Back to Journals » Breast Cancer: Targets and Therapy » Volume 9
Structure-based virtual screening and molecular docking for the identification of potential multi-targeted inhibitors against breast cancer
Authors Yousuf Z, Iman K, Iftikhar N, Mirza MU ![]()
Received 17 January 2017
Accepted for publication 3 May 2017
Published 14 June 2017 Volume 2017:9 Pages 447—459
DOI https://doi.org/10.2147/BCTT.S132074
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Pranela Rameshwar
Zeeshan Yousuf,1 Kanzal Iman,1 Nauman Iftikhar,2 Muhammad Usman Mirza3,4
1Institute of Molecular Biology and Biotechnology, The University of Lahore, Lahore, 2National Institute for Genomics and Advanced Biotechnology, National Agricultural Research Centre, Islamabad, 3Centre for Research in Molecular Medicine, The University of Lahore, Lahore, Pakistan; 4Medicinal Chemistry, Department of Pharmaceutical and Pharmacological Sciences, Rega Institute for Medical Research, University of Leuven, Leuven, Belgium
Abstract: Breast cancer is characterized by an uncontrolled growth of cells in breast tissue. Genes that foster cell growth in breast cells are overexpressed, giving rise to breast tumors. The identification of effective inhibitors represents a rational chemopreventive strategy. The current in silico study provides a pharmacoinformatic approach for the identification of active compounds against a co-chaperone HSP90 and the human epidermal growth factor receptors EGFR and HER2/neu receptor. The elevated levels of expression of these target proteins have been documented in breast cancer. The utilization of drug-likeness filters helped to evaluate the pharmacological activity of potential lead compounds. Those fulfilling this criterion were subjected to energy minimization for 1000 steepest descent steps at a root means square gradient of 0.02 with an Amber ff12SB force field. Based on molecular docking results and binding interaction analysis, this study represents five chemical compounds (S-258282355, S-258012947, S-259417539, S-258002927, and S-259411474) that indicate high binding energies that range between −8.7 to −10.3 kcal/mol. With high cytochrome P inhibitory promiscuity activity, these multi-targeted potential hits portray not only good physiochemical interactions but also an excellent profile of absorption, distribution, metabolism, excretion, and toxicity, which hypothesizes that these compounds can be developed as anticancer drugs in the near future.
Keywords: EGFR, metastasis, TNBC, HER2, heat shock proteins
Introduction
Breast cancer is characterized by the uncontrolled growth of cells forming a hard painless lump in the breast tissue, usually in the milk ducts or lobules that provide them with milk.1,2 The most common practice to classify breast tumors is according to the status of three specific cell surface receptors: the estrogen receptor (ER), the progesterone receptor, and the human epidermal growth factor (EGF) receptor HER2/neu receptor. Approximately 75% of all breast cancers are hormone receptor-positive.3 HER2-positive breast cancer accounts for 20–30% of hormone receptor-positive breast cancer that relates to the overexpression of HER2/neu protein. A rare form of breast cancer, triple-negative breast cancer (TNBC), comprises tumors cells that lack receptors for estrogen and progesterone and do not overexpress the HER2 protein.
One of the factors that has been shown to increase a woman’s risk of developing breast cancer is age.4 Breast cancer in women has been observed to occur at or over the age of 50. A positive personal or family history is also known to pose an increased risk of developing breast cancer. In addition, a long menstrual life or the use of hormone replacement therapy after menopause increases the risk of developing breast cancer.5
Growth factors and their receptors play pivotal roles in the regulation of epithelial cell growth and differentiation.6 The HER/erbB family of receptors includes the EGF receptor (EGFR, HER1, and erbB-1), the orphan HER2/neu (erbB-2), and the neuregulin/neuregulin receptors HER3 (erbB-3) and HER4 (erbB-4). Increased knowledge about the pathophysiological mechanisms of breast cancer has led to exponential growth in the identification of biomolecular markers. In this study, a rational approach has been utilized to research the more widely recognized molecular targets in breast cancer. Although there are FDA-approved drugs for breast cancer treatment, their use is accompanied by various side effects. Therefore, it is important to devise an alternative treatment that not only treats the tumor but also has no side effects.
Members of EGF family specifically interact with the EGF receptor (EGFR), which is a cell surface receptor.7 Binding of specific ligands such as EGF and transforming growth factor α activates EGFR. PI3 kinase, Ras-Raf-MAPK, JNK, and PLCγ mediate the major EGFR signaling pathways.8,9 Loss of cell polarization is one of the main features observed when most of the members of the EGFR family are activated, leading to scattering and invasion of breast epithelial cells.10 Metastasis and angiogenesis processes have been found to be linked with the dysregulation of EGFR pathways. Furthermore, in many human malignancies, this has been observed to be the reason behind the poor prognosis.11–13 One of the mechanisms of EGFR overexpression is an amplification of the EGFR gene, which has been implicated in oligodendroglioma,14 glioblastoma, lung cancer,15 gastric cancer, and breast cancer.16 In breast cancers, EGFR gene amplification has been observed in 0.8–14% of tumors.17,18 However, gene amplification has been shown in ~25% of cases of metaplastic breast cancer – a specific phenotype of TNBC.19,20
The neu gene encodes a protein HER2, similar in structure as human EGFR.21,22 The HER2 receptor plays a significantly important role in the cell growth and differentiation process, an overexpression of which is associated with the development of human cancers including breast, ovarian, and gastrointestinal (GI) tract cancers.23,24 HER2 is overexpressed in 15–30% of invasive breast cancers, which has both prognostic and predictive implications. A 40- to 100-fold increase in the expression of HER2 protein results in the tumor cell surface containing ~2 million receptors. Approximately 20–50 HER2-gene copies have been found in breast cancers.25 HER2 gene amplification is associated with shorter disease-free and overall survival in breast cancer. The prognostic significance of HER2 amplification has been established in 189 human breast cancers.26 HER2 amplification is one of the early events in human breast tumorigenesis. As per evidence, during progression to invasive disease and nodal or distant metastasis, HER2 status is maintained.27 An increased resistance to certain hormonal agents, enhanced propensity to metastasize to the brain, and high sensitivity to certain cytotoxic chemotherapeutic agents have been found to be associated with breast cancers resulting from an overexpression of HER2.28
Heat shock proteins (HSPs) are members of the molecular chaperones that play an essential role in the folding of cellular proteins.29,30 Furthermore, through inhibition of programmed cell death and cell senescence during hyperthermia, HSPs directly participate in cell survival.31–33 In carcinogenesis, HSPs have a role in helping cells (a) to escape tumor suppression pathways, (b) in becoming treatment resistant, (c) in progressing to an advanced stage of cancer, and (d) for facilitating metastasis.34 Tumor suppressor protein (p53), ER, HIF-1alpha, Raf-1 MAP kinase, and a variety of receptor tyrosine kinases, such as HER2, bind with HSP90. These interactions play a key role in breast neoplasia.35 Breast ductal carcinomas indicate an increased HSP90 expression,36–38 whereas lobular carcinomas and lobular neoplasia manifest significantly decreased HSP90 expression.39 Breast cancer cells become resistant to stress stimuli through HSP90 overexpression. Therefore, therapeutic opportunities in treating cancer can be devised by the pharmacological inhibition of these targets.
