of the membrane where hexose is also present, and vice versa. Rosenberg and Wilbrandt argued that the phenomenon was due to two different transport systems present in the red cell, which Naftalin and Holman referred to as a two binding site carrier.92,112 Furthermore, it was believed that hexoses could exchange freely between these two sites within the carrier cavity, a process that Naftalin termed a geminate exchange.92,99,100 Alternatively, others argued that the carrier contains only one binding site, and the carrier can move from outward facing to inward facing with and without substrate binding. It is the empty carrier return from the trans side to the cis side of the membrane that limits the rate of transport. Therefore, trans-acceleration occurs when hexose is presented on the trans side, which allows the carrier to return faster to the cis side with a bound substrate.84,100
Currently, hexose/hexose trans-acceleration has only been shown in GLUT1, GLUT3, and GLUT9,29,100,113 and few studies have probed the mechanism at the molecular level. One study indicated that the ATP-binding site of GLUT1 contains residues that are necessary for glucose trans-acceleration to occur114 and proposed that mutations in the ATP-binding sites would alter the tertiary structure of GLUT1, and thus restrict the flexibility of the transporter for sensing substrates on both sides of the membrane. A second study employed chimeras constructed from GLUT1 and GLUT4 expressed in human embryo kidney (HEK-293) cells.100 It found that the TM6 of GLUT1 contains residues necessary for trans-acceleration with glucose-glucose exchange and is responsible for constraining the relaxation of GLUT1, but not direct binding or translocation of substrate during transport. Hence, this study was not able to distinguish between the simple carrier model or the two-site model to explain trans-acceleration. GLUT2 and GLUT4 do not show trans-acceleration of hexose transport; instead, they show symmetrical transport in both oocyte and mammalian cell systems.103,115
Human GLUT9 is the only Class II GLUT reported to show not only hexose/hexose trans-acceleration activity but also hexose/urate trans-acceleration when using the Xenopus oocyte expression system.29 GLUT5, GLUT7, GLUT11, and Class III GLUTs have not been subjected to detailed molecular examination of their transport mechanisms or substrate-binding pockets.
Subcellular trafficking of GLUT transporters
In the 1990s it was discovered that while GLUT1 is primarily present in the plasma membrane, GLUT4 is mostly localized to intracellular compartments.116–121 The C-terminus is primarily responsible for the differential targeting of GLUTs122–126 to the plasma membrane or intracellular vesicles that can translocate these proteins to other sites.127–131 GLUT1 and GLUT4 are believed to be recycled by exocytosis and endocytosis between the plasma membrane and cytosol, but to varying degrees. Insulin promotes their surface expression, but the regulatory mechanisms are thought to be different for the two proteins.123,127,128,130
GLUT1
The cytoskeleton and microtubules within cells are important in regulating the trafficking of GLUT1. For example, GLUT1 carrying intracellular vesicles are upregulated by protein kinase C and casein kinase substrate 3 (PACSIN 3) in adipocytes and downregulated by Syntaxin 1C (STX1C) in a lung epithelial cell line.132,133 STX1C is a soluble syntaxin suppressing the stability of microtubules and vesicle transport mobility, whereas PACSIN 3 is an adaptor protein involved in regulation of cellular cytoskeletal elements and the clathrin-coated pit pathway. Trafficking of GLUT1 from an intracellular pool to the plasma membrane is also increased by AMP kinase in murine brain microvasculature endothelium bEnd.3 cells.134 In the human megakaryocytic leukemia M07e cell line, GLUT1 trafficking is also influenced by cytokine stem cell factor and cholesterol depletion induced by methyl-b-cyclodextrin.135
The most recent studies of subcellular trafficking of GLUT1 have focused on the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt) pathway.129,136 Increased PI3K/Akt activity regulates GLUT1 trafficking and glucose uptake in T-cells/B-cells.137 Factors that activate the Akt pathway include the growth factors interleukin-7 and interleukin-3, granulocyte/macrophage colony-stimulating factor, the inhibitor of NF-kB-kinase b, CD28, and Kaposi sarcoma-associated herpes virus infection.137–139
GLUT4
The targeting, trafficking, and recycling of GLUT4 between the intracellular compartments and the plasma membrane has been reviewed extensively elsewhere.6,98,131,140–142 Consequently, we will simply outline some of the key features of the GLUT4 trafficking process. Cushman and Wardzala143 proposed the first schematic mechanism of how insulin stimulates GLUT translocation to and from the membrane in the rat adipose cell in 1980. This study hypothesized that vesicles containing glucose transporters were stimulated to bind and fuse with the plasma membrane by undefined “secondary messengers” upon association of insulin with its receptor, and that when insulin disassociates from the receptor, the process reversed. Subsequently, numerous potential pathways involved in the insulin-dependent pathway have been identified.140,142 They include insulin receptor substrate, PI3K and its regulatory subunit, P85 and catalytic P110, protein kinase C, ADP-ribosylation factors, phosphoinositide-dependent protein kinase 1, Golgi-localized, g-ear-containing ADP-ribosylation factor-binding proteins and protein kinase B (or Akt) and its substrate protein of 160 kDa (AS160).144–154
GLUT4 trafficking and recycling are also regulated independently from the insulin signal, which is referred to as a general (insulin-independent) pathway and that the movement of GLUT4 is also controlled by muscle contraction. Muscle contraction activates several signaling messengers, including calcium, nitric oxide, and reactive oxygen species within the muscle cells themselves, that regulate glucose homeostasis endogenously.142,155–157
Subcellular trafficking of other GLUTs
GLUT2 and GLUT3
There is good evidence that GLUT2 can be trafficked to the small intestinal enterocyte apical membrane under the influence of protein kinase C bII, which is activated by phorbol 12-myristate 13-acetate PMA, extracellular signal-regulated kinase p38, and glucagon-like-peptide 2 in the rat intestine.158–163 In contrast, stress, a high-fat diet, and glucocorticoids inhibit GLUT2 trafficking to the brush border membrane.164–166 Most recently, subcellular localization studies using live cell imaging demonstrated that GLUT2 is endocytosed through a caveolae-dependent mechanism, which is partially recovered in Rab11A-positive recycling endosomes.99 However, what is still not entirely clear is the role that this transporter plays while it is transiently expressed at the cell surface. It has been proposed that this transporter provides a high capacity entry pathway for glucose and fructose at the start of a meal when the luminal concentrations are high, and is then withdrawn as the meal progresses, leaving SGLT1 to bring in the remaining hexose upward into the cells. Another theory is that apical GLUT2 provides a shunt, moving glucose back out into the lumen and providing an osmotic control during the absorptive process.167
Trafficking of GLUT3 is mediated by intracellular vesicles with a SNARE complex in the neuronal PC12 cell line, and is possibly regulated by Rab11 in neurons from mice with Huntington’s disease.168,169 However, only a few studies have been conducted on the actual processes for these trafficking pathways.
