Back to Archived Journals » Advances and Applications in Bioinformatics and Chemistry » Volume 10
Structural and functional characterization of the Helicobacter pylori cytidine 5'-monophosphate-pseudaminic acid synthase PseF: molecular insight into substrate recognition and catalysis mechanism
Authors Wahid SUH ![]()
Received 16 April 2017
Accepted for publication 4 August 2017
Published 6 October 2017 Volume 2017:10 Pages 79—88
DOI https://doi.org/10.2147/AABC.S139773
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Juan Fernandez-Recio
Syeda Umme Habiba Wahid
Department of Microbiology, University of Chittagong, Chittagong, Bangladesh
Abstract: The bacterium Helicobacter pylori is a human gastric pathogen that can cause a wide range of diseases, including chronic gastritis, peptic ulcer and gastric carcinoma. It is classified as a definitive (class I) human carcinogen by the International Agency for Research on Cancer. Flagella-mediated motility is essential for H. pylori to initiate colonization and for the development of infection in human beings. Glycosylation of the H. pylori flagellum with pseudaminic acid (Pse; 5,7-diacetamido-3,5,7,9-tetradeoxy-l-glycero-l-manno-nonulosonic acid) is essential for flagella assembly and function. The sixth step in the Pse biosynthesis pathway, activation of Pse by addition of a cytidine 5′-monophosphate (CMP) to generate CMP-Pse, is catalyzed by a metal-dependent enzyme pseudaminic acid biosynthesis protein F (PseF) using cytidine 5′-triphosphate (CTP) as a cofactor. No crystal–structural information for PseF is available. This study describes the first three-dimensional model of H. pylori PseF obtained using biocomputational tools. PseF harbors an α/β-type hydrolase fold with a β-hairpin (HP) dimerization domain. Comparison of PseF with other structural homologs allowed identification of crucial residues for substrate recognition and the catalytic mechanism. This structural information would pave the way to design novel therapeutics to combat bacterial infection.
Keywords: H. pylori, motility, glycosylation, homology modeling, pseudaminic acid
Introduction
Motility is crucial for the bacterium Helicobacter pylori to colonize in the human stomach and to develop an infection. More than half of the global population is infected with H. pylori.1 H. pylori can cause acute gastritis, duodenal ulcers, mucosa-associated lymphoid tissue lymphoma, gastric carcinoma2–6 and other extra-gastrointestinal diseases,7 including iron-deficiency anemia,8 idiopathic thrombocytopenic purpura,9 cardiovascular diseases,10 liver illness,11 pancreatic cancer,12 respiratory illness,13 skin diseases14 and diabetes.15 This is the first bacterium to be classified as a definitive human carcinogen by the International Agency for Research on Cancer.16 The current treatment for H. pylori eradication is the simultaneous use of a proton pump inhibitor with two or more broad-spectrum antibiotics. However, the emergence of multiple antibiotic resistance has narrowed down the choice of treatment.17 Therefore, identification and development of novel therapeutics to treat H. pylori infection is an urgent need.
H. pylori uses flagella-mediated motility to facilitate colonization in mucosa in the human stomach. H. pylori flagella need to be O-glycosylated with an unusual nine-carbon sugar pseudaminic acid (Pse) for proper assembly and function.18 Pse is synthesized in H. pylori via five enzymatic steps and then activated by addition of a cytidine 5′-monophosphate (CMP) before being transferred onto the flagellin. The activation reaction is catalyzed by a metal-dependent enzyme CMP-Pse synthase (also known as pseudaminic acid biosynthesis protein F [PseF], EC 2.7.7.38) using cytidine 5′-triphosphate (CTP) as a cofactor. This enzyme belongs to the nucleotide–diphosphate–sugar transferases superfamily.19 Insertional inactivation of the PseF gene in H. pylori resulted in the loss of formation of functional flagella and rendered the bacterium nonmotile.18 This suggests the importance of PseF for biosynthesis of flagella and bacterial virulence.18 Therefore, the Pse biosynthesis pathway is a potential target for the development of novel drugs targeting bacterial motility.
A functional homolog from Neisseria meningitidis CMP-5-N-acetylneuraminic acid synthetase (NmCNS; 24% sequence identity with H. pylori PseF [HpPseF]) catalyzes activation of N-acetylneuraminic acid (NeuAc) to form a CMP-neuraminic acid (CMP-Neu5Ac) in the presence of Mg2+ using CTP as a cofactor.20 The CMP-NeuAc is then transferred onto the sugar moiety that forms part of lipopolysaccharide and capsule, which are essential for bacterial virulence.21 These glycoproteins serve an important role in cellular development, immunity and other functions.22,23 Structural analysis of NmCNS structure revealed that it exists as a domain-swapped homodimer architecture consisting of a catalytic hydrolase fold with a short α-hairpin (HP) domain.20
The CMP-5-N-acetylneuraminic acid synthetase (CNS) enzymes follow ordered–sequential kinetic mechanism where the CTP binds first, which is followed by the sialic acid to catalyze the reaction.24 Horsfall et al24 reported that CNS enzymes require two Mg2+ ions for proper orientation of the substrates and activating the α-phosphate of CTP. An ordered solvent molecule has been proposed to serve as the general base for the reaction.20
Currently, to the best of knowledge, no crystallographic three-dimensional (3D) structural information on HpPseF is available. In the current study, the physicochemical, structural and functional properties of HpPseF along with its protein–protein interaction (PPI) network information were obtained via different in silico approaches. In addition, the modeled 3D structure is compared with other structurally characterized hydrolyzing enzymes to study the structure–activity relationship and identify crucial residues for substrate recognition and catalytic mechanism.