In silico approaches have paved the way to solve many biological problems,40–42 which have led to the identification of novel inhibitors against numerous diseases.43–47 In this investigation, active compounds against these three targets were screened by structure-based virtual screening (VS) to identify potential virtual hits. The molecular docking tool, AutoDockVina (AD Vina),48 was used to dock 50 filtered compounds against EGFR, HER2, and HSP90. The ligands were also analyzed for their profile of absorption, distribution, metabolism, excretion, and toxicity (ADMET), which determined the ADMET efficiency of the drug. Potential hits that indicate good pharmacokinetic (PK) and pharmacodynamic (PD) properties have a better chance to be future drugs. The results of the current study concluded that five multi-targeted compounds with high binding energies as well as a good ADMET profile against all three targets be taken into consideration, suggesting them as potential hits for drug development against breast cancer after testing through in vitro experiments.
Materials and methods
Binding-site analysis
High-resolution crystals with experimentally identified drug sites for EGFR, HSP90, and HER2 were used. Binding pockets of these proteins were examined from crystal structures and were further evaluated using the CASTp server (Computed Atlas of Surface Topography of Proteins).49 It locates all likely binding pockets, and the algorithm critically determines the binding pocket and possible cavities in a solvent-accessible surface area.
Protein dataset
X-ray crystallographic structures of EGFR (Protein Data Bank [PDB] ID: 1M17) in complex with the 4-anilinoquinazoline inhibitor erlotinib (resolution: 2.6 Å; R-value free: 0.295), HSP90 complexed with ganetespib (PDB ID: 3TUH; resolution: 1.8 Å; R-value free: 0.213), and HER2 in complex with TAK-285 (PDB ID: 3RCD; resolution: 3.21 Å; R-value free: 0.294) were retrieved from PDB. The criteria for choosing PDBs were (a) minimum resolution and (b) conformation of docked ligand being the same as in the crystallized structure after the redocking procedure.
Protein preparation
To prepare the selected proteins for docking, co-crystallized water molecules, small molecules, nonpolar hydrogens, lone pairs, and nonstandard residues were deleted, and hydrogens and Gasteiger charges were added and merged. Energy minimization and geometry optimization of all structures were performed using Dock Prep, a built-in tool for preparing structures before docking, in UCSF Chimera 10.1 (Resource for Biocomputing, Visualization, and Informatics, University of California San Francisco, San Francisco, CA, USA). The energy was minimized for the 1000 steepest descent steps at a root means square gradient of 0.02, an update interval of 10, and with an Amber ff12SB force field using UCSF Chimera 10.1.50 Furthermore, incomplete side chains were repaired using the Dunbrack rotamer library. Hydrogens were added to reasonably generate protonation states at physiological pH. Potentially ambiguous or rare protonation states, especially in binding sites and nonstandard residues, were verified and corrected before charges were added. Charges were added to assign partial charges to atoms. Partial charges were assigned as an atom attribute named charge and included in the Mol2 file (molecule model 3D atomic coordinate files and metadata).
Preparation of chemical library
E-molecule databases and chemical companies including PubChem and Active ZINC were ultilized for literature analysis. All information on chemical compounds was merged, thus obtaining an overall database of ~3 million chemical compounds. Duplicated structures were removed to acquire new scaffolds through InChlKey generated by open babel. Drugs presenting poor PK properties failed to continue progression in the drug development process. The compounds were checked for their oral bioavailability alongside PK parameters such as the blood–brain barrier (BBB), better human intestinal absorption (HIA), good solublity, and low toxicity through the application of Lipinski’s Ro5 (LRo5).40 Compounds violating this rule were eliminated from the study. Using Open Bable, the two-dimensional (2D) files of structures that followed this criteria were transformed into three-dimensional (3D) format and saved as molecule files. Discovery Studio® (DS) visualizer (Accelrys Inc., San Diego, CA, USA)51 was used for energy minimization during the preparation of ligand molecules.
Virtual screening
VS is a computational technique used to screen potential compounds against specific target protein from chemical compound libraries. The aim was to reduce the number of druggable compounds, inhibiting target proteins more efficiently, to a manageable number. Structure-based virtual screening (SBVS) was utilized to screen substantial databases in the quest of finding novel and potential lead compounds. SBVS estimates the likelihood of a ligand to bind efficiently to the target protein. It involves the application of a scoring function preceeding docking of candidate ligands into a protein target with high binding energies.
In the current study, the drug discovery platform Mcule was utilized to employ SBVS.52 For this, an Mcule database of filtered compounds was created manually. Mcule48 has a built-in AD Vina tool, which was used to perform molecular docking. In SBVS, molecular docking predicts the binding conformation of small-molecule ligands to the binding site of the selected target. Characterization of the binding behavior plays an essential role in the rational design of drugs as well as to elucidate fundamental biochemical processes. Before VS, an in-depth analysis of the protein structures was performed to understand the binding pocket of target proteins. Using the AutoGrid program of Chimera, a grid map with dimensions of 30 × 30 × 30 Å was generated that covered the binding pocket of each protein. For SBVS, each ligand was docked in the respective binding site of EGFR, HER2, and HSP90, and was scored by maximum binding affinity.
In silico ADMET assessment
Prediction of the ADMET profile for drug candidates and environmental chemicals plays a major role in drug discovery and environmental hazard assessment. Top virtual hits from VS against each protein were further filtered through PK and PD parameters and evaluation of favorable ADMET parameters. To identify the possible adverse effects of these compounds in humans, the ADMET properties of the filtered compounds were predicted using various tools that include the OSIRIS property explorer (Actelion Pharmaceuticals Ltd., Allschwil, Switzerland) which highlights undesired effects if posed by the compound being analyzed, admetSAR (http://lmmd.ecust.edu.cn:8000/) for prediction of quantitative structure–activity relationship-based ADMET properties, and Molsoft (Molsoft LLC, La Jolla, CA, USA) for calculation of drug-likeness properties. ADMET parameters include the BBB, HIA, P-glycoprotein substrates and inhibitors, renal organic cation transporter (ROCT), cytochrome P (CYP) inhibitory promiscuity, and toxicity risks.
Results and discussion
The aim of the current study was to elucidate alternative inhibitory compounds against EGFR, HER2, and HSP90 proteins. In silico VS of ~3 million compounds was performed by establishing a pipeline of LRo5 and PK properties to assess drug likeness. Compounds that showed a strong binding affinity for EGFR, HER2, and HSP90 proteins were selected for further investigations. Further, the ADMET profile was analyzed to filter top hits to identify suitable virtual hit candidate compounds. The current in silico study was undertaken to identify efficient anti-breast cancer compounds. The five multi-targeted top compounds were also evaluated for their PK and PD properties. The virtual hits identified in this study can be used as an alternative targeting agent for breast cancer after being tested through in vitro experiments.