GLUT8
Trafficking of GLUT8 is different from that of the other GLUTs. GLUT8 expression levels at the plasma membrane are controlled by glucose and insulin, and a t-SNARE protein, syntaxin 4, which has also been found in blastocyts to be necessary for the fusion of GLUT8 carrying vesicles with the plasma membrane.170–172 Other targeting/trafficking studies showed that GLUT8 distributes more to the intracellular compartment in neurons and blastocysts than at the cell surface.172–174 Piroli et al pointed out that, in rat hippocampal neurons, GLUT8 rapidly translocated to the rough endoplasmic reticulum following peripheral glucose administration.175 Therefore, they hypothesized that GLUT8 transports glucose out of the rough endoplasmic reticulum into the cytosol to maintain cellular glucose homeostasis in neurons.172,173 On the other hand, it has been observed that a highly conserved dileucine-containing motif (DEXXXLLI) was critical for GLUT8 sorting to the late endosomal/lysosomal compartments in mouse blastocytes.172–176
GLUT12
Similar to GLUT8, GLUT12 has the conserved DEXXXLLI in its sequence; however, this motif affects the cell surface expression level instead of directing GLUT12 to the intracellular compartment as seen for GLUT8 in mouse blastocytes.172,173 In the case of MCF-7 cells, GLUT12 was localized perinuclearly when insulin was absent.38 However, the acute change in subcellular distribution of GLUT12 was not detected after insulin treatments. Another study indicated that GLUT12 trafficking is influenced by the mTOR-raptor signaling pathway.39
Pathophysiologies associated with GLUT proteins
Given the essential roles played by this family of proteins in cellular metabolism, it is not surprising that there are really no common diseases associated with genetic mutations in the GLUTs. In most situations, such mutations would be lethal for the embryo and development could not occur. However, it is now becoming appreciated that there are a number of disease states in which the expression patterns of hexose transporters are altered, providing an opportunity for diagnosis or even treatment. Therefore, this section covers the few documented GLUT-specific genetic diseases and then looks at recent advances in diagnosis and treatment.
GLUT1: deficiency syndrome
A limited number of patients (~250) have been described since 1991 with autosomal dominant haplo-insufficiency mutations in their hSLC2A1 gene leading to reduced the concentrations of glucose in cerebrospinal fluid.177 This mutation in turn results in seizures, delayed development, and small brain size. The types of mutation are numerous and could affect functional activity of the protein directly, its ability to form dimers or tetramers, or its trafficking to the plasma membrane.177–180
GLUT2: Fanconi–Bickel syndrome
This is an extremely rare glycogen storage disease, for which a little over 100 affected patients have been reported worldwide, the first described in 1949.181 Three mutations in hSLC2A2 responsible for this autosomal recessive disease were identified in 1997, and they appeared to result in a truncated non-functional protein.182 This would fit with a number of the clinical symptoms associated with the syndrome, including glucosuria, presumably as a consequence of significantly impaired glucose reabsorption across the renal proximal convoluted tubular basolateral membrane where GLUT2 is expressed.182 The diarrhea could be explained by poor hexose absorption in the small intestine, again resulting from lack of GLUT2 in the epithelial basolateral membrane. Patients also suffer from poor regulation of blood glucose and galactose levels, which after a meal, rise rapidly and are maintained for some time. This condition could result from both the inability of the liver to take up hexoses and store them as glycogen and also probably from impaired insulin release by pancreatic beta cells as GLUT2 forms part of the normal blood glucose-sensing mechanism.183,184 Similarly, after a meal, the liver would be unable to release glucose back into the circulation, which could be responsible for the severe accumulation of glycogen in this organ seen in many patients. The lack of renal reabsorption of glucose would further exacerbate the situation, leading to hypoglycemia. Patients with a variety of mutations in GLUT2 continue to be reported, some with a full-length but non-functional transporter, while in others the defective protein fails to be targeted to the plasma membrane.184,185
GLUT9: association with urate metabolism disorders
While GLUT9 can transport glucose and fructose, its primary physiological substrate appears to be the organic anion, urate. This observation is supported by reports of patients, primarily from Japan, who have mutations in the hSLC2A9 gene.28 Unlike the majority of mammals, humans maintain high plasma levels of urate, as a result of their modified metabolic pathway that is missing the enzyme uricase. Consequently, this metabolite is retained in the body as opposed to being broken down to allantoin and lost in the urine. Plasma levels of urate are regulated within narrow limits (250–300 μM) by secretion from the liver mediated by GLUT9 and by reabsorption in the proximal convoluted tubule of the kidney. Transport across the renal epithelium is achieved by the organic anion exchanger URAT1 in the apical membrane and by GLUT9a in the basolateral membrane. Loss of function of GLUT9 results in hypouricemia, presumably as a consequence of both a reduced release of urate from the liver and poor reabsorption from the urine in the kidney.186–192
Elevated levels of plasma urate have also been associated with hypertension, gout, and metabolic disease. A number of genome-wide association studies have identified polymorphisms in the hSLC2A9 gene.29 Consequently, it has been proposed that GLUT9 in the kidney and liver may well play a role in these disease processes. However, to date, few, if any, single nucleotide polymorphisms have been demonstrated to have direct effects on the function of the protein when it is expressed in vitro.187–192 It is more likely that such mutations have subtle effects on levels of expression or interactions with other proteins in the cell.
GLUT expression in relation to disease
The most common and well described change of GLUT expression is in cancer cells that switch their metabolism primarily to glycolysis, which is far less energy-efficient and requires far more substrate. This rapid metabolic change is often referred to as the Warburg effect.193,194 To supply the greatly elevated need for glucose, tumor cells increase their expression of some GLUT proteins, particularly GLUT1. This observation has been used to develop imaging tools, such as positron emission tomography, to detect some cancers.195,196 Patients are injected with fluorodeoxy-d-glucose, a radioactively labeled glucose analog, which is rapidly taken up by the tumor cells and then metabolically trapped after phosphorylation.197–199 Research efforts are also focusing on breast cancer cells, wherein some subtypes of the disease, the cells overexpress GLUT5 rather than GLUT1, suggesting that fructose-based probes may be of value.199,200 However, this positron emission tomography technique is expensive and the probes are very short-lived. This has led to efforts to develop fluorescently labeled hexose analogs that would be preferentially taken up by tumor cells. There are no reports as yet of clinical trials for such compounds, but a number of reports have shown promising results obtained in vitro.201,202
Other studies observed that the GLUT1 gene rather than that for GLUT4 was predominantly expressed in the failing human heart, resembling the fetal stage heart.40 Several studies have also reported a similar phenomenon in the rodent heart.203–205 However, the actual cause of the heart failure in humans has not been directly determined. Some recent studies indicate that upregulated GLUT1 expression is related to HIV infection in T-cells.206–208 It was observed that increased expression of the GLUT1 gene in interleukin-7-induced T-cells rendered CD4 T-cells and thymocytes susceptible to HIV-1 infection.206,207 These reports suggest that the GLUT1-mediated metabolic pathway is the major regulator in HIV-infected cells.207 Thus, GLUT1 could be a potential marker of a diseased heart and inflammation in HIV-infected subjects.40,202,207
Moreover, other GLUTs, such as GLUT4, have been reported to be expressed on the myeloma cell membrane, where it is responsible for cell glucose consumption.209 It was also found that GLUT4 expression levels were increased and GLUT2 expression levels were decreased in senescent hepatic cells and chronically diseased human liver tissues, which suggests that GLUT4 may play an important role in liver cirrhosis.210 In many studies, GLUT4 was also found to be associated with type 2 diabetes.142 While the expression level of GLUT4 did not change in type 2 diabetic rodents, trafficking of GLUT4 was affected in these animals.210,211
It has also been reported that the pattern of GLUT expression in cartilage changes in the presence of joint disease, offering a possible tool for diagnosis and ultimately treatment. Proinflammatory cytokines have been shown to alter the pattern of GLUT expression in primary cultured articular chondrocytes, and the patterns are very different under the anaerobic conditions that are normal for these cells.212
Summary
The SLC2A family of transporter proteins are essential for the handling of hexoses and a variety of other substrates. They are expressed in numerous cell types, some almost ubiquitously, while others have a very specialized cell localization and work with other protein families to perform a wide variety of functions. The recently reported first crystal structure for GLUT1 will undoubtedly lead to a better understanding of just how these proteins bind their substrates and move them across the cell membrane. Major genetic mutations are uncommon, underlining the critical roles played by GLUTs in metabolism; however, their patterns of expression can change significantly in disease. There is now a focus developing on profiling those changes for use as a diagnostic tool or even to develop treatments for a variety of conditions. Thus, it may prove possible in the future to develop inhibitors or activity modulators for GLUTs based on a detailed knowledge of their binding sites and how transport is mediated. However, the future development of therapies employing GLUT proteins will need to take account of their multi-tissue expression patterns which could easily result in unforeseen side effects.