Materials and methods
Sequence retrieval, physicochemical properties and secondary structure analysis
The amino acid sequence of PseF from H. pylori P12 (UniProtKB id: B6JKQ2, 229 amino acids) was retrieved from the protein database of National Center for Biotechnology Information (NCBI). The ExPASy ProtParam tool was used to predict the physicochemical characteristics of HpPseF, and the results are listed in Table 1.25 The computed physicochemical parameters included molecular mass, isoelectric point, extinction coefficient, instability index, aliphatic index and grand average of hydropathicity (GRAVY). The CYS_REC tool from SoftBerry and the CONCORD26 server were used to predict the existence of disulfide bridges and secondary structural components of HpPseF, respectively.
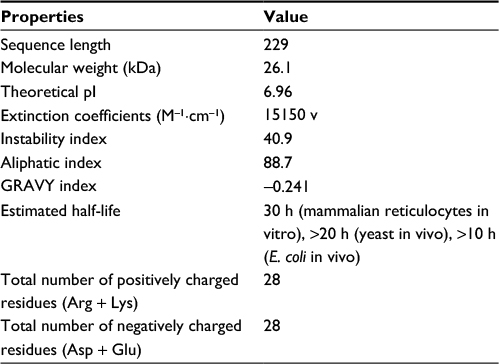 | Table 1 Physicochemical properties of the HpPseF Abbreviations: HpPseF, Helicobacter pylori pseudaminic acid biosynthesis protein F; pI, isoelectric point; GRAVY, grand average of hydropathicity; E. coli, Escherichia coli. |
TM helix prediction and subcellular localization
The subcellular localization of HpPseF was predicted by the consensus web server PSORTb.27 Membrane topology and transmembrane (TM) helix of HpPseF were determined by the TOPCONS server.28
Model building, refinement and validation
The HpPseF model was predicted with the following servers: IntFOLD,29 I-TASSER,30 SWISS-MODEL,31 RaptorX,32 Phyre233 and M4T.34 The quality of the predicted models was determined by the protein structure and model assessment tools in the SWISS-MODEL workspace.31,35 The best model was selected according to the QMEAN4 Z-score, which is a linear combination of four geometrical descriptors of protein.36 Model refinement was performed by the 3Drefine web server.37 The final model was validated using the ProQ38 and Verify3D programs.39 The accuracy and stereochemical features of the final 3D model were checked by the RAMPAGE40 and ERRAT41 servers. PPI networks analysis was carried out by the STRING database.42 The HpPseF model was achieved in the Protein Model Database under the access code of PM0080987. Structure figures were prepared using PYMOL (http://www.pymol.org/).
Ligand docking
The HpPseF model was used for detailed structural analysis and prediction of the ligand-binding site using the 3DLigandSite43 and I-TASSER30 web servers. The CTP was modeled in the ligand-binding pocket. The program LigPlot+ was used to depict ligand-binding residues of HpPseF.44
Results and discussion
Physicochemical and functional characterization
Analysis of the physicochemical characteristics of HpPseF revealed that the protein is neutral (estimated isoelectric point [pI] = 7.0) with a molecular weight of 26115 Da (Table 1). Analysis of the amino acid composition of HpPseF revealed that leucine is the most predominant amino acid (12.2%). The instability index of the protein is >40, which suggests that the protein would be less stable in solution. The protein is expected to be cytoplasmic and harbors no TM helix. The computed negative GRAVY index (-0.24) suggests that HpPseF is hydrophobic and most likely to be insoluble in water. Therefore, expression of the protein for X-ray crystallographic studies would be difficult. No disulfide bonding pattern among cysteine (Cys) residues has been predicted in HpPseF. Estimation of the secondary structure element revealed that HpPseF is an α/β protein and consists of mainly α-helices (45%), followed by the random coil (20%), extended β-strands (27%) and β-turn (8%).
Model building and refinement
The HpPseF models were predicted using different web servers (Table 2). The best model was selected based on the global QMEAN4 Z-score. The global QMEAN4 Z-score is an estimate of the absolute quality of a model and calculated based on the structure comparison between the modeled structure and high-resolution X-ray structures obtained from the protein database. Structure with QMEAN4 Z-score equivalent to 1 is similar to X-ray-diffracted crystal structure. The detailed analysis of the predicted models is presented in Table 2. RaptorX generated a model with a QMEAN4 Z-score of -3.10 (Table 2).29 The RaptorX model 1 was further refined and optimized by the 3Drefine web tool.37 The estimated QMEAN4 Z-score of the refined HpPseF model was -2.65 (Figure 1).