Binding-site evaluation
The crystal structure of the kinase domain of the EGFR in complex with 4-anilinoquinazoline inhibitor erlotinib (PDB ID: 1M17) was obtained from PDB. It was found that residues Leu694, Leu768, Met769, Gly772, and Leu820 formed significant interactions with the inhibitor. The crystal structure of HER2 complexed with TAK-285 (PDB ID: 3RCD) indicated Ala751, Leu800, Met801, Leu852, and Asp863 to be among the significant interacting residues. The X-ray co-crystallized structure of TAK-285 with HER2 demonstrated that it interacts with the expected residues in the respective ATP pocket. HSP90 complexed with ganetespib (PDB ID: 3TUH) indicated that the residues Asn51, Lys58, Asp93, Gly97, and Thr184 form significant interactions at the binding site. The binding-site residues of all three target proteins were also confirmed by using CASTp. Binding-site interacting residues, as reported in the PDB, are shown in Figure 1.
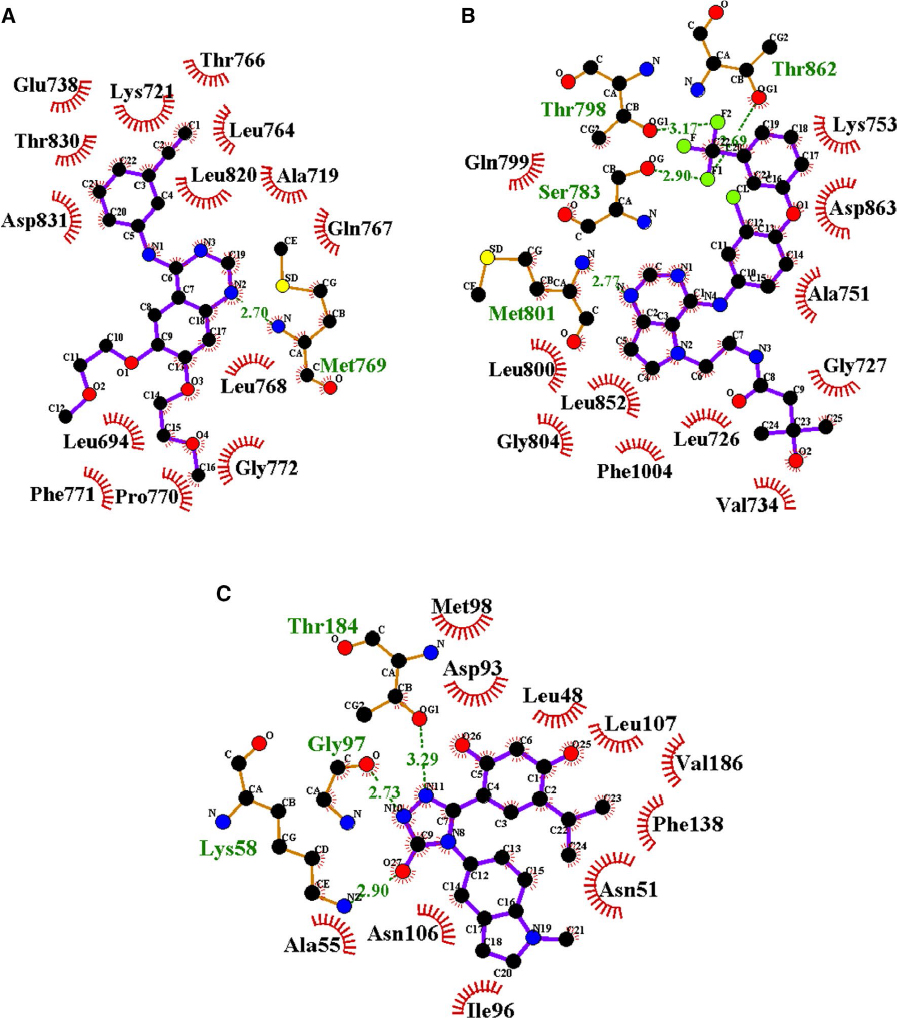 | Figure 1 (A) Epidermal growth factor receptor (EGFR) tyrosine kinase domain with 4-anilinoquinazoline inhibitor erlotinib. (B) Novel HER2/EGFR dual. Inhibitors bearing a pyrrolo[3,2-d] pyrimidine scaffold. (C) The crystal structure of the N-terminal domain of an HSP90 in the presence of an inhibitor ganetespib. Notes: Green dashed lines indicate hydrogen bonds with distance in Angstrom (Å), spoked red arcs indicate hydrophobic contacts, atoms are shown in black for carbon, blue for nitrogen, red represents oxygen, yellow represents sulfur, and green represents fluorine. |
Structure-based virtual screening
To identify anti-breast cancer compounds, a database of ~3 million compounds was screened by the application of a series of filters (LRo5, drug likeness, PK filters), which led us to select the top 66, 832 best compounds. These compounds were subjected to SBVS against EGFR, HER2, and HSP90 separately to identify multi-targeted compounds common among all these targets. The flowchart of multistep VS is shown in Figure 2. The docking compounds were ranked based on their highest binding energy with the corresponding protein. To identify potential hits having binding affinities greater than the reported co-crystallized inhibitor, binding energy cutoffs of −8.9, −8.5, and −8.3 kcal/mol were set against EGFR, HER2, and HSP90, respectively, after redocking with the reported co-crystallized inhibitor as deposited in the PDB to select only those hits which were above that specific threshold. Only top-ranked hits with higher binding energy for all three proteins were considered and investigated further.
 | Figure 2 Schematic workflow summarizing the screening of active compounds against breast cancer. Abbreviations: AD Vina, AutoDockVina; PD, pharmacodynamic; PDB, Protein Data Bank; PK, pharmacokinetic. |
Binding energy cutoff value was applied for VS to select only the high-ranked compounds. A total of 96 hits with binding energies lower than −8.9 kcal/mol for EGFR, 114 hits with <−8.5 kcal/mol for HER2, and 79 hits with <−8.3 kcal/mol for HSP90 were chosen based upon the AD Vina docking score (∆G). Visual inspection was carried out to screen these 289 compounds further. As a result, 71 compounds with promising high binding energies against all three targets were selected. An extensive ADMET analysis was performed to screen these virtual hits, and their molecular interactions with binding-site residues were carefully analyzed.
Prediction of ADMET properties
Molecular descriptors are the deciding factor for PK properties and toxicity of a compound. ADMET properties predicted in silico identify the likelihood of compounds to be used as human therapeutic agent.53 The admetSAR online server was utilized for calculating the ADMET properties of 71 common compounds of all three proteins. It is important for compounds to have a promising ADMET profile. The BBB,54 HIA,55 aqueous solubility,56 Caco-2 cell permeability, CYP450 inhibition,57 and Ames toxicity were calculated for 71 compounds. It was observed that only 15 virtual hits were successful at passing through these ADMET filters. These 15 virtual hits were further filtered through a series of drug safety profiling parameters. Undesirable moieties and substructures causing potential toxicity were eliminated through a series of PAINS (pan assay interference compounds) filters. Among the 15, only five compounds were unidentified by PAINS-1, -2, and -3.
The admetSAR predictions indicated that the ability to penetrate the BBB and HIA was portrayed by all compounds; in addition, all the compounds were revealed as non-inhibitors of the P-gp inhibitor except S-258002927. None of the compounds indicated any inhibitory effects on the ROCT. CYP enzymes, including various CYP450 substrates and inhibitors, play a fundamental role in drug metabolism. The results showed that most compounds were non-inhibitors of CYP450 enzymes. A significant finding was observed for compounds S-258012947 and S-258002927 that indicated low CYP-inhibitory promiscuity, being non-inhibitors of all CYP450 enzymes including 1A2, 2C9, 2D6, 2C19, and 3A4. Further drug metabolism analyses showed these compounds were non-substrates of two CYP450 substrates (2C9 and 2D6). The ADMET properties of the top five best virtual hits as common against all three targets are tabulated in Table 1, and their 2D structures are presented in Figure 3.