Acknowledgments
This work was supported in part by a grant from the Canadian Institutes of Health and the Canadian Breast Cancer Foundation.
Disclosure
The authors report no conflicts of interest in this work.
References
Brett KE, Ferraro ZM, Holcik M, Adamo KB. Prenatal physical activity and diet composition affect the expression of nutrient transporters and mTOR signaling molecules in the human placenta. Placenta. 2015;36:204–212. | |
Hedicer A, Budarf ML, Emanuel BS, Mohandas TK, Wright EM. Assignment of the human intestinal Na+/glucose cotransporter transporter. Genomics. 1989;4:297–300. | |
Wright EM, Turk E, Hager K, et al. The Na+/glucose cotransporter (SGLT1). Acta Physiol Scand Suppl. 1992;607:201–207. | |
Pao SS, Paulsen IT, Saier MH. Major facilitator superfamily. Microbiol Mol Biol Rev. 1998;62:1–34. | |
Saier MH, Beatty JT, Goffeau A, et al. The major facilitator superfamily. J Mol Microbiol Biotechnol. 1999;1:257–279. | |
Cura AJ, Carruthers AJ. The role of monosaccharide transport proteins in carbohydrate assimilation, distribution, metabolism and homeostasis. Compr Physiol. 2013;2:863–914. | |
Manolescu AR, Witkowska K, Kinnaird A, Cessford T, Cheeseman C. Facilitated hexose transporters: new perspectives on form and function. Physiology. 2007;22:234–240. | |
Thorens B, Mueckler M. Glucose transporters in the 21st century. Am J Physiol Endocrinol Metab. 2010;298:141–145. | |
Kasahara M, Hinkle PC. Reconstitution and purification of the D-glucose transporter from human erythrocytes. J Biol Chem. 1977;252: 7384–7390. | |
Sogin DC, Hinkle PC. Characterization of the glucose transporter from human erythrocytes. J Supramol Struct. 1978;8:447–453. | |
Mueckler M, Caruso C, Baldwin SA, et al. Sequence and structure of a human glucose transporter. Science. 1985;229:941–945. | |
Thorens B, Sarkar HK, Kaback HR, Lodish HF. Cloning and functional expression in bacteria of a novel glucose transporter present in liver, intestine, kidney, and beta-pancreatic islet cells. Cell. 1988;55: 281–290. | |
Fukumoto H, Kayano T, Buse JB, et al. Cloning and characterization of the major insulin-responsive glucose transporter expressed in human skeletal muscle and other insulin-responsive tissues. J Biol Chem. 1989;264:7776–7779. | |
Cheeseman CI. GLUT2 is the transporter for fructose across the rat intestinal basolateral membrane. Gastroenterology. 1993;105: 1050–1056. | |
Colville CA, Seatter MJ, Jess TJ, Gould GW, Thomas HM. Kinetic analysis of the liver-type (GLUT2) and brain-type (GLUT3) glucose transporters in Xenopus oocytes: substrate specificities and effects of transport inhibitors. Biochem J. 1993;290 Pt 3:701–706. | |
Craik JD, Elliott KR. Kinetics of 3-O-methyl-D-glucose transport in isolated rat hepatocytes. Biochem J. 1979;182:503–508. | |
Kayanos T, Fukumotoo H, Eddyl RL, et al. Evidence for a family of human glucose transporter-like proteins. Biol Chem. 1988;263: 15245–15248. | |
Yano H, Seino Y, Inagaki N, et al. Tissue distribution and species difference of the brain type glucose transporter (GLUT3). Biochem Biophys Res Commun. 1991;174:470–477. | |
Shepherd PR, Gould GW, Colville CA, McCoid SC, Gibbs EM, Kahn BB. Distribution of GLUT3 glucose transporter protein in human tissues. Biochem Biophys Res Commun. 1992;188:149–154. | |
Wu, X, Freeze HH. GLUT14, a duplicon of GLUT3, is specially expressed in testis as alternative splice forms. Genomics. 2002;80:553–557. | |
Clancy BM, Czech MP. Hexose transport stimulation and membrane redistribution of glucose transporter isoforms in response to cholera toxin, dibutyryl cyclic AMP, and insulin in 3T3-L1 adipocytes. J Biol Chem. 1990;265:12434–12443. | |
Kayano T, Burant CF, Fukumoto H, et al. Human facilitative glucose transporters. Isolation, functional characterization, and gene localization of cDNAs encoding an isoform (GLUT5) expressed in small intestine, kidney, muscle, and adipose tissue and an unusual glucose transporter pseudogene-like. J Biol Chem. 1990;265:13276–13282. | |
Corpe CP, Basaleh MM, Affleck J, Gould G, Jess TJ, Kellett GL. The regulation of GLUT5 and GLUT2 activity in the adaptation of intestinal brush-border fructose transport in diabetes. Pflugers Arch. 1996;432:192–201. | |
Waddell ID, Zomerschoe AG, Voice MW, Burchell A. Cloning and expression of a hepatic microsomal glucose transport protein. Comparison with liver plasma-membrane glucose-transport protein GLUT 2. Biochem J. 1992;286 Pt 1:173–177. | |
Burchell A. A re-evaluation of GLUT 7. Biochem J. 1998;331 Pt 3:973. | |
Li Q, Manolescu A, Ritzel M, et al. Cloning and functional characterization of the human GLUT7 isoform SLC2A7 from the small intestine. Am J Physiol Gastrointest Liver Physiol. 2004;287:G236–G242. | |
Phay JE, Hussain HB, Moley JF. Cloning and expression analysis of a novel member of the facilitative glucose transporter family, SLC2A9 (GLUT9). Genomics. 2000;9:217–220. | |
Kimura T, Takahashi M, Yan K, Sakurai H. Expression of SLC2A9 isoforms in the kidney and their localization in polarized epithelial cells. PLoS One. 2014;9:e84996. | |
Caulfield MJ, Munroe PB, O’Neill D, et al. SLC2A9 is a high-capacity urate transporter in humans. PLoS Med. 2008;5:1509–1523. | |
Doege H, Bocianski A, Joost HG, Schürmann A. Activity and genomic organization of human glucose transporter 9 (GLUT9), a novel member of the family of sugar-transport facilitators predominantly expressed in brain and leucocytes. Biochem J. 2000;350 Pt 3:771–776. | |
Lisinski I, Schürmann A, Joost HG, Cushman SW, Al-Hasani H. Targeting of GLUT6 (formerly GLUT9) and GLUT8 in rat adipose cells. Biochem J. 2001;358:517–522. | |
Vitart V, Rudan I, Hayward C, et al. SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nat Genet. 2008;40:437–442. | |
Doege H, Bocianski A, Scheepers A, et al. Characterization of human glucose transporter (GLUT) 11 (encoded by SLC2A11), a novel sugar-transport facilitator specifically expressed in heart and skeletal muscle. Biochem J. 2001;359:443–449. | |
Doege H, Schürmann A, Bahrenberg G, Brauers A, Joost HG. GLUT8, a novel member of the sugar transport facilitator family with glucose transport activity. J Biol Chem. 2000;275:16275–16280. | |
Ibberson M, Uldry M, Thorens B. GLUTX1, a novel mammalian glucose transporter expressed in the central nervous system and insulin-sensitive tissues. J Biol Chem. 2000;275:4607–4612. | |