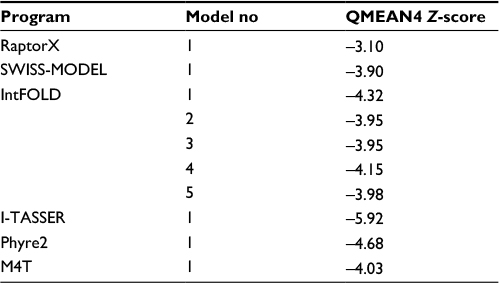 | Table 2 Comparison of the predicted models of the HpPseF Abbreviation: HpPseF, Helicobacter pylori pseudaminic acid biosynthesis protein F. |
 | Figure 1 Quality analysis of the refined 3D model of PseF from H. pylori. Notes: (A) The QMEAN quality analysis of the HpPseF 3D model (presented by a red star) is compared to the X-ray crystallographically determined structures of similar size proteins. (B) The QMEAN4 Z-score of the final HpPseF model. Abbreviations: 3D, three-dimensional; PseF, pseudaminic acid biosynthesis protein F; H. pylori, Helicobacter pylori; HpPseF, Helicobacter pylori pseudaminic acid biosynthesis protein F; PDB, Protein Data Bank. |
Model validation
The stereochemical properties of the HpPseF model was analyzed by using two web servers, ProQ38 and Verify3D.39 The HpPseF model showed a ProQ-LG score of 4.48, which suggests that the quality of the refined model is very good. Analysis with Verify3D revealed that >79% of the residues in the model had scored ≥0.2 in the 3D/one-dimensional (1D) profile, indicating that residues are in the favorable region (Figure 2).
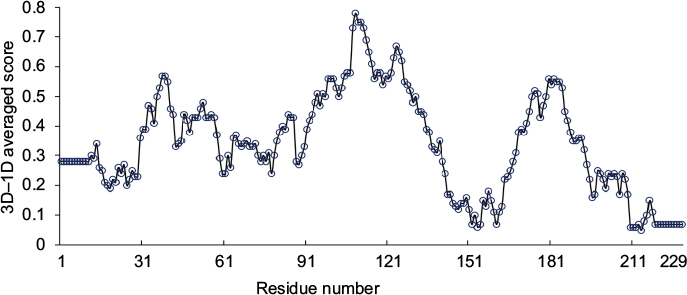 | Figure 2 The 3D scores of the HpPseF model calculated by the Verify3D server. Abbreviations: 3D, three-dimensional; HpPseF, Helicobacter pylori pseudaminic acid biosynthesis protein F; 1D, one-dimensional. |
The HpPseF model was further evaluated for overall quality, backbone torsion angles and side chain interaction energies. The overall quality of the model was assessed by the ProSA-web server.45 The estimated Z-score of -6.83 is in the range of structures of proteins of similar size determined by X-ray crystallography. The distribution of backbone torsion angles was checked by the RAMPAGE (Figure 3).40 A total of 95% of the residues (n = 215) were in the favored region, and the other 5% of the residues (n = 12) were present in the allowed region. Errors in non-bonded interactions were calculated by the ERRAT server,41 and the obtained score of 94.11 means that 94% of the residues in the model were below the rejection limit (experimental structures had scores ~90%; Figure 4). Altogether, this suggests that the final HpPseF model is of a good quality and could be used for further studies.
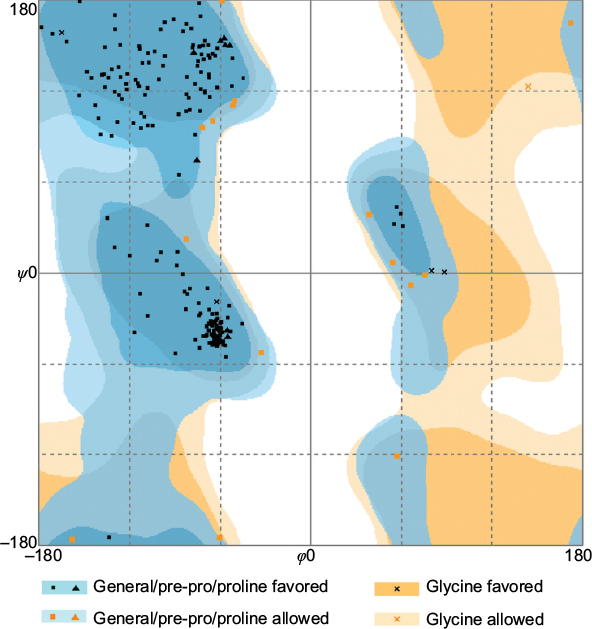 | Figure 3 Ramachandran plot for the modeled PseF from H. pylori. Note: The dihedral angles ϕ and ψ were measured in degrees. Abbreviations: PseF, pseudaminic acid biosynthesis protein F; H. pylori, Helicobacter pylori. |
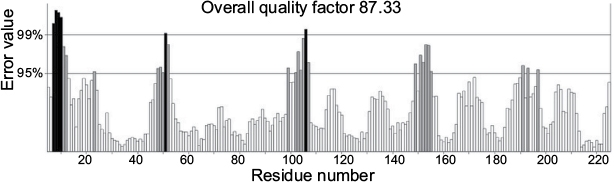 | Figure 4 ERRAT result shows the overall quality factor of the HpPseF model (error-axis represents the error values to reject regions that exceed error value). Abbreviation: HpPseF, Helicobacter pylori pseudaminic acid biosynthesis protein F. |
Overall structure of HpPseF and comparison to other homologs