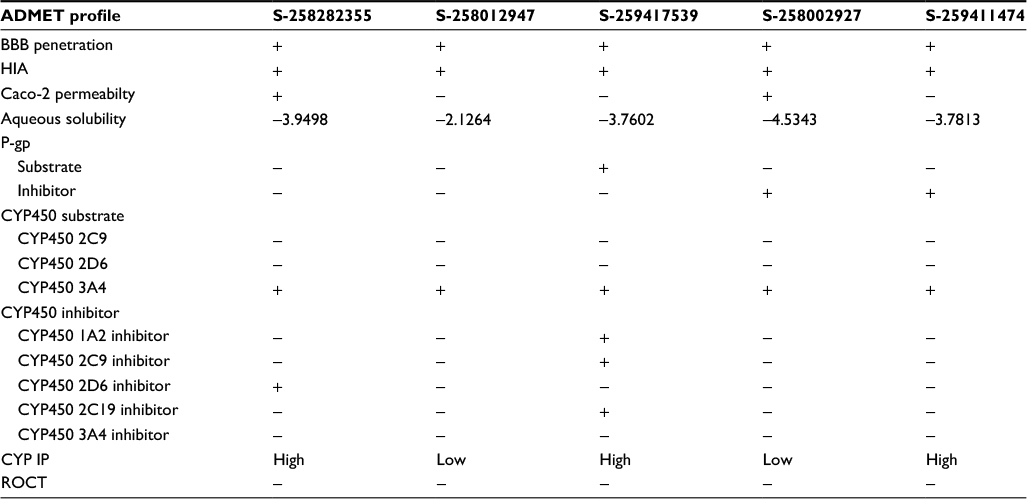 | Table 1 ADMET profile of potential five multi-targeted virtual hits Abbreviations: ADMET, absorption, distribution, metabolism, excretion, and toxicity; BBB, blood–brain barrier; HIA, human intestinal absorption; CYP, cytochrome P; IP, inhibitory promiscuity; ROCT, renal organic cation transporter; +, present; −, not present. |
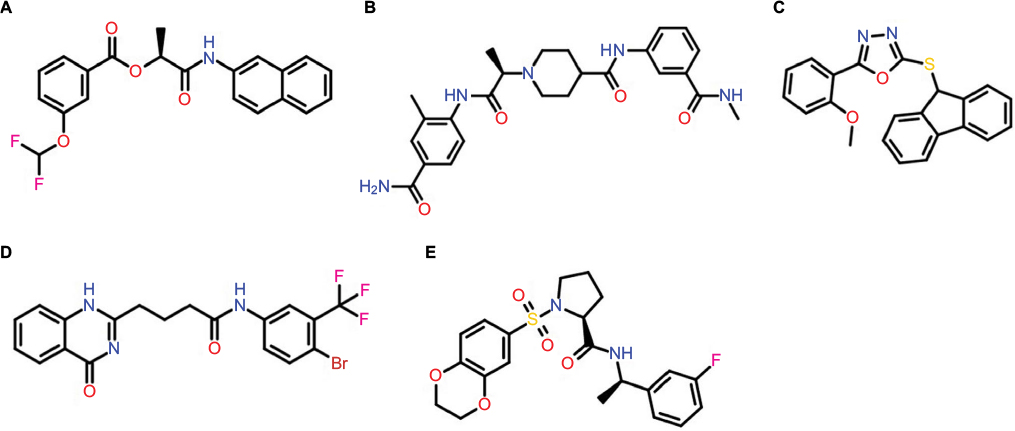 | Figure 3 Chemical structures of lead compounds: (A) S-258002927; (B) S-258012947; (C) S-258282355; (D) S-259411474; (E) S-259417539. |
Molecular docking studies of EGFR
All five compounds showed high binding energies with EGFR ranging from −8.9 to −9.7 kcal/mol with AD Vina, as presented in Table 2. All compounds established a network of molecular interactions (H-bonds, Van der Waals [VdW], alkyl, π-alkyl, and π-sigma bonds) with the active-site residues of EGFR when analyzed in 2D plots as shown in Figure 4. S-259417539 formed various binding interactions including H-bonds (Thr766), π-alkyl (Ala719), alkyl (Val702), and VdW (Leu694, Cys751, Asp831, Lys721, Leu764, Met742, Glu738, Asn818, Arg817). S-259411474 established H-bond and alkyl bond with Met769 and Leu820, π-alkyl with Ala719 and Val702, and VdW interactions with Ile720, Ile765, Leu768, Leu764, Leu753, Thr830, Asp831, Gly695, Glu738, Cys773, Phe699, and Gly772, respectively. Likewise, S-258012947 was involved in various binary interactions including H-bond (Asp831), π-alkyl (Leu694), and VdW (Ile720, Ile765, Cys773, Leu834, Gly833, and Thr765), whereas S-258282355 and S-258002927 were found to interact with Asn818 and Asp831 through H-bond, with residues Leu694, Leu820, and Val702 by forming alkyl bonds, and with residues Arg817, Thr766, Gln767, Leu768, Met769, Gly772, and Cys773, and Gly695, Gly772, Arg817, Cys773, Leu820, Ala719, Met769, Leu768, Phe699, Thr830, Leu764, Glu738, Gln767, and Met742 through VdW interactions, respectively.