McVie-Wylie AJ, Lamson DR, Chen YT. Molecular cloning of a novel member of the GLUT family of transporters, SLC2a10 (GLUT10), localized on chromosome 20q13.1: a candidate gene for NIDDM susceptibility. Genomics. 2001;72:113–117. | |
Dawson PA, Mychaleckyj JC, Fossey SC, Mihic SJ, Craddock AL, Bowden DW. Sequence and functional analysis of GLUT10: a glucose transporter in the type 2 diabetes-linked region of chromosome 20q12–13.1. Mol Genet Metab. 2001;74:186–199. | |
Rogers S, Macheda M, Docherty S, et al. Identification of a novel glucose transporter-like protein GLUT-12. Appl Phys Endocrinol Metab. 2002;283:E733–E738. | |
Wilson-O’Brien AL, DeHaan CL, Rogers S. Mitogen-stimulated and rapamycin-sensitive glucose transporter 12 targeting and functional glucose transport in renal epithelial cells. Endocrinology. 2008;149: 917–924. | |
Razeghi P, Young ME, Alcorn JL, Moravec CS, Frazier OH, Taegtmeyer H. Metabolic gene expression in fetal and failing human heart. Circulation. 2001;104:2923–2931. | |
Rogers S, Docherty SE, Slavin JL, Henderson MA, Best JD. Differential expression of GLUT12 in breast cancer and normal breast tissue. Cancer Lett. 2003;193:225–233. | |
Gude NM, Stevenson JL, Rogers S, et al. GLUT12 expression in human placenta in first trimester and term. Placenta. 2003;24:566–570. | |
Rogers S, Chandler JD, Clarke AL, Petrou S, Best JD. Glucose transporter GLUT12-functional characterization in Xenopus laevis oocytes. Biochem Biophys Res Commun. 2003;308:422–426. | |
Uldry M, Ibberson M, Horisberger JD, Chatton JY, Riederer BM, Thorens B. Identification of a mammalian H+-myo-inositol symporter expressed predominantly in the brain. EMBO J. 2001;20: 4467–4477. | |
Hresko RC, Kruse M, Strube M, Mueckler M. Topology of the GLUT1 glucose transporter deduced from glycosylation scanning mutagenesis. J Biol Chem. 1994;269:20482–20488. | |
Mueckler M. Facilitative glucose transporters. Eur J Biochem. 1994; 219: 713–725. | |
Cairns MT, Alvarez J, Panico M, et al. Investigation of the structure and function of the human erythrocyte glucose transporter by proteolytic dissection. Biochim Biophys Acta. 1987;905:295–310. | |
Zhao F-Q, Keating AF. Functional properties and genomics of glucose transporters. Curr Genomics. 2007;8:113–128. | |
Mori H, Hashiramoto M, Clark AE, et al. Substitution of tyrosine 293 of GLUT1 locks the transporter into an outward facing conformation. J Biol Chem. 1994;269:11578–11583. | |
Moris H, Muraokas A. Substitution at Pro385 of GLUTl perturbs the glucose transport function by reducing conformational flexibility. J Biol Chem. 1994;269:2982–2986. | |
Schürmann A, Doege H, Ohnimus H, Monser V, Buchs A, Joost HG. Role of conserved arginine and glutamate residues on the cytosolic surface of glucose transporters for transporter function. Biochemistry. 1997;36:12897–12902. | |
Olson AL, Pessin JE. Structure, function, and regulation of the mammalian facilitative glucose transporter gene family. Annu Rev Nutr. 1996;16:235–256. | |
Mueckler M, Weng W, Kruse M. Glutamine 161 of GLUT1 glucose transporter is critical for transport activity and exofacial ligand binding. J Biol Chem. 1994;269:20533–20538. | |
Doege H, Schürmann A, Ohnimus H, Monser V, Holman GD, Joost HG. Serine-294 and threonine-295 in the exofacial loop domain between helices 7 and 8 of glucose transporters (GLUT) are involved in the conformational alterations during the transport process. Biochem J. 1998;329 Pt 2:289–293. | |
Garcia JC, Strube M, Leingang K, Keller K, Mueckler MM. Amino acid substitutions at tryptophan 388 and tryptophan 412 of the HepG2 (Glut1) glucose transporter inhibit transport activity and targeting to the plasma membrane in Xenopus oocytes. J Biol Chem. 1992;267: 7770–7776. | |
Schürmann A, Keller K, Monden I, et al. Glucose transport activity and photolabelling with 3-[125I]iodo-4-azidophenethylamido-7-O-succinyldeacetyl (IAPS)-forskolin of two mutants at tryptophan-388 and -412 of the glucose transporter GLUT1: dissociation of the binding domains of forskolin and glucose. Biochem J. 1993;290 Pt 2:497–501. | |
Seatter MJ, De La Rue SA, Porter LM, Gould GW. QLS motif in transmembrane helix VII of the glucose transporter family interacts with the C-1 position of D-glucose and is involved in substrate selection at the exofacial binding site. Biochemistry. 1998;37:1322–1326. | |
Joost H, Thorens B. The extended GLUT-family of sugar/polyol transport facilitators: nomenclature, sequence characteristics, and potential function of its novel members. Mol Membr Biol. 2001;18:257–264. | |
Manolescu AR, Augustin R, Moley K, Cheeseman C. A highly conserved hydrophobic motif in the exofacial vestibule of fructose transporting SLC2A proteins acts as a critical determinant of their substrate selectivity. Mol Membr Biol. 2007;24:455–463. | |
Manolescu A, Salas-Burgos AM, Fischbarg J, Cheeseman CI. Identification of a hydrophobic residue as a key determinant of fructose transport by the facilitative hexose transporter SLC2A7 (GLUT7). J Biol Chem. 2005;280:42978–42983. | |
Long W, Panwar P, Witkowska K, et al. Critical roles of two hydrophobic residues within human glucose transporter 9 (hSLC2A9) in substrate selectivity and urate transport. J Biol Chem. 2015;290: 15292–15303. | |
Alvarez J, Lee DC, Baldwin SA, Chapman D. Fourier transform infrared spectroscopic study of the structure and conformational changes of the human erythrocyte glucose transporter. J Biol Chem. 1987;262: 3502–3509. | |
Chin JJ, Jung EK, Chen V, Jung CY. Structural basis of human erythrocyte glucose transporter function in proteoliposome vesicles: circular dichroism measurements. Proc Natl Acad Sci U S A. 1987;84: 4113–4116. | |
Heinze M, Monden I, Keller K. Cysteine-scanning mutagenesis of transmembrane segment 1 of glucose transporter GLUT1: extracellular accessibility of helix positions. Biochemistry. 2004;43:931–936. | |
Mueckler M, Roach W, Makepeace C. Transmembrane segment 3 of the GLUT1 glucose transporter is an outer helix. J Biol Chem. 2004;279:46876–46881. | |
Mueckler M, Makepeace C. Cysteine-scanning mutagenesis and substituted cysteine accessibility analysis of transmembrane segment 4 of the GLUT1 glucose transporter. J Biol Chem. 2005;280:39562–39568. | |
Mueckler M, Makepeace C. Transmembrane segment 5 of the Glut1 glucose transporter is an amphipathic helix that forms part of the sugar permeation pathway. J Biol Chem. 1999;274:10923–10926. | |
Mueckler M, Makepeace C. Transmembrane segment 6 of the Glut1 glucose transporter is an outer helix and contains amino acid side chains essential for transport activity. J Biol Chem. 2008;283:11550–11555. | |