The HpPseF has an α/β-hydrolase domain with an ~35-residue inserted β-HP domain. The core domain is a αβα three-layer sandwich architecture consisting of seven β-strands with the topological order (↑β3-↑β2-↑β1-↑β4-↓β8-↑β5-↑β9; Figure 5). The central β-sheet is flanked by four α-helices (α1, α2, α5 and α9) on one side and three α-helices (α3, α4 and α8) on the other side. The loop between strand β1 and helix α1 is known as P-loop (Figure 5), which interacts with the pyrophosphate arm of the cofactor CTP in other structural homologs.20,46 The HP domain protrudes out from the central β-sheet and contains two antiparallel β-strands (β6 and β7) and two α-helices (α6 and α7). In other structurally characterized CNS enzymes, the HP domain plays an important role in the formation of a homodimer.20,46
 | Figure 5 The overall structural fold of HpPseF. Notes: (A) Stereo diagram of the HpPseF structure. β-strands and α-helices are colored as green and red, respectively. The P-loop is colored cyan. The figure was prepared using PyMOL. (B) Secondary structure profile of PseF from H. pylori. Abbreviations: HpPseF, Helicobacter pylori pseudaminic acid biosynthesis protein F; PseF, pseudaminic acid biosynthesis protein F; H. pylori, Helicobacter pylori; HP, hairpin. |
Comparison of PseF against the published structures deposited in the Protein Data Bank that have been described in the literature, using the Dali server,47 revealed that mammal (Mus musculus) CMP-5-N-acetylneuraminic acid synthetase (MmCNS; root-mean-square deviation [RMSD] = 1.3, sequence = 23%, Z-score = 32.5, PDB code: 1QWJ46) showed the highest similarity followed by NmCNS (RMSD = 2.2, sequence = 22%, Z-score = 28.1, PDB code: 1EYR;20 Figures 6 and 7). Detailed analysis of the superimpositions revealed that structural similarity extends over the entire fold and includes all the secondary structural elements and the HP domain.
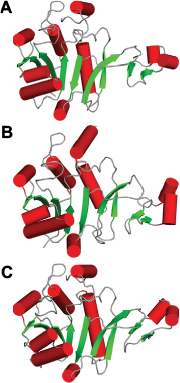 | Figure 6 Comparison of HpPseF with other hydrolyzing homologous enzymes. Notes: (A) HpPseF. (B) NmCNS. (C) MmCNS. α-helices, loops and β-strands are colored red, gray and green, respectively. Abbreviations: HpPseF, Helicobacter pylori pseudaminic acid biosynthesis protein F; NmCNS, Neisseria meningitidis CMP-5-N-acetylneuraminic acid synthetase; MmCNS, Mus musculus CMP-5-N-acetylneuraminic acid synthetase; CMP, cytidine 5′-monophosphate. |
Prediction of the ligand-binding pocket and ligand docking
The CASTp server48 predicted the putative ligand-binding site in the core of the hydrolase domain. The calculated surface area and volume of the pocket are ~438.2 Å2 and ~1711.6 Å3, respectively. Prediction of the ligand-binding residues in HpPseF was performed using the 3DLigandSite43 and I-TASSER servers.30 The ligand CTP was modeled at the putative pocket. Figure 8 shows residues that form interactions with the CTP-modeled structure. The pyrophosphate moiety interacts with the residues of the P-loop between β1 and α1, while the sugar moiety is accommodated by the residue from the β3–α3 region. The CTP forms hydrogen bonds and van der Waals interactions with six (Lys14, Lys19, Arg70, Ala75, Asp76 and Tyr104) and five residues (Leu8, Arg10, Ser13, Asn20 and Asp209), respectively. Prediction of the metal-binding site has revealed that Tyr104, Asp207 and Asp209 would likely interact with the metal cofactor. Sequence alignment of HpPseF with other homologs shows that residues involved in CTP binding are conserved among other homologous enzymes (Figure 7).