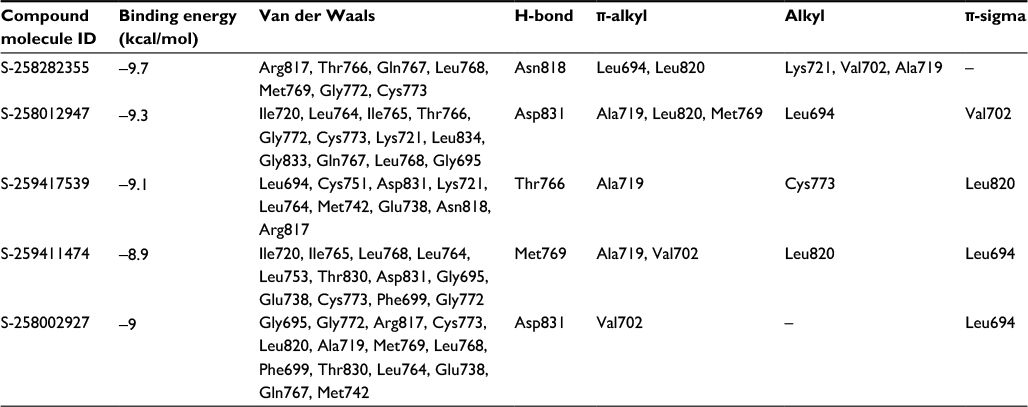 | Table 2 Binding energy and molecular interaction of top five multi-targeted compounds with EGFR |
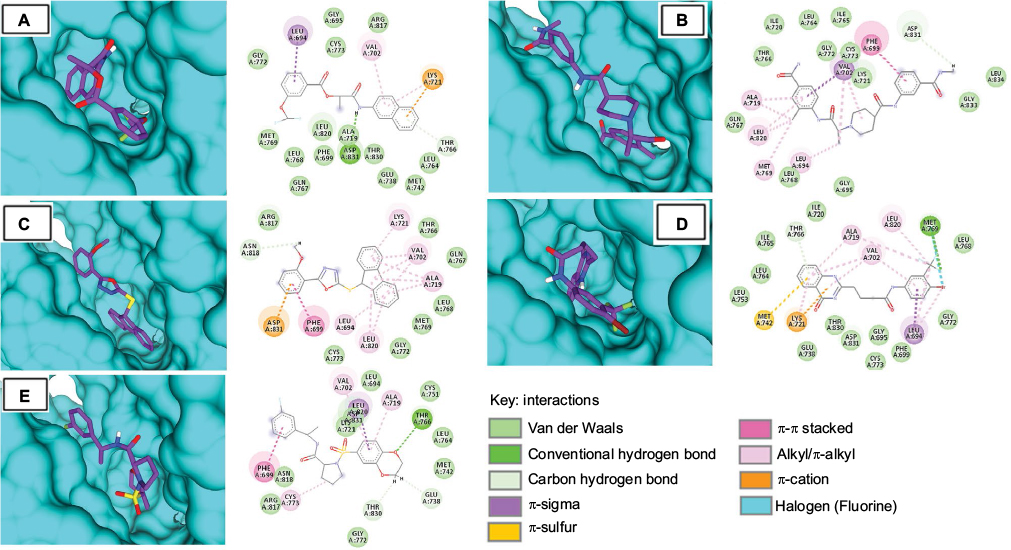 | Figure 4 Molecular surface representation of EGFR with top-ranked multi-targeted virtual hits: (A) S-258002927; (B) S-258012947; (C) S-258282355; (D) S-259411474; (E) S-259417539. Notes: Molecular surface representation of the EGFR binding pocket (in blue) with respective ligands in stick format (in purple). Alongside each 3D complex, 2D interaction plots indicate important binding-site interactions between respective ligands and binding-site residues. |
Molecular docking studies of HER2
The top hits represented high binding energies with HER2 ranging from −9.4 to −10.3 kcal/mol, as shown in Table 3. Post-docking analysis of HER2 revealed that all five compounds (S-259417539, S-259411474, S-258012947, S-258002927, and S-258282355) were also found to bind inside the kinase domain, surrounded by interacting residues with noticeably high binding energy value that ranged from −9.4 to −10.3 kcal/mol (Figure 5). The ligand S-258282355 was found not to form any H-bond, but alkyl bonds with Leu694 and Leu820 were observed as were π-alkyl bonds with Lys721, Val702, and Ala719 residues, whereas VdW with Ile752, Leu726, Leu800, Thr798, Ser783, Arg784, Thr862, Leu852, Asp863, Phe864, Leu755, Gly865, and Glu770 were found. The S-258012947 established H-bonds (Asp863), π-alkyl (Leu694), alkyl (Ala719, Leu820, and Met769), and VdW (Asn850, Phe731, Arg849, Gly729, Phe864, Gly727, Ser783, Cys805, Gly804, Leu800, Met801, Thr798, Leu785, Val797, and Ile752). Similarly, H-bond for S-258002927 was found to form H-bonds with Asp863 and Cys805, π-alkyl with Ala719, π-sulfur with Met774, alkyl bonds with Leu764 and Lys721, and VdW with Ile752, Thr798, Thr862, Ser783, Arg784, Gln799, Val734, Met801, Leu800, Leu726, Phe1004, and Gly804. S-259411474 and S-259417539 were found to form H-bonds with Thr862, Thr798, Leu785, Ser783, and Asp863, and Lys753, respectively, π-alkyl with Val702, Ala719, and Cys773, alkyl with Leu820, Val702, Ala719, and VdW with Met801, Gly804, Leu800, Ile752, Val734, Asp863, Lys753, Val797, and Asn850, and Phe731, and Arg849, Gly729, Phe864, Gly727, Ser783, Cys805, Gly804, Leu800, Met801, Thr798, Leu785, Val797, and Ile752 residues, respectively.
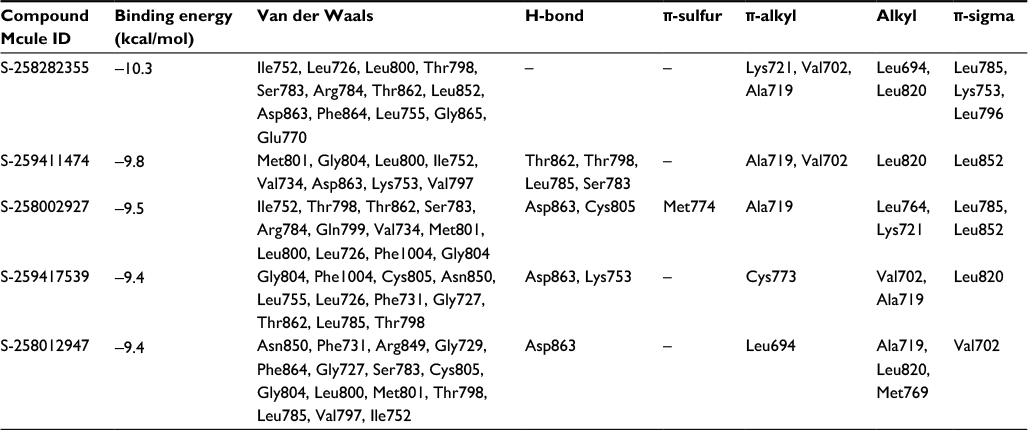 | Table 3 Binding energy and molecular interaction of top five multi-targeted compounds with HER2 |
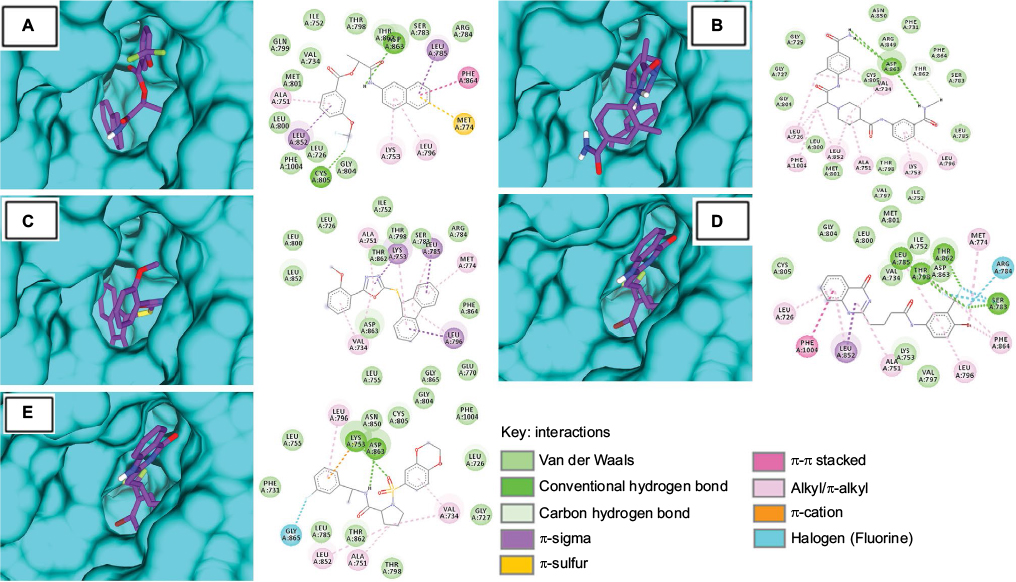 | Figure 5 Molecular surface representation of HER2 with respective ligands: (A) S-258002927; (B) S-258012947; (C) S-258282355; (D) S-259411474; (E) S-259417539. Notes: Molecular surface representation of HER2 binding pocket (in blue) with respective ligands in stick format (in purple). Alongside each 3D complex, 2D interaction plots indicate important binding-site interactions between respective ligands and binding-site residues. |
Molecular docking studies of HSP90