Hruz PW, Mueckler MM. Cysteine-scanning mutagenesis of transmembrane segment 7 of the GLUT1 glucose transporter. J Biol Chem. 1999;274:36176–36180. | |
Mueckler M, Makepeace C. Model of the exofacial substrate-binding site and helical folding of the human Glut1 glucose transporter based on scanning mutagenesis. Biochemistry. 2009;48:5934–5942. | |
Hruz PW, Mueckler MM. Cysteine-scanning mutagenesis of transmembrane segment 11 of the GLUT1 facilitative glucose transporter. Biochemistry. 2000;39(51):9367–9372. | |
Mueckler M, Makepeace C. Transmembrane segment 12 of the Glut1 glucose transporter is an outer helix and is not directly involved in the transport mechanism. J Biol Chem. 2006;281(48):36993–36998. | |
Mueckler M, Makepeace C. Analysis of Transmembrane Segment 8 of the GLUT1 Glucose Transporter by Cysteine-scanning Mutagenesis and Substituted Cysteine Accessibility. J Biol Chem. 2003;279: 10494–10499. | |
Mueckler M, Makepeace C. Analysis of transmembrane segment 10 of the Glut1 glucose transporter by cysteine scanning mutagenesis and substituted cysteine accessibility. J Biol Chem. 2002;277(5):3498–3503. | |
Salas-Burgos A, Iserovich P, Zuniga F, Vera JC, Fischbarg J. Predicting the three-dimensional structure of the human facilitative glucose transporter GLUT1 by a novel evolutionary homology strategy: insights on the molecular mechanism of substrate migration, and binding sites for glucose and inhibitory molecules. Biophys J. 2004;87: 2990–2999. | |
Saier MH. Phylogenetic approaches to the identification and characterization of protein families and superfamilies. Microb Comp Genomics. 1996;1:129–150. | |
Abramson J, Smirnova I, Kasho V, Verner G, Kaback HR, Iwata S. Structure and mechanism of the lactose permease of Escherichia coli. Science. 2003;301:610–615. | |
Lemieux MJ. Eukaryotic major facilitator superfamily transporter modeling based on the prokaryotic GlpT crystal structure. Mol Membr Biol. 2007;24:333–341. | |
Dwyer DS. Model of the 3-D structure of the GLUT3 glucose transporter and molecular dynamics simulation of glucose transport. Proteins. 2001;42:531–541. | |
Mohan SS, Perry JJ, Poulose N, Nair BG, Anilkumar G. Homology modeling of GLUT4, an insulin regulated facilitated glucose transporter and docking studies with ATP and its inhibitors. J Biomol Struct Dyn. 2009;26:455–464. | |
Madej MG, Sun L, Yan N, Kaback HR. Functional architecture of MFS D-glucose transporters. Proc Natl Acad Sci U S A. 2014;111: E719–E727. | |
Clémençon B, Lüscher BP, Fine M, et al. Expression, purification, and structural insights for the human uric acid transporter, GLUT9, using the Xenopus laevis oocytes system. PLoS One. 2014;9:e108852. | |
Chang G, Spencer RH, Lee AT, Barclay MT, Rees DC. Structure of the MscL homolog from Mycobacterium tuberculosis: a gated mechanosensitive ion channel. Science. 1998;282:2220–2226. | |
Deng D, Xu C, Sun P, et al. Crystal structure of the human glucose transporter GLUT1. Nature. 2014;510:121–125. | |
Sun L, Zeng X, Yan C, et al. Crystal structure of a bacterial homologue of glucose transporters GLUT1–4. Nature. 2012;490:361–366. | |
Iancu CV, Zamoon J, Woo SB, Aleshin A, Choe JY. Crystal structure of a glucose/H+ symporter and its mechanism of action. Proc Natl Acad Sci U S A. 2013;110:17862–17867. | |
Deng D, Sun P, Yan C, et al. Molecular basis of ligand recognition and transport by glucose transporters. Nature. July 15, 2015. doi:10.1038/nature14655. | |
Widdas W. Inability of diffusion to account for placental glucose transfer in the sheep and consideration of the kinetics of a possible carrier transfer. J Physiol. 1952;118:23–39. | |
Fisher RB, Parsons DS. Glucose movements across the wall of the rat small intestine. J Physiol. 1953;119:210–223. | |
Fisher RB, Parsons DS. Galatose absorption from the surviving small intestine of the rat. J Physiol. 1953;119:224–232. | |
Neame KD, Richards TG, editors. Elementary Kinetics of Membrane Carrier Transport. 1st ed. London, UK: Blackwell Scientific Publications; 1972. | |
Naftalin RJ, Holman GD. Transport of sugars in human red cells. In: Ellory JC, Lew V, editors. Membrane Transport in Red Cells. New York, NY, USA: Academic Press; 1977. | |
Regen DM, Tarpley HL. Anomalous transport kinetics and the glucose carrier hypothesis. Biochim Biophys Acta. 1974;339:218–233. | |
Eilam Y. Two-carrier models for mediated transport. 1. Theoretical analysis of several two-carrier models. Biochim Biophys Acta. 1975; 401: 349–363. | |
Naftalin RJ. Model for sugar transport across red cell membrane without carriers. Biochim Biophys Acta. 1970;211:65–78. | |
Lieb WR, Stein WD. Quantitative predictions of a noncarrier model for glucose transport across the human red cell membrane. Biophys J. 1970;10:585–609. | |
LeFevre PG. A model for erythrocyte sugar transport based on substrate-conditioned “introversion” of binding sites. J Membr Biol. 1973;11:1–19. | |
Mueckler M, Thorens B. The SLC2 (GLUT) family of membrane transporters. Mol Asp Med. 2013;34:121–138. | |
Naftalin RJ. Alternating carrier models of asymmetric glucose transport violate the energy conservation laws. Biophys J. 2008;95: 4300–4314. | |
Vollers SS, Carruthers A. Sequence determinants of GLUT1-mediated accelerated-exchange transport: analysis by homology-scanning mutagenesis. J Biol Chem. 2012;287:42533–42544. | |
Carruthers A, DeZutter J, Ganguly A, Devaskar SU. Will the original glucose transporter isoform please stand up! Am J Physiol Endocrinol Metab. 2009;297:E836–E848. | |
Toyoda N, Flanagan JE, Kono T. Reassessment of insulin effects on the V(max) and K(m) values of hexose transport in isolated rat epididymal adipocytes. J Biol Chem. 1987;262:2737–2745. | |
Taylor LP, Holman GD. Symmetrical kinetic parameters for 3-O-methyl-D-glucose transport in adipocytes in the presence and in the absence of insulin. Biochim Biophys Acta. 1981;642:325–335. | |
Pessino A, Hebert DN, Woon CW, et al. Evidence that functional erythrocyte-type glucose transporters are oligomers. J Biol Chem. 1991;266:20213–20217. | |
Zottola RJ, Cloherty EK, Coderre PE, Hansen A, Hebert DN, Carruthers A. Glucose transporter function is controlled by transporter oligomeric structure. A single, intramolecular disulfide promotes GLUT1 tetramerization. Biochemistry. 1995;34:9734–9747. | |
Cloherty EK, Levine KB, Carruthers A. The red blood cell glucose transporter presents multiple, nucleotide-sensitive sugar exit sites. Biochemistry. 2001;40:15549–15561. | |