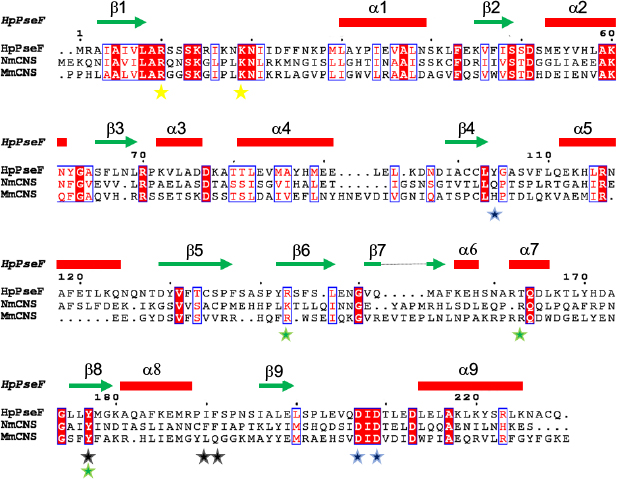 | Figure 7 A sequence alignment of HpPseF, NmCNS and MmCNS. Notes: The elements of the secondary structure and sequence numbering for HpPseF are shown above the alignment. Conserved residues are highlighted in red. Metal binding and catalytic residue are presented as blue stars. Yellow stars represent residues that interact with pyrophosphate arm of cofactor CTP. Residues that form a hydrophobic pocket to determine substrate specificity and contribute in proper positioning of the substrate NeuAc in NmCNS are represented as black and green stars. Abbreviations: HpPseF, Helicobacter pylori pseudaminic acid biosynthesis protein F; NmCNS, Neisseria meningitidis CMP-5-N-acetylneuraminic acid synthetase; MmCNS, Mus musculus CMP-5-N-acetylneuraminic acid synthetase; CMP, cytidine 5′-monophosphate; CTP, cytidine 5′-triphosphate; NeuAc, N-acetylneuraminic acid. |
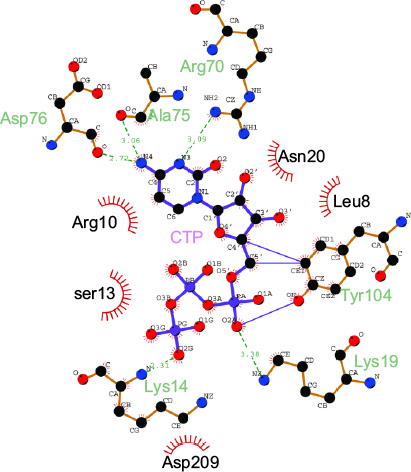 | Figure 8 Ligplot+ results showing the residues of PseF from H. pylori interacting with the modeled CTP molecule. Note: Residues making van der Waal’s contacts with CTP are indicated by red arcs with spokes radiating toward the ligand moieties they contact. Abbreviations: PseF, pseudaminic acid biosynthesis protein F; H. pylori, Helicobacter pylori; CTP, cytidine 5′-triphosphate. |
Analysis of the acceptor substrate-binding site and catalysis mechanism
The closest bacterial functional homolog NmCNS was solved in complex with CDP,20 while the murine homolog (MmCNS) was solved in complex with the reaction product CMP-NeuAc.46 Both CNS proteins form a homodimer, where the HP domain from one monomer interacts with the catalytic domain of the opposite monomer and contributes to the formation of the active site. Detailed structural analysis of the NmCNS active site revealed that the enzyme has a hydrophobic pocket formed by the residues (Tyr179, Phe192 and Phe193) to aid binding of the methyl group of the N-acetyl moiety of acceptor substrate Neu5Ac and thus determines substrate specificity.24 Alanine substitution of these residues resulted in the greatest loss of enzyme activity and thus confirmed their crucial role in substrate recognition.24 The HpPseF model also adopts a hydrophobic pocket consisting of Y177, I190 and F191, which would likely play a crucial role in the determination of the substrate specificity (Figures 7 and 9).
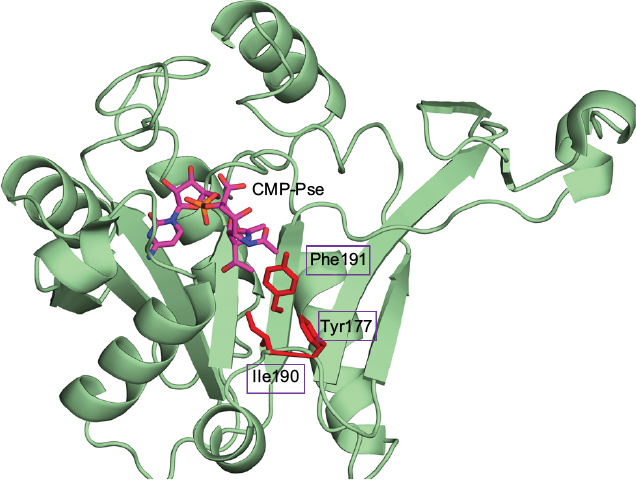 | Figure 9 Substrate binding pocket of HpPseF. Notes: CMP-Pse, modeled in the active site of PseF, is colored by atom type with the carbon set as magenta. Predicted residues that would likely determine substrate specificity are represented as red sticks. Abbreviations: HpPseF, Helicobacter pylori pseudaminic acid biosynthesis protein F; CMP, cytidine 5′-monophosphate; Pse, pseudaminic acid; PseF, pseudaminic acid biosynthesis protein F. |
The CNS enzymes require two metal ions (preferably, manganese Mg2+) to catalyze the condensation reaction.24 In NmCNS and MmCNS, two Asp residues (D211 and D209) are involved in binding the catalytic Mg2+ ion,20 while another Gln residue (Q104 in NmCNS and Q141 in MmCNS) is believed to play a crucial role in binding the second metal ion at the intermediate stage of the reaction. A mutagenesis study has confirmed their role in catalysis.24 Structural and sequence alignment analyses have shown that HpPseF harbors two Asp residues (D207 and D209) and one Tyr residue (Y104) in the corresponding positions (Figures 7, 10 and 11).