The binding energies of top hits with HSP90 ranged from −8.7 to −9.4 kcal/mol, as represented in Table 4, and binary interactions are displayed in Figure 6. The binding interactions of S-258002927 with HSP90 included H-bond with Thr184, π-alkyl with Ala55, π-sulfur with Met98, and VdW with residues Val186, Leu48, Val150, Ser52, Leu107, Phe138, Asn51, Val136, Tyr139, Gly135, Gly97, Ile96, Lys58, Asp54, Asn106, Lys112, Ala111, and Ser113. H-bond for S-258012947 was found with Ala111, alkyl bond with Ala111 residue, and VdW with Ile91, Ile26, Val186, Ala55, Thr184, Met98, Ser113, Tyr139, Gly135, Val150, Asn106, and Leu107 residues. S-258282355 did not form any H-bonds; π-alkyl bond was formed with residues Met98 and Ala55, alkyl with Val136 and Ala111, π-sulfur with Met98, and VdW with Thr109, Ile110, Lys112, Leu48, Ser113, Ile26, Leu107, Tyr139, Gly137, Val186, Phe138, Thr184, Asn106, Lys58, Ile96, and Gly97. S-259411474 and S-259417539 were observed to form H-bonds with Asn51 and Lys58, respectively, π-alkyl with Ala111, alkyl bond with Leu107, Phe138, Val186, Leu48, and Ala55, π-sulfur with Met98, and VdW with Ile26, Thr109, Tyr139, Val150, Val136, Asn106, Gly135, Ser113, Ile110, Lys112, Lys58, Gly97, Ser52, Asp93, Ile96 and Val186, and Ser52, Ala55, Met98, Val150, Asn51, Asp54, Thr184, Asp93, Phe138, Leu107, Gly137, Val136, Tyr139, Ser113, Lys112, and Ile110, respectively.
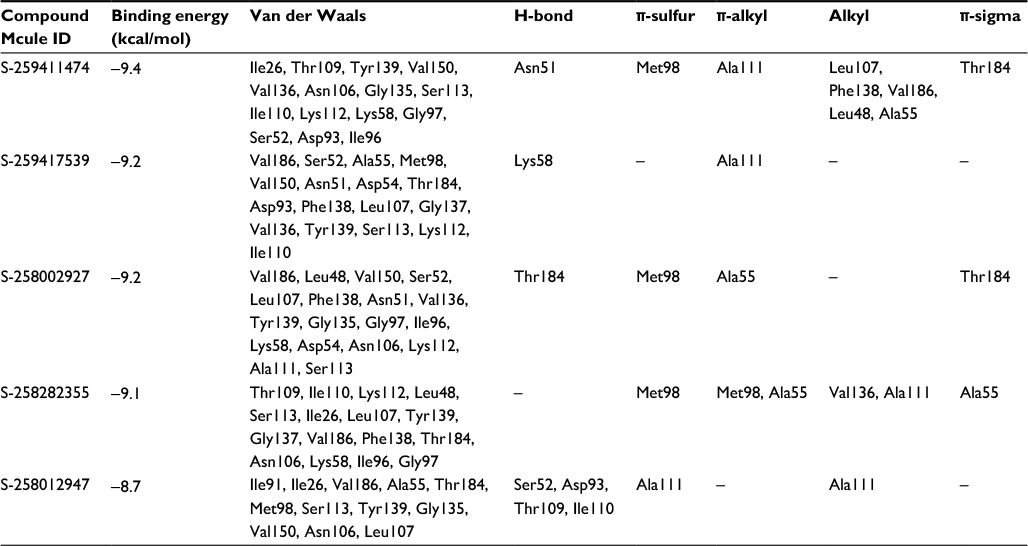 | Table 4 Binding energy and molecular interaction of top five multi-targeted compounds with HSP90 |
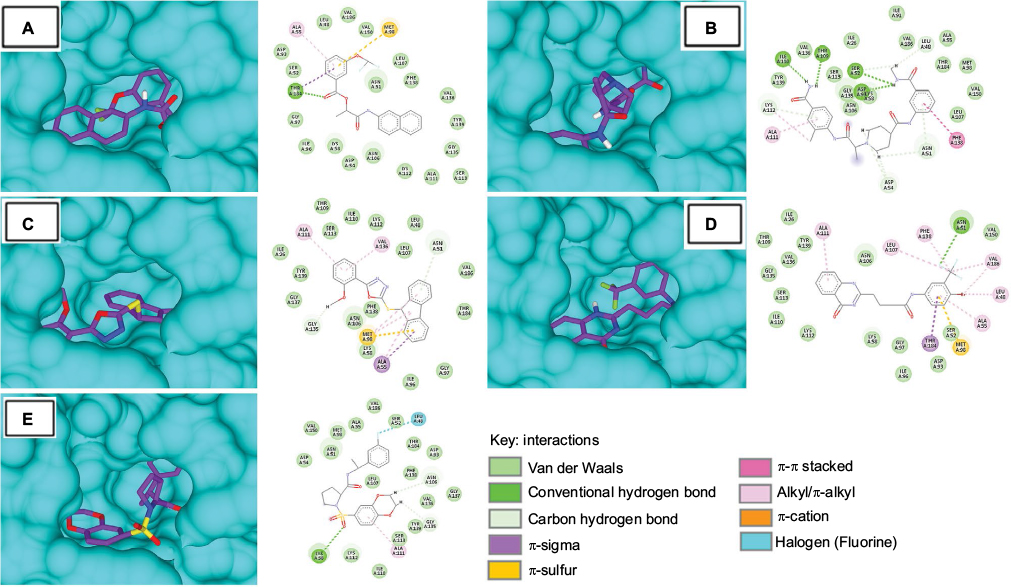 | Figure 6 Molecular surface representation of HSP90 with respective ligands: (A) S-258002927; (B) S-258012947; (C) S-258282355; (D) S-259411474; (E) S-259417539. Notes: Molecular surface representation of HSP90 binding pocket (in blue) with respective ligands in stick format (in purple). Alongside each 3D complex, 2D interaction plots indicate important binding-site interactions between respective ligands and binding-site residues. |
The current study is an effort to identify anti-breast cancer compounds that may be considered for drug development to treat breast cancer. The SBVS of ~3 million compounds against three EGFR, HER2, and HSP90 proteins has assisted in robust screenings of most potential virtual hits. Based on the experimentally determined EGFR, HSP90, and HER2 inhibitors from previous studies, binding energy cutoff to select for possible 96, 79, and 114 virtual hits that could act on EGFR, HSP0, and HER2 proteins, respectively, was set. These virtual hits followed: (a) LRo5 and drug likeness; the compounds violating more than one drug-like parameter indicated that they might lead to issues with bioavailability, and hence, such compounds were not included in the study; (b) good HIA and BBB for moderately lipophilic compounds to cross the BBB, whereas polar molecules are poor central nervous system drugs; and (c) comprehensive ADMET profile. The best possible orientations forming stable ligand–target protein complexes, through the process of molecular docking, were achieved.