De Zutter JK, Levine KB, Deng D, Carruthers A. Sequence determinants of GLUT1 oligomerization: analysis by homology-scanning mutagenesis. J Biol Chem. 2013;288:20734–20744. | |
Lemieux MJ, Huang Y, Wang da N. Crystal structure and mechanism of GlpT, the glycerol-3-phosphate transporter from E. coli. J Electron Microsc (Tokyo). 2005;54 Suppl 1:43–46. | |
Dang S, Sun L, Huang Y, et al. Structure of a fucose transporter in an outward-open conformation. Nature. 2010;467:734–738. | |
Jardetzky O. Simple allosteric model for membrane pumps. Nature. 1966;211:969–970. | |
Park CR, Crofford OB, Kono T. Mediated (nonactive) transport of glucose in mammalian cells and its regulation. J Gen Physiol. 1968;52: 296–318. | |
Rosenberg T, Wilbrandt W. Uphill transport induced by counterflow. J Gen Physiol. 1957;41:289–296. | |
Maher F, Davies-Hill TM, Simpson IA. Substrate specificity and kinetic parameters of GLUT3 in rat cerebellar granule neurons. J Biol Chem. 1996;315:827–831. | |
Liu Q, Vera JC, Peng H, Golde DW. The predicted ATP-binding domains in the hexose transporter GLUT1 critically affect transporter activity. Biochemistry. 2001;40:7874–7881. | |
Sweet IR, Matschinsky FM. Are there kinetic advantages of GLUT2 in pancreatic glucose sensing? Diabetologia. 1997;40: 112–119. | |
Clark AE, Holman GD, Kozka IJ. Determination of the rates of appearance and loss of glucose transporters at the cell surface of rat adipose cells. Biochem J. 1991;278 Pt 1:235–241. | |
Haney PM, Slot JW, Piper RC, James DE, Mueckler M. Intracellular targeting of the insulin-regulatable glucose transporter (GLUT4) is isoform specific and independent of cell type. J Cell Biol. 1991;114: 689–699. | |
Hudson AW, Ruiz M, Birnbaum MJ. Isoform-specific subcellular targeting of glucose transporters in mouse fibroblasts. J Cell Biol. 1992;116:785–797. | |
Hudson AW, Fingar DC, Seidner GA, Griffiths G, Burke B, Birnbaum MJ. Targeting of the “insulin-responsive” glucose transporter (GLUT4) to the regulated secretory pathway in PC12 cells. J Cell Biol. 1993;122:579–588. | |
Piper RC, Tai C, Slot JW, et al. The efficient intracellular sequestration of the insulin-regulatable glucose transporter (GLUT-4) is conferred by the NH2 terminus. J Cell Biol. 1992;117:729–743. | |
Shibasaki Y, Asano T, Lin JL, et al. Two glucose transporter isoforms are sorted differentially and are expressed in distinct cellular compartments. Biochem J. 1992;281 Pt 3:829–834. | |
Verhey KJ, Hausdorff SF, Birnbaum MJ. Identification of the carboxy terminus as important for the isoform-specific subcellular targeting of glucose transporter proteins. J Cell Biol. 1993;123:137–147. | |
Riskin A, Nannegari VH, Mond Y. Acute effectors of GLUT1 glucose transporter subcellular targeting in CIT3 mouse mammary epithelial cells. Pediatr Res. 2008;63:56–61. | |
Czech MP, Chawla A, Woon CW, et al. Exofacial epitope-tagged glucose transporter chimeras reveal COOH-terminal sequences governing cellular localization. J Cell Biol. 1993;123:127–135. | |
Andrisse S, Patel GD, Chen JE, et al. ATM and GLUT1-S490 phosphorylation regulate GLUT1 mediated transport in skeletal muscle. PLoS One. 2013;8:e66027. | |
Inukai K, Shewan AM, Pascoe WS, Katayama S, James DE, Oka Y. Carboxy terminus of glucose transporter 3 contains an apical membrane targeting domain. Mol Endocrinol. 2004;18:339–349. | |
Yang J, Holman GD. Comparison of GLUT4 and GLUT1 subcellular trafficking in basal and insulin-stimulated 3T3-L1 cells. J Biol Chem. 1993;268:4600–4603. | |
Yang J, Clark AE, Harrison R, Kozka IJ, Holman GD. Trafficking of glucose transporters in 3T3-L1 cells trafficking proteins. 1992;281:809–817. | |
Egert S, Nguyen N, Schwaiger M. Myocardial glucose transporter GLUT1: translocation induced by insulin and ischemia. J Mol Cell Cardiol. 1999;31:1337–1344. | |
El-Jack AK, Kandror KV, Pilch PF. The formation of an insulin-responsive vesicular cargo compartment is an early event in 3T3-L1 adipocyte differentiation. Mol Biol Cell. 1999;10:1581–1594. | |
Mueckler M. The molecular biology of mammalian glucose transporters. Curr Opin Nephrol Hypertens. 1992;1:12–20. | |
Roach W, Plomann M. PACSIN3 overexpression increases adipocyte glucose transport through GLUT1. Biochem Biophys Res Commun. 2007;355:745–750. | |
Nakayama T, Kamiguchi H, Akagawa K. Syntaxin 1C, a soluble form of syntaxin, attenuates membrane recycling by destabilizing microtubules. J Cell Sci. 2012;125:817–830. | |
Cura AJ, Carruthers A. AMP kinase regulation of sugar transport in brain capillary endothelial cells during acute metabolic stress. Am J Physiol Cell Physiol. 2012;303:C806–C814. | |
Caliceti C, Zambonin L, Prata C, et al. Effect of plasma membrane cholesterol depletion on glucose transport regulation in leukemia cells. PLoS One. 2012;7:e41246. | |
Fang J, Zhou SH, Fan J, Yan SX. Roles of glucose transporter-1 and the phosphatidylinositol 3-kinase/protein kinase B pathway in cancer radioresistance. Mol Med Rep. 2015;11:1573–1578. | |
Wofford JA, Wieman HL, Jacobs SR, Zhao Y, Rathmell JC, Jeffrey C. IL-7 promotes Glut1 trafficking and glucose uptake via STAT5-mediated activation of Akt to support T cell survival. Blood. 2008;111: 2101–2112. | |
Zambrano A, Jara E, Murgas P, et al. Cytokine stimulation promotes increased glucose uptake via translocation at the plasma membrane of GLUT1 in HEK293 cells. J Cell Biochem. 2010;110:1471–1480. | |
Sommermann TG, O’Neill K, Plas DR, Cahir-McFarland E. IKK² and NF-ºB transcription govern lymphoma cell survival through AKT-induced plasma membrane trafficking of GLUT1. Cancer Res. 2011;71:7291–7300. | |
Stockli J, Fazakerley DJ, James DE. GLUT4 exocytosis. J Cell Sci. 2011;124:4147–4159. | |
Belman JP, Habtemichael EN, Bogan JS. A proteolytic pathway that controls glucose uptake in fat and muscle. Rev Endocr Metab Disord. 2014;15:55–66. | |
Alvim RO, Cheuhen MR, Machado SR. General aspects of muscle glucose uptake. 2015;87:351–368. | |
Cushman SW, Wardzala LJ. Potential mechanism of insulin action on glucose transport in the isolated rat adipose cell. Apparent translocation of intracellular transport systems to the plasma membrane. J Biol Chem. 1980;255(10):4758–4762. | |
Millar CA, Powell KA, Hickson GR, Bader MF, Gould GW. Evidence for a role for ADP-ribosylation factor 6 in insulin-stimulated glucose transporter-4 (GLUT4) trafficking in 3T3-L1 adipocytes. J Biol Chem. 1999;274:17619–17625. | |