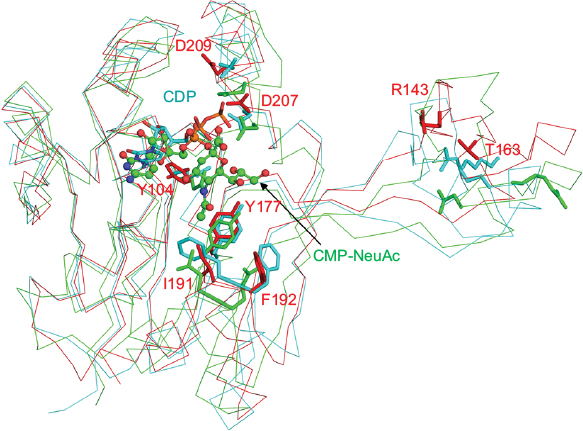 | Figure 10 Cartoon representation of the superimposed structures of PseF from H. pylori (red), CNS from N. meningitis (Cyan) and CNS from M. musculus (green). Notes: The CDP cofactor is drawn as sticks, whereas the reaction product, CMP-NeuAc, is shown using ball-and-stick representation. The predicted important residues for substrate recognition and catalysis in the HpPseF model are shown as red sticks. Corresponding residues of NmCNS and MmCNS are shown as cyan and green sticks, respectively. Abbreviations: PseF, pseudaminic acid biosynthesis protein F; H. pylori, Helicobacter pylori; CNS, CMP-5-N-acetylneuraminic acid synthetase; N. meningitis, Neisseria meningitidis; M. musculus, Mus musculus; CMP, cytidine 5′-monophosphate; NeuAc, N-acetylneuraminic acid; HpPseF, Helicobacter pylori pseudaminic acid biosynthesis protein F; NmCNS, Neisseria meningitidis CMP-5-N-acetylneuraminic acid synthetase; MmCNS, Mus musculus CMP-5-N-acetylneuraminic acid synthetase. |
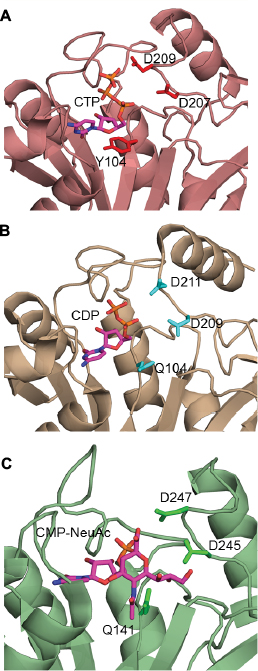 | Figure 11 Metal-binding residues of the HpPseF and other enzymes. Notes: (A) HpPseF (CTP modeled). (B) NmCNS in complex with CDP and (C) M. musculus CNS in complex with CMP-Neu5Ac. CTP, CDP and CMP-Neu5Ac in the active sites are colored by atom type with the carbon set as magenta. Abbreviations: HpPseF, Helicobacter pylori pseudaminic acid biosynthesis protein F; CTP, cytidine 5′-triphosphate; NmCNS, Neisseria meningitidis CMP-5-N-acetylneuraminic acid synthetase; M. musculus, Mus musculus; CNS, CMP-5-N-acetylneuraminic acid synthetase, CMP-Neu5Ac, CMP 5-N-acetylneuraminic acid; CMP, cytidine 5′-monophosphate. |
The CNS enzymes follow an ordered bi–bi sequential kinetic mechanism where binding of a CTP with the enzyme is followed by sialic acid to generate CMP-NeuAc.24,46 The role of both Mg2+ ions is believed to be in proper orientation of the substrates and activation of the α-phosphate moiety of CTP. The sugar hydroxyl group of NeuAc is activated by the catalytic Mg2+ ion.24 The structural similarities among HpPseF and other well-characterized homologs (NmCNS and MmCNS) suggest that HpPseF would likely follow the same sequential catalytic mechanism (Michaelis–Menten equation) and catalyze the reaction via formation of a tetrahedral intermediate.
Protein network analysis
Analysis of PPI networks is necessary for better understanding of intricate molecular mechanisms and identification of novel therapeutics to combat multidrug-resistant bacteria. Figure 12 shows PPI partner proteins of HpPseF derived from the STRING database. The functional partners are mainly flagella glycosylation pathway proteins (PseB, PseC, PseH, HpPseG, PseI)19,49 and flagellar basal body L-ring protein (FlgH). In addition, HpPseF interacts with other proteins including polysialic acid capsule expression protein (KpsF), 50S ribosomal protein L34 (RpmH) and three hypothetical proteins (HP0465; HP0114, HP1570).