The binding energy of the ligand and its half-life quantifies the efficiency of the ligand–protein complex. The rate of dissociation is rapid for weaker interactions; however, the rate of dissociation slows down with an increase in the value of binding energy.58 Compounds with a longer half-life have strong binding energies, and hence, they take a longer time to dissociate. Strong interactions between residues imply that binding with inhibitory compounds may be stable, leading to an inhibitory reaction. The top five best virtual hits in complex with EGFR, HER2, and HSP0 revealed strong binding affinities and highlighted several H-bonds and hydrophobic interactions between functional groups, and side chains of essential residues. Post-docking analysis of EGFR, HER2, and HSP90 with these hits disclosed more in-depth details. The EGFR that complexed with S-259429764 showed the highest binding energy (−10.7 kcal/mol). Other compounds showed binding energy in the range from −10.7 to −8.7 kcal/mol. Moreover, common interacting residues that displayed VdW and H-bond with different compounds were Gln767, Leu768, Gly772, Cys773, Leu764, Gly695, and Thr830, respectively (Table 2). However, the HER2 complexed with S-258282355 showed the highest binding energy (−10.3 kcal/mol), and other compounds showed binding energy in the range from −9.8 to −8.9 kcal/mol. Together with this, the common interacting residues that displayed VdW and H-bond with different compounds were Ile752, Leu726, Leu800, Thr798, Ser783, Thr862, Met801, and Asp863, respectively (Table 3). Likewise, HSP90 complexed with S-259411474 showed the highest binding energy (−9.4 kcal/mol) and displayed H-bonds with Asn106 and Ser50. Other compounds showed binding energy in the range from −9.4 to −8.3 kcal/mol. Interestingly, all compounds interacted equally in terms of VdW interactions with Tyr139 and Ser113 of HSP90. Other common interacting residues were Val150, Asn106, Lys112, Val136, Gly135, Ile110, Lys58, Ser52, and Gly97 that displayed VdW interactions with different compounds (Table 4).
Furthermore, all screened virtual hits predicted promising ADMET profile, and their strong binding affinity stipulated the multi-targeted potential of these top virtual hits.
Conclusion
Breast cancer is one of the leading cancer types in women across the globe. Computational tools have been widely used for drug development and discovery of multi-targeted inhibitors of many overexpressed proteins induced in breast cancer. This study reveals five multi-targeted compounds that possess high binding energies against most common target proteins that are involved in breast cancer. With excellent PK and PD properties as predicted, these virtual hits may be considered for early drug development against breast cancer after being tested through in vitro and in vivo studies.
Disclosure
The authors report no conflicts of interest in this work.
References
Salomon DS, Brandt R, Ciardiello F, Normanno N. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol. 1995;19(3):183–232. | ||
Goldsmith YB, Roistacher N, Baum MS. Capecitabine-induced coronary vasospasm. J Clin Oncol. 2008;26(22):3802–3804. | ||
DeSantis C, Ma J, Bryan L, Jemal A. Breast cancer statistics, 2013. CA Cancer J Clin. 2014;64(1):52–62. | ||
Li CI, Beaber EF, Tang MT, Porter PL, Daling JR, Malone KE. Reproductive factors and risk of estrogen receptor positive, triple-negative, and HER2-neu overexpressing breast cancer among women 20-44 years of age. Breast Cancer Res Treat. 2013;137(2):579–587. | ||
Anderson KN, Schwab RB, Martinez ME. Reproductive risk factors and breast cancer subtypes: a review of the literature. Breast Cancer Res Treat. 2014;144(1):1–10. | ||
Aaronson SA. Growth factors and cancer. Science. 1991;254(5035):1146–1153. | ||
Herbst RS, Johnson DH, Mininberg E, et al. Phase I/II trial evaluating the anti-vascular endothelial growth factor monoclonal antibody bevacizumab in combination with the HER-1/epidermal growth factor receptor tyrosine kinase inhibitor erlotinib for patients with recurrent non-small-cell lung cancer. J Clin Oncol. 2005;23(11):2544–2555. | ||
Downward J, Yarden Y, Mayes E, et al. Close similarity of epidermal growth factor receptor and v-erb-B oncogene protein sequences. Nature. 1984;307(5951):521–527. | ||
Schulze WX, Deng L, Mann M. Phosphotyrosine interactome of the ErbB-receptor kinase family. Mol Syst Biol. 2005;1:2005.0008. | ||
Wang F, Weaver VM, Petersen OW, et al. Reciprocal interactions between beta1-integrin and epidermal growth factor receptor in three-dimensional basement membrane breast cultures: a different perspective in epithelial biology. Proc Natl Acad Sci U S A. 1998;95(25):14821–14826. | ||
Yarden Y, Schlessinger J. Epidermal growth factor induces rapid, reversible aggregation of the purified epidermal growth factor receptor. Biochemistry. 1987;26(5):1443–1451. | ||
Lurje G, Lenz HJ. EGFR signaling and drug discovery. Oncology. 2009;77(6):400–410. | ||
Martinazzi M, Crivelli F, Zampatti C, Martinazzi S. Relationships between epidermal growth factor receptor (EGF-R) and other predictors of prognosis in breast carcinomas. An immunohistochemical study. Pathologica. 1993;85(1100):637–644. | ||
Fallon KB, Palmer CA, Roth KA, et al. Prognostic value of 1p, 19q, 9p, 10q, and EGFR-FISH analyses in recurrent oligodendrogliomas. J Neuropathol Exp Neurol. 2004;63(4):314–322. | ||
Giaccone G. Epidermal growth factor receptor inhibitors in the treatment of non-small-cell lung cancer. J Clin Oncol. 2005;23(14):3235–3242. | ||
Al-Kuraya K, Schraml P, Torhorst J, et al. Prognostic relevance of gene amplifications and coamplifications in breast cancer. Cancer Res. 2004;64(23):8534–8540. | ||
Ro J, North SM, Gallick GE, Hortobagyi GN, Gutterman JU, Blick M. Amplified and overexpressed epidermal growth factor receptor gene in uncultured primary human breast carcinoma. Cancer Res. 1988;48(1):161–164. | ||
Spyratos F, Delarue JC, Andrieu C, et al. Epidermal growth factor receptors and prognosis in primary breast cancer. Breast Cancer Res Treat. 1990;17(2):83–89. | ||
Yatabe Y, Kosaka T, Takahashi T, Mitsudomi T. EGFR mutation is specific for terminal respiratory unit type adenocarcinoma. Am J Surg Pathol. 2005;29(5):633–639. | ||