van Dam EM, Govers R, James DE. Akt activation is required at a late stage of insulin-induced GLUT4 translocation to the plasma membrane. Mol Endocrinol. 2005;19:1067–1077. | |
Backer JM, Myers MG, Shoelson SE, et al. Phosphatidylinositol 3′-kinase is activated by association with IRS-1 during insulin stimulation. EMBO J. 1992;11:3469–3479. | |
White MF, Kahn CR. The insulin signaling system. J Biol Chem. 1994;269:1–4. | |
Zorzano A, Munoz P, Camps M, Mora C, Testar X, Palacin M. Insulin-induced redistribution of GLUT4 glucose carriers in the muscle fiber: in search of GLUT4 trafficking pathways. Diabetes. 1996;45 Suppl 1: S70–S81. | |
Steinbusch LK, Schwenk RW, Ouwens DM, Diamant M, Glatz JF, Luiken JJ. Subcellular trafficking of the substrate transporters GLUT4 and CD36 in cardiomyocytes. Cell Mol Life Sci. 2011;68:2525–2538. | |
Capilla E, Suzuki N, Pessin JE, Hou JC. The glucose transporter 4 FQQI motif is necessary for Akt substrate of 160-kilodalton-dependent plasma membrane translocation but not Golgi-localized (gamma)-ear-containing Arf-binding protein-dependent entry into the insulin-responsive storage compartment. Mol Endocrinol. 2007;21:3087–3099. | |
Khan AH, Capilla E, Hou JC, Watson RT, Smith JE, Pessin JE. Entry of newly synthesized GLUT4 into the insulin-responsive storage compartment is dependent upon both the amino terminus and the large cytoplasmic loop. J Biol Chem. 2004;279:37505–37511. | |
Tsuchiya A, Kanno T, Nishizaki T. Diacylglycerol promotes GLUT4 translocation to the cell surface in a PKCµ-dependent and PKC»/¹ and -¶- independent manner. Life Sci. 2013;93:240–246. | |
Asano T, Kanda A, Katagiri H, et al. P110² is up-regulated during differentiation of 3T3-L1 cells and contributes to the highly insulin-responsive glucose transport activity. J Biol Chem. 2000;275:17671–17676. | |
Waller A, Kohler K, Burns T, Mudge M, Belknap J, Lacombe V. Naturally- occurring compensated insulin resistance selectively alters glucose transporters in visceral and subcutaneous adipose tissue without change in AS 160 activation. Biochim Biophys Acta. 2011;1812:1098–1103. | |
Tsuchiya Y, Hatakeyama H, Emoto N, Wagatsuma F, Matsushita S, Kanzaki M. Palmitate-induced down-regulation of sortilin and impaired GLUT4 trafficking in C2C12 myotubes. J Biol Chem. 2010;285: 34371–34381. | |
Hong YH, Betik AC, McConell GK. Role of nitric oxide in skeletal muscle glucose uptake during exercise. Exp Physiol. 2014;99:1569–1573. | |
Li Q, Zhu X, Ishikura S, et al. Ca2+ signals promote GLUT4 exocytosis and reduce its endocytosis in muscle cells. Am J Physiol Endocrinol Metab. 2014;307:e209–e224. | |
Kellett GL, Helliwell PA. The diffusive component of intestinal glucose absorption is mediated by the glucose-induced recruitment of GLUT2 to the brush-border membrane. Biochem J. 2000;350:155–162. | |
Tobin V, Le Gall M, Fioramonti X, et al. Insulin internalizes GLUT2 in the enterocytes of healthy but not insulin-resistant mice. Diabetes. 2008;57:555–562. | |
Helliwell P, Richardson M, Affleck J, Kellett G. Stimulation of fructose transport across the intestinal brush-border membrane by PMA is mediated by GLUT2 and dynamically regulated by protein kinase C. Biochem J. 2000;350:149–154. | |
Mace OJ, Lister N, Morgan E, et al. An energy supply network of nutrient absorption coordinated by calcium and T1R taste receptors in rat small intestine. J Physiol. 2009;587 Pt 1:195–210. | |
Cheeseman CI, O’Neill D. Basolateral D-glucose transport activity along the crypt-villus axis in rat jejunum and upregulation induced by gastric inhibitory peptide and glucagon-like peptide-2. Exp Physiol. 1998;83:605–616. | |
Shepherd EJ, Helliwell PA, Mace OJ, Morgan EL, Patel N, Kellett GL. Stress and glucocorticoid inhibit apical GLUT2-trafficking and intestinal glucose absorption in rat small intestine. J Physiol. 2004;560 Pt 1: 281–290. | |
Habold C, Foltzer-Jourdainne C, Le Maho Y, Lignot J-H, Oudart H. Intestinal gluconeogenesis and glucose transport according to body fuel availability in rats. J Physiol. 2005;566 Pt 2:575–586. | |
Wis´niewski J, Friedrich A, Keller T, Mann M, Koepsell H. The impact of high-fat diet on metabolism and immune defense in small intestine mucosa. J Proteome Res. 2015;14:353–365. | |
Cohen M, Kitsberg D, Tsytkin S, Shulman M, Aroeti B, Nahmias Y. Live imaging of GLUT2 glucose-dependent trafficking and its inhibition in polarized epithelial cysts. Open Biol. 2014;4:140091. | |
Greenlee W, Heather M, Uemura E, Carpenter SL, Doyle RT, Buss JE. Glucose uptake in PC12 cells: GLUT3 vesicle trafficking and fusion as revealed with a novel GLUT3-GFP fusion protein. J Neurosci Res. 2003;73:518–525. | |
McClory H, Williams D, Sapp E, et al. Glucose transporter 3 is a rab11-dependent trafficking cargo and its transport to the cell surface is reduced in neurons of CAG140 Huntington’s disease mice. Acta Neuropathol Commun. 2014;2:1–9. | |
Pinto AB, Carayannopoulos MO, Hoehn A, Dowd L, Moley KH. Glucose transporter 8 expression and translocation are critical for murine blastocyst survival. Biol Reprod. 2002;66:1729–1733. | |
Carayannopoulos MO, Chi MM, Cui Y, et al. GLUT8 is a glucose transporter responsible for insulin-stimulated glucose uptake in the blastocyst. Proc Natl Acad Sci U S A. 2000;97:7313–7318. | |
Wyman AH, Chi M, Riley J, et al. Syntaxin 4 expression affects glucose transporter 8 translocation and embryo survival. Mol Endocrinol. 2003;17:2096–2102. | |
Piroli GG, Grillo CA, Hoskin EK, et al. Peripheral glucose administration stimulates the translocation of GLUT8 glucose transporter to the endoplasmic reticulum in the rat hippocampus. J Comp Neurol. 2002;452:103–114. | |
Augustin R, Riley J, Moley KH. GLUT8 contains [DE]XXXL[LI] sorting motif and localizes to a late endomosal/lysosomal compartment. Traffic. 2005;6:1196–1212. | |
Flessner L, Moley K. Similar [DE]XXX[LI] motif differentially target GLUT8 and GLUT12 in Chinese hamster ovary cells. Traffic. 2010;10:324–333. | |
Piroli GG, Grillo C a., Hoskin EK, et al. Peripheral glucose administration stimulates the translocation of GLUT8 glucose transporter to the endoplasmic reticulum in the rat hippocampus. J Comp Neurol. 2002;452(2):103–114. | |
Aerni-Flessner L, Otu M, Moley K. The amino acids upstream of NH(2)-terminal dileucine motif play a role in regulating the intracellular sorting of the Class III transporters GLUT8 and GLUT12. Mol Membr Biol. 2011;28:30–41. | |