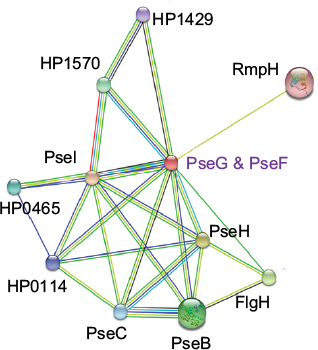 | Figure 12 PPI network of HpPseF derived from the STRING database. Abbreviations: PPI, protein–protein interaction; HpPseF, Helicobacter pylori pseudaminic acid biosynthesis protein F. |
Conclusion
The present study presents physicochemical properties, structural and functional analysis and crucial residues of substrate recognition and the catalytic mechanism of HpPseF derived by using a broad range of biocomputational tools. The deduced structural information would serve as a fundamental basis to design in vivo biochemical and X-ray crystallographic assays for a better understanding of the catalytic mechanism and molecular biology of Pse and sialic acid synthase enzymes. The HpPseF 3D model would pave the way for the design and production of inhibitors and mimetics of Pse and sialic acid biosynthesis pathway enzymes targeting motility to combat drug-resistant bacterial infection in the near future.
Disclosure
The author reports no conflicts of interest in this work.
References
Dunn B, Cohen H, MJ B. Helicobacter pylori. Clin Microbiol Rev. 1997;10(4):720–741. | ||
Marshall B, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet. 1984;323(8390):1311–1315. | ||
Wroblewski LE, Peek RM, Wilson KT. Helicobacter pylori and gastric cancer: factors that modulate disease risk. Clin Microbiol Rev. 2010;23(4):713–739. | ||
Kusters JG, van Vliet AH, Kuipers EJ. Pathogenesis of Helicobacter pylori infection. Clin Microbiol Rev. 2006;19(3):449–490. | ||
Ernst PB, Gold BD. The disease spectrum of Helicobacter pylori: the immunopathogenesis of gastroduodenal ulcer and gastric cancer. Annu Rev Microbiol. 2000;54(1):615–640. | ||
Testerman TL, Morris J. Beyond the stomach: an updated view of Helicobacter pylori pathogenesis, diagnosis, and treatment. World J Gastroenterol. 2014;20(36):12781–12808. | ||
Goni E, Franceschi F. Helicobacter pylori and extragastric diseases. Helicobacter. 2016;21(S1):45–48. | ||
Capurso G, Lahner E, Marcheggiano A, et al. Involvement of the corporal mucosa and related changes in gastric acid secretion characterize patients with iron deficiency anaemia associated with Helicobacter pylori infection. Aliment Pharmacol Ther. 2001;15(11):1753–1761. | ||
Pellicano R, Franceschi F, Saracco G, Fagoonee S, Roccarina D, Gasbarrini A. Helicobacters and extragastric diseases. Helicobacter. 2009;14(s1):58–68. | ||
Vafaeimanesh J, Hejazi SF, Damanpak V, Vahedian M, Sattari M, Seyyedmajidi M. Association of Helicobacter pylori infection with coronary artery disease: is Helicobacter pylori a risk factor? ScientificWorldJournal. 2014;2014:516354. | ||
Pirouz T, Zounubi L, Keivani H, Rakhshani N, Hormazdi M. Detection of Helicobacter pylori in paraffin-embedded specimens from patients with chronic liver diseases, using the amplification method. Dig Dis Sci. 2009;54(7):1456–1459. | ||
Schulte A, Pandeya N, Fawcett J, et al. Association between Helicobacter pylori and pancreatic cancer risk: a meta-analysis. Cancer Causes Control. 2015;26(7):1027–1035. | ||
Wang L, Guan Y, Li Y, et al. Association between chronic respiratory diseases and Helicobacter pylori: a meta-analysis. Arch Bronconeumol. 2015;51(6):273–278. | ||
Argenziano G, Donnarumma G, Iovene MR, Arnese P, Assunta Baldassarre M, Baroni A. Incidence of anti-Helicobacter pylori and anti-CagA antibodies in rosacea patients. Int J Dermatol. 2003;42(8):601–604. | ||
Zhou X, Zhang C, Wu J, Zhang G. Association between Helicobacter pylori infection and diabetes mellitus: a meta-analysis of observational studies. Diabetes Res Clin Pract. 2013;99(2):200–208. | ||
IARC. Infection with Helicobacter pylori. IARC Monogr Eval Carcinog Risks Hum. 1994;61:177–240. | ||
Graham DY, Fischbach L. Helicobacter pylori treatment in the era of increasing antibiotic resistance. Gut. 2010;59(8):1143–1153. | ||
Schirm M, Soo E, Aubry A, Austin J, Thibault P, Logan S. Structural, genetic and functional characterization of the flagellin glycosylation process in Helicobacter pylori. Mol Microbiol. 2003;48(6):1579–1592. | ||
Schoenhofen IC, McNally DJ, Brisson JR, Logan SM. Elucidation of the CMP-pseudaminic acid pathway in Helicobacter pylori: synthesis from UDP-N-acetylglucosamine by a single enzymatic reaction. Glycobiology. 2006;16(9):8C–14C. | ||