Weber F, Fukino K, Sawada T, et al. Variability in organ-specific EGFR mutational spectra in tumour epithelium and stroma may be the biological basis for differential responses to tyrosine kinase inhibitors. Br J Cancer. 2005;92(10):1922–1926. | ||
Coussens L, Yang-Feng TL, Liao YC, et al. Tyrosine kinase receptor with extensive homology to EGF receptor shares chromosomal location with neu oncogene. Science. 1985;230(4730):1132–1139. | ||
King CR, Kraus MH, Aaronson SA. Amplification of a novel v-erbB-related gene in a human mammary carcinoma. Science. 1985; 229(4717):974–976. | ||
Hynes NE, Stern DF. The biology of erbB-2/neu/HER-2 and its role in cancer. Biochem Biophys Acta. 1994;1198(2–3):165–184. | ||
Klapper LN, Kirschbaum MH, Sela M, Yarden Y. Biochemical and clinical implications of the ErbB/HER signaling network of growth factor receptors. In: Klein G, Woude V, editors. Advances in Cancer Research. New York: Academic Press; 2000:25–79. | ||
Kallioniemi A, Kallioniemi OP, Sudar D, et al. Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science. 1992;258(5083):818–821. | ||
Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235(4785):177–182. | ||
Park IH, Ro J, Lee KS, Nam BH, Kwon Y, Shin KH. Trastuzumab treatment beyond brain progression in HER2-positive metastatic breast cancer. Ann Oncol. 2009;20(1):56–62. | ||
Gabos Z, Sinha R, Hanson J, et al. Prognostic significance of human epidermal growth factor receptor positivity for the development of brain metastasis after newly diagnosed breast cancer. J Clin Oncol. 2006;24(36):5658–5663. | ||
Lindquist S, Craig EA. The heat-shock proteins. Ann Rev Genet. 1988;22:631–677. | ||
Bukau B, Weissman J, Horwich A. Molecular chaperones and protein quality control. Cell. 2006;125(3):443–451. | ||
Tang D, Khaleque MA, Jones EL, et al. Expression of heat shock proteins and heat shock protein messenger ribonucleic acid in human prostate carcinoma in vitro and in tumors in vivo. Cell Stress Chaperones. 2005;10(1):46–58. | ||
Jones SE, Erban J, Overmoyer B, et al. Randomized phase III study of docetaxel compared with paclitaxel in metastatic breast cancer. J Clin Oncol. 2005;23(24):5542–5551. | ||
Gabai VL, Meriin AB, Yaglom JA, Volloch VZ, Sherman MY. Role of Hsp70 in regulation of stress-kinase JNK: implications in apoptosis and aging. FEBS Lett. 1998;438(1–2):1–4. | ||
Calderwood SK, Ciocca DR. Heat shock proteins: stress proteins with Janus-like properties in cancer. Int J Hyperthermia. 2008;24(1):31–39. | ||
Beliakoff J, Whitesell L. Hsp90: an emerging target for breast cancer therapy. Anticancer Drugs. 2004;15(7):651–662. | ||
Conroy SE, Latchman DS. Do heat shock proteins have a role in breast cancer? Br J Cancer. 1996;74(5):717–721. | ||
Yano M, Naito Z, Tanaka S, Asano G. Expression and roles of heat shock proteins in human breast cancer. Jpn J Cancer Res. 1996;87(9):908–915. | ||
Pick E, Kluger Y, Giltnane JM, et al. High HSP90 expression is associated with decreased survival in breast cancer. Cancer Res. 2007; 67(7):2932–2937. | ||
Zagouri F, Sergentanis TN, Nonni A, et al. Hsp90 in the continuum of breast ductal carcinogenesis: evaluation in precursors, preinvasive and ductal carcinoma lesions. BMC Cancer. 2010;10:353. | ||
Mirza MU, Ghori NU, Ikram N, Adil AR, Manzoor S. Pharmacoinformatics approach for investigation of alternative potential hepatitis C virus nonstructural protein 5B inhibitors. Drug Des Devel Ther. 2015; 9:1825–1841. | ||
Mirza MU, Ikram N. Integrated computational approach for virtual hit identification against Ebola viral proteins VP35 and VP40. Int J Mol Sci. 2016;17(11):pii: E1748. | ||
Mirza MU, Rafique S, Ali A, et al. Towards peptide vaccines against Zika virus: immunoinformatics combined with molecular dynamics simulations to predict antigenic epitopes of Zika viral proteins. Sci Rep. 2016; 6:37313. | ||
Mirza MU, Mirza AH, Ghori NU, Ferdous S. Glycyrrhetinic acid and E.resveratroloside act as potential plant derived compounds against dopamine receptor D3 for Parkinson’s disease: a pharmacoinformatics study. Drug Des Devel Ther. 2014;9:187–198. | ||
Ferdous S, Mirza MU, Saeed U. Docking studies reveal phytochemicals as the long searched anticancer drugs for breast cancer. Int J Comp Appl. 2013;67(25):1–5. | ||
Sehgal SA, Khattak NA, Mir A. Structural, phylogenetic and docking studies of D-amino acid oxidase activator (DAOA), a candidate schizophrenia gene. Theor Biol Med Model. 2013;10:3. | ||
Sehgal SA, Tahir RA, Shafique S, Hassan M, Rashid S. Molecular modeling and docking analysis of CYP1A1 associated with head and neck cancer to explore its binding regions. J Theor Comput Sci. 2014;1(112):2. | ||
Ahmed B, Ali Ashfaq U, Usman Mirza M. Medicinal plant phytochemicals and their inhibitory activities against pancreatic lipase: molecular docking combined with molecular dynamics simulation approach. Nat Prod Res. Epub 2017 Apr 26. | ||
Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. | ||
Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, Liang J. CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006;34(Web Server issue):W116–W118. | ||
Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. | ||
Accelrys Software Inc. Discovery Studio Modeling Environment (release 4.0). San Diego, CA: Accelrys Software Inc.; 2013. | ||
Kiss R, Sandor M, Szalai FA. http://Mcule.com: a public web service for drug. Drug Discov. 2012;4(1):17. | ||
Moroy G, Martiny VY, Vayer P, Villoutreix BO, Miteva MA. Toward in silico structure-based ADMET prediction in drug discovery. Drug Discov Today. 2012;17(1–2):44–55. | ||
Oldendorf WH. Lipid solubility and drug penetration of the blood brain barrier. Proc Soc Exp Biol Med. 1974;147(3):813–815. | ||
Egan WJ, Merz KM Jr, Baldwin JJ. Prediction of drug absorption using multivariate statistics. J Med Chem. 2000;43(21):3867–3877. | ||
Cheng A, Merz KM Jr. Prediction of aqueous solubility of a diverse set of compounds using quantitative structure-property relationships. J Med Chem. 2003;46(17):3572–3580. | ||
Susnow RG, Dixon SL. Use of robust classification techniques for the prediction of human cytochrome P450 2D6 inhibition. J Chem Inf Comput Sci. 2003;43(4):1308–1315. | ||
Boyer S, Arnby CH, Carlsson L, Smith J, Stein V, Glen RC. Reaction site mapping of xenobiotic biotransformations. J Chem Inf Model. 2007;47(2):583–590. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.