De Vivo DC, Trifiletti RR, Jacobson RI, Ronen GM, Behmand RA, Harik SI. Defective glucose transport across the blood-brain barrier as a cause of persistent hypoglycorrhachia, seizures, and development delay. N Engl J Med. 1991;325:703–709. | |
Sen S, Keough K, Gibson J. Clinical reasoning: novel GLUT1-DS mutation refractory seizures and ataxia. Am Acad Neurol. 2015;84:e111–e114. | |
Raja M, Kinne RK. Pathogenic mutations causing glucose transport defects in GLUT1 transporter: the role of intermolecular forces in protein structure-function. Biophys Chem. 2015;200–201:9–17. | |
Yang H, Wang D, Engelstad K, et al. Glut1 deficiency syndrome and erythrocyte glucose uptake assay. Ann Neurol. 2011;70:996–1005. | |
Rohr K. Familial panmyelophthisis, Fanconi syndrome in adults. Blood. 1949;4:130–141. | |
Santer R, Schneppenheim R, Dombrowske A, Gotzer H, Steinmann B, Schaub J. Mutation in GLUT2, the gene for the liver-type glucose transporter, in patients with Fanconi-Bickel syndrome. Nat Genet. 1997;15:57–61. | |
De los Angeles García M, Millán C, Balmaceda-Aguilera C, et al. Hypothalamic ependymal-glial cells express the glucose transporter GLUT2, a protein involved in glucose sensing. J Neurochem. 2003;86: 709–724. | |
Ohtsubo K, Takamatsu S, Gao C, Korekane H, Kurosawa TM, Taniguchi N. N-glycosylation modulates the membrane sub-domain distribution and activity of glucose transporter 2 in pancreatic beta cells. Biochem Biophys Res Comm. 2013;434:346–351. | |
Abbasi F, Azizi F, Javaheri M, Mosallanejad A, Ebrahim-Habibi A, Ghafouri-Fard S. Segregation of a novel homozygous 6 nucleotide deletion in GLUT2 gene in a Fanconi–Bickel syndrome family. Gene. 2015;557:103–105. | |
Matsuo H, Chiba T, Nagamori S, et al. Mutations in glucose transporter 9 gene SLC2A9 cause renal hypouricemia. Am J Hum Genet. 2008;83: 744–751. | |
Jeannin G, Chiarelli N, Gaggiotti M, et al. Recurrent exercise-induced acute renal failure in a young Pakistani man with severe renal hypouricemia and SLC2A9 compound heterozygosity. BMC Med Genet. 2014:15:1–8. | |
Mou L, Jiang L, Hu Y. A novel homozygous GLUT9 mutation cause recurrent exercise-induced acute renal failure and posterior reversible encephalopathy syndrome. J Nephrol. 2015;28:387–392. | |
Stiburkova B, Ichida K, Sebesta I. Novel homozygous insertion in SLC2A9 gene caused renal hypouricemia. Mol Genet Metab. 2011;102: 430–435. | |
Dinour D, Gray NK, Ganon L, et al. Two novel homozygous SLC2A9 mutations cause renal hypouricemia type 2. Nephrol Dial Transplant. 2012;27:1035–1041. | |
Stiburkova B, Taylor J, Marinaki AM, Sebesta I. Acute kidney injury in two children caused by renal hypouricaemia type 2. Pediatr Nephrol. 2012;27:1411–1415. | |
Dinour D, Gray NK, Campbell S, et al. Homozygous SLC2A9 mutations cause severe renal hypouricemia. J Am Soc Nephrol. 2010;21:64–72. | |
Warburg O. On the origin of cancer cells. Oncologia. 1956;9:75–83. | |
Racker E, Spector M. Warburg effect revisited: merger of biochemistry and molecular biology. Science. 1981;213:303–307. | |
Wang H, Li D, Liu S, et al. Small animal PET imaging of pancreatic cancer xenografts using 64Cu labeled monoclonal antibody MAb159. J Nucl Med. 2015:56:908–913. | |
Kepka L, Socha J. PET-CT use and the occurrence of elective nodal failure in involved field radiotherapy for non-small cell lung cancer: a systematic review. Radiother Oncol. 2015;115:151–156. | |
Koo HR, Park JS, Kang KW, Han W, Park IA, Moon WK. Correlation between 18F-FDG uptake on PET/CT and prognostic factors in triple-negative breast cancer. Eur Radiol. April 23, 2015. [Epub ahead of print.] | |
Weber WA, Gatsonis CA, Mozley PD, et al. Repeatability of 18F-FDG PET/CT in advanced non-small cell lung cancer: prospective assessment in two multicenter trials. J Nucl Med. April 23, 2015. [Epub ahead of print.] | |
Levi J, Cheng Z, Gheysens O, et al. Fluorescent fructose derivatives for imaging breast cancer cells. Bioconjug Chem. 2007;18:628–634. | |
Zamora-León SP, Golde D, Concha I, et al. Expression of the fructose transporter GLUT5 in human breast cancer. Proc Natl Acad Sci U S A. 1996;93:1847–1852. | |
Wuest M, Trayner BJ, Grant TN, et al. Radiopharmacological evaluation of 6-deoxy-6-[18F]fluoro-d-fructose as a radiotracer for PET imaging of GLUT5 in breast cancer. Nucl Med Biol. 2011;38:461–475. | |
Trayner BJ, Grant TN, West FG, Cheeseman CI. Synthesis and characterization of 6-deoxy-6-fluoro-d-fructose as a potential compound for imaging breast cancer with PET. Bioorg Med Chem. 2009;17: 5488–5495. | |
Neves FA, Cortez E, Bernardo AF, et al. Heart energy metabolism impairment in Western-diet induced obese mice. J Nutr Biochem. 2014;25:50–57. | |
Aerni-Flessner L, Abi-Jaoude M, Koenig A, Payne M, Hruz PW. GLUT4, GLUT1, and GLUT8 are the dominant GLUT transcripts expressed in the murine left ventricle. Cardiovasc Diabetol. 2012;11:63–73. | |
Pereira RO, Wende AR, Olsen C, et al. Inducible overexpression of GLUT1 prevents mitochondrial dysfunction and attenuates structural remodeling in pressure overload but does not prevent left ventricular dysfunction. J Am Heart Assoc. 2013;2:e000301. | |
Craveiro M, Clerc I, Sitbon M, Taylor N. Metabolic pathways as regulators of HIV infection. Curr Opin HIV AIDS. 2013;8:182–189. | |
Loisel-Meyer S, Swainson L, Craveiro M, et al. Glut1-mediated glucose transport regulates HIV infection. Proc Natl Acad Sci U S A. 2012;109:2549–2554. | |
Palmer CS, Anzinger JJ, Zhou J, et al. Glucose transporter 1 expressing proinflammatory monocytes are elevated in combination antiretroviral therapy treated and untreated HIV+ subjects. J Immunol. 2014;193: 5595–5603. | |
McBrayer SK, Cheng JC, Singhal S, Krett NL, Rosen ST, Shanmugam M. Multiple myeloma exhibits novel dependence on GLUT4, GLUT8, and GLUT11: implications for glucose transporter- directed therapy. Blood. 2012;119:4686–4697. | |
Aravinthan A, Challis B, Shannon N, Hoare M, Heaney J, Alexander GJ. Selective insulin resistance in hepatocyte senescence. Exp Cell Res. 2015;331:38–45. | |
Garvey WT, Maianu L, Zhu JH, Brechtel-Hook G, Wallace P, Baron AD. Evidence for defects in the trafficking and translocation of GLUT4 glucose transporters in skeletal muscle as a cause of human insulin resistance. J Clin Invest. 1998;101:2377–2386. | |
Favaretto F, Milan G, Collin GB, et al. GLUT4 defects in adipose tissue are early signs of metabolic alterations in alms1GT/GT, a mouse model for obesity and insulin resistance. PLoS One. 2014;9:e109540. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.