Mosimann SC, Gilbert M, Dombroswki D, To R, Wakarchuk W, Strynadka NC. Structure of a sialic acid-activating synthetase, CMP-acylneuraminate synthetase in the presence and absence of CDP. J Biol Chem. 2001;276(11):8190–8196. | ||
Bravo IG, Garcı́a-Vallvé S, Romeu A, Reglero Á. Prokaryotic origin of cytidylyltransferases and α-keto acid synthases. Trends Microbiol. 2004;12(3):120–128. | ||
Sellmeier M, Weinhold B, Münster-Kühnel A. CMP-sialic acid synthetase: the point of constriction in the sialylation pathway. SialoGlyco Chemistry and Biology I. Berlin Heidelberg: Springer; 2013:139–167. | ||
Traving C, Schauer R. Structure, function and metabolism of sialic acids. Cell Mol Life Sci. 1998;54(12):1330–1349. | ||
Horsfall LE, Nelson A, Berry A. Identification and characterization of important residues in the catalytic mechanism of CMP-Neu5Ac synthetase from Neisseria meningitidis. FEBS J. 2010;277(13):2779–2790. | ||
Gasteiger E, Hoogland C, Gattiker A, et al. Protein identification and analysis tools on the ExPASy server. In: Walker JM, editor. The Proteomics Protocols Handbook. Totowa, NJ: Humana Press; 2005:571–607. | ||
Wei Y, Thompson J, Floudas C. CONCORD: a consensus method for protein secondary structure prediction via mixed integer linear optimization. Proc R Soc A. 2011;468:831–850. | ||
Nancy YY, Wagner JR, Laird MR, et al. PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics. 2010;26(13):1608–1615. | ||
Tsirigos KD, Peters C, Shu N, Kall L, Elofsson A. The TOPCONS web server for consensus prediction of membrane protein topology and signal peptides. Nucleic Acids Res. 2015;43(W1):W401–W407. | ||
McGuffin LJ, Atkins JD, Salehe BR, Shuid AN, Roche DB. IntFOLD: an integrated server for modelling protein structures and functions from amino acid sequences. Nucleic Acids Res. 2015;43(W1):W169–W173. | ||
Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5(4):725–738. | ||
Biasini M, Bienert S, Waterhouse A, et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42(Web Server issue):gku340. | ||
Källberg M, Margaryan G, Wang S, Ma J, Xu J. RaptorX server: a resource for template-based protein structure modeling. Methods Mol Biol. 2014;1137(17–27):17–27. | ||
Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10(6):845–858. | ||
Fernandez-Fuentes N, Madrid-Aliste CJ, Rai BK, Fajardo JE, Fiser A. M4T: a comparative protein structure modeling server. Nucleic Acids Res. 2007;35(Suppl 2):W363–W368. | ||
Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22(2):195–201. | ||
Benkert P, Künzli M, Schwede T. QMEAN server for protein model quality estimation. Nucleic Acids Res. 2009;37:W510–W514. | ||
Bhattacharya D, Cheng J. 3Drefine: consistent protein structure refinement by optimizing hydrogen bonding network and atomic-level energy minimization. Proteins. 2013;81(1):119–131. | ||
Wallner B, Elofsson A. Can correct protein models be identified? Protein Sci. 2003;12(5):1073–1086. | ||
Eisenberg D, Lüthy R, Bowie JU. Verify3D: assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997;277:396–404. | ||
Lovell S, Davis I, Arendall W, et al. Structure validation by Cα geometry: φ/ψ and Cβ deviation. Proteins. 2002;50:437–450. | ||
Colovos C, Yeates T. ERRAT: an empirical atom-based method for validating protein structures. Protein Sci. 1993;2:1511–1519. | ||
Szklarczyk D, Franceschini A, Wyder S, et al. STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43(D1):D447–D452. | ||
Wass MN, Kelley LA, Sternberg MJ. 3DLigandSite: predicting ligand-binding sites using similar structures. Nucleic Acids Res. 2010;38(Web Server issue):W469–W473. | ||
Laskowski RA, Swindells MB. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51(10):2778–2786. | ||
Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35(Suppl 2):W407–W410. | ||
Krapp S, Münster-Kühnel AK, Kaiser JT, et al. The crystal structure of murine CMP-5-N-acetylneuraminic acid synthetase. J Mol Biol. 2003;334(4):625–637. | ||
Holm L, Rosenström P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38(Web Server issue):W545–W549. | ||
Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, Liang J. CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006;34(Suppl 2):W116–W118. | ||
Liu F, Tanner ME. PseG of pseudaminic acid biosynthesis a UDP-sugar hydrolase as a masked glycosyltransferase. J Biol Chem. 2006;281(30):20902–20909. